
Antisense Oligonucleotide-Based Rescue of Aberrant Splicing Defects Caused by 15 Pathogenic Variants in ABCA4

 ,
,  , and
, and
Abstract
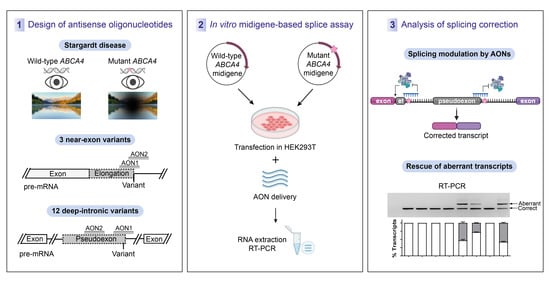
:
1. Introduction
2. Results
2.1. Design of Antisense Oligonucleotides
2.2. AON-Driven Splicing Modulation for Near-Exon Variants
2.3. AON-Driven Splicing Modulation for Deep-Intronic Variants
3. Discussion
4. Materials and Methods
4.1. AON Design
4.2. Generation of ABCA4 Mutant Midigenes
4.3. In Vitro Splice Assay in HEK293T Cells
4.4. In Vitro AON Rescue Studies Using HEK293T Cells
4.5. RNA Isolation and RT-PCR Analysis
4.6. Statistical Analysis
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Westeneng-van Haaften, S.C.; Boon, C.J.; Cremers, F.P.; Hoefsloot, L.H.; den Hollander, A.I.; Hoyng, C.B. Clinical and genetic characteristics of late-onset Stargardt’s disease. Ophthalmology 2012, 119, 1199–1210. [Google Scholar] [CrossRef]
- Lambertus, S.; Lindner, M.; Bax, N.M.; Mauschitz, M.M.; Nadal, J.; Schmid, M.; Schmitz-Valckenberg, S.; Hollander, A.I.D.; Weber, B.H.F.; Holz, F.G.; et al. Progression of Late-Onset Stargardt Disease. Investig. Ophthalmol. Vis. Sci. 2016, 57, 5186–5191. [Google Scholar] [CrossRef]
- Zernant, J.; Lee, W.; Collison, F.T.; A Fishman, G.; Sergeev, Y.V.; Schuerch, K.; Sparrow, J.R.; Tsang, S.H.; Allikmets, R. Frequent hypomorphic alleles account for a significant fraction of ABCA4 disease and distinguish it from age-related macular degeneration. J. Med. Genet. 2017, 54, 404–412. [Google Scholar] [CrossRef]
- Runhart, E.H.; Sangermano, R.; Cornelis, S.S.; Verheij, J.B.G.M.; Plomp, A.S.; Boon, C.J.F.; Lugtenberg, D.; Roosing, S.; Bax, N.M.; Blokland, E.A.W.; et al. The Common ABCA4 Variant p.Asn1868Ile Shows Nonpenetrance and Variable Expression of Stargardt Disease When Present in trans with Severe Variants. Investig. Ophthalmol. Vis. Sci. 2018, 59, 3220–3231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zernant, J.; Lee, W.; Nagasaki, T.; Collison, F.T.; Fishman, G.A.; Bertelsen, M.; Rosenberg, T.; Gouras, P.; Tsang, S.H.; Allikmets, R. Extremely hypomorphic and severe deep intronic variants in the ABCA4 locus result in varying Stargardt disease phenotypes. Mol. Case Stud. 2018, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Runhart, E.H.; Valkenburg, D.; Cornelis, S.S.; Khan, M.; Sangermano, R.; Albert, S.; Bax, N.M.; Astuti, G.D.N.; Gilissen, C.; Pott, J.-W.R.; et al. Late-Onset Stargardt Disease Due to Mild, Deep-Intronic ABCA4 Alleles. Investig. Ophthalmol. Vis. Sci. 2019, 60, 4249–4256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cremers, F.P.; Lee, W.; Collin, R.W.; Allikmets, R. Clinical spectrum, genetic complexity and therapeutic approaches for retinal disease caused by ABCA4 mutations. Prog. Retin. Eye Res. 2020, 79, 100861. [Google Scholar] [CrossRef] [PubMed]
- Illing, M.; Molday, L.L.; Molday, R.S. The 220-kDa Rim Protein of Retinal Rod Outer Segments Is a Member of the ABC Transporter Superfamily. J. Biol. Chem. 1997, 272, 10303–10310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasiliou, V.; Vasiliou, K.; Nebert, D.W. Human ATP-binding cassette (ABC) transporter family. Hum. Genom. 2008, 3, 281–290. [Google Scholar] [CrossRef]
- Molday, R.S. Insights into the Molecular Properties of ABCA4 and Its Role in the Visual Cycle and Stargardt Disease. Prog. Mol. Biol. Transl. Sci. 2015, 134, 415–431. [Google Scholar] [CrossRef]
- Sparrow, J.R.; Wu, Y.; Kim, C.Y.; Zhou, J. Phospholipid meets all-trans-retinal: The making of RPE bisretinoids. J. Lipid Res. 2010, 51, 247–261. [Google Scholar] [CrossRef] [Green Version]
- Taveau, N.; Cubizolle, A.; Guillou, L.; Pinquier, N.; Moine, E.; Cia, D.; Kalatzis, V.; Vercauteren, J.; Durand, T.; Crauste, C.; et al. Preclinical pharmacology of a lipophenol in a mouse model of light-induced retinopathy. Exp. Mol. Med. 2020, 52, 1090–1101. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Msc, S.S.C.; Msc, M.D.P.-V.; Whelan, L.; Runhart, E.H.; Mishra, K.; Bults, F.; AlSwaiti, Y.; AlTalbishi, A.; De Baere, E.; et al. Resolving the dark matter of ABCA4 for 1054 Stargardt disease probands through integrated genomics and transcriptomics. Genet. Med. 2020, 22, 1235–1246. [Google Scholar] [CrossRef]
- Bax, N.M.; Sangermano, R.; Roosing, S.; Thiadens, A.A.; Hoefsloot, L.H.; Born, L.I.V.D.; Phan, M.; Klevering, B.J.; Haaften, C.W.-V.; Braun, T.A.; et al. Heterozygous Deep-Intronic Variants and Deletions inABCA4in Persons with Retinal Dystrophies and One ExonicABCA4Variant. Hum. Mutat. 2014, 36, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Cornelis, S.S.; Bax, N.M.; Zernant, J.; Allikmets, R.; Fritsche, L.G.; Dunnen, J.T.D.; Ajmal, M.; Hoyng, C.B.; Cremers, F.P. In SilicoFunctional Meta-Analysis of 5,962ABCA4Variants in 3,928 Retinal Dystrophy Cases. Hum. Mutat. 2017, 38, 400–408. [Google Scholar] [CrossRef]
- Sangermano, R.; Khan, M.; Cornelis, S.S.; Richelle, V.; Albert, S.; Garanto, A.; Elmelik, D.; Qamar, R.; Lugtenberg, D.; Born, L.I.V.D.; et al. ABCA4 midigenes reveal the full splice spectrum of all reported noncanonical splice site variants in Stargardt disease. Genome Res. 2017, 28, 100–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fadaie, Z.; Khan, M.; Del Pozo-Valero, M.; Cornelis, S.S.; Ayuso, C.; Cremers, F.P.M.; Roosing, S. The ABCA4 study group Identification of splice defects due to noncanonical splice site or deep-intronic variants in ABCA4. Hum. Mutat. 2019, 40, 2365–2376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sangermano, R.; Garanto, A.; Khan, M.; Runhart, E.H.; Bauwens, M.; Bax, N.M.; Born, L.I.V.D.; Khan, M.I.; Msc, S.S.C.; Verheij, J.B.G.M.; et al. Deep-intronic ABCA4 variants explain missing heritability in Stargardt disease and allow correction of splice defects by antisense oligonucleotides. Genet. Med. 2019, 21, 1751–1760. [Google Scholar] [CrossRef] [Green Version]
- Zernant, J.; Xie, Y.; Ayuso, C.; Riveiro-Alvarez, R.; Lopez-Martinez, M.-A.; Simonelli, F.; Testa, F.; Gorin, M.B.; Strom, S.P.; Bertelsen, M.; et al. Analysis of the ABCA4 genomic locus in Stargardt disease. Hum. Mol. Genet. 2014, 23, 6797–6806. [Google Scholar] [CrossRef]
- Jaganathan, K.; Panagiotopoulou, S.K.; McRae, J.F.; Darbandi, S.F.; Knowles, D.; Li, Y.I.; Kosmicki, J.A.; Arbelaez, J.; Cui, W.; Schwartz, G.B.; et al. Predicting Splicing from Primary Sequence with Deep Learning. Cell 2019, 176, 535–548.e24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collin, R.W.; Hollander, A.I.D.; van der Velde-Visser, S.D.; Bennicelli, J.; Bennett, J.; Cremers, F.P. Antisense Oligonucleotide (AON)-based Therapy for Leber Congenital Amaurosis Caused by a Frequent Mutation in CEP290. Mol. Ther.-Nucleic Acids 2012, 1, e14. [Google Scholar] [CrossRef]
- Gerard, X.; Perrault, I.; Hanein, S.; Silva, E.; Bigot, K.; Defoort-Delhemmes, S.; Rio, M.; Munnich, A.; Scherman, D.; Kaplan, J.; et al. AON-mediated Exon Skipping Restores Ciliation in Fibroblasts Harboring the Common Leber Congenital Amaurosis CEP290 Mutation. Mol. Ther.-Nucleic Acids 2012, 1, e29. [Google Scholar] [CrossRef] [PubMed]
- Garanto, A.; Chung, D.C.; Duijkers, L.; Corral-Serrano, J.C.; Messchaert, M.; Xiao, R.; Bennett, J.; Vandenberghe, L.H.; Collin, R.W.J. In vitro and in vivo rescue of aberrant splicing in CEP290-associated LCA by antisense oligonucleotide delivery. Hum. Mol. Genet. 2016, 25, 2552–2563. [Google Scholar] [PubMed] [Green Version]
- Parfitt, D.A.; Lane, A.; Ramsden, C.M.; Carr, A.F.; Munro, P.M.; Jovanovic, K.; Schwarz, N.; Kanuga, N.; Muthiah, M.N.; Hull, S.; et al. Identification and Correction of Mechanisms Underlying Inherited Blindness in Human iPSC-Derived Optic Cups. Cell Stem Cell 2016, 18, 769–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duijkers, L.; Born, L.I.V.D.; Neidhardt, J.; Bax, N.M.; Pierrache, L.H.M.; Klevering, B.J.; Collin, R.W.J.; Garanto, A. Antisense Oligonucleotide-Based Splicing Correction in Individuals with Leber Congenital Amaurosis due to Compound Heterozygosity for the c.2991+1655A>G Mutation in CEP290. Int. J. Mol. Sci. 2018, 19, 753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dulla, K.; Aguila, M.; Lane, A.; Jovanovic, K.; Parfitt, D.A.; Schulkens, I.; Chan, H.L.; Schmidt, I.; Beumer, W.; Vorthoren, L.; et al. Splice-Modulating Oligonucleotide QR-110 Restores CEP290 mRNA and Function in Human c.2991+1655A>G LCA10 Models. Mol. Ther.-Nucleic Acids 2018, 12, 730–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Slijkerman, R.W.; Vaché, C.; Dona, M.; García-García, G.; Claustres, M.; Hetterschijt, L.; A Peters, T.; Hartel, B.P.; Pennings, R.J.; Millan, J.M.; et al. Antisense Oligonucleotide-based Splice Correction for USH2A-associated Retinal Degeneration Caused by a Frequent Deep-intronic Mutation. Mol. Ther.-Nucleic Acids 2016, 5, e381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garanto, A.; Van Der Velde-Visser, S.D.; Cremers, F.P.M.; Collin, R.W.J. Antisense Oligonucleotide-Based Splice Correction of a Deep-Intronic Mutation in CHM Underlying Choroideremia. Adv. Exp. Med. Biol. 2018, 1074, 83–89. [Google Scholar] [CrossRef]
- Bonifert, T.; Menendez, I.G.; Battke, F.; Theurer, Y.; Synofzik, M.; Schöls, L.; Wissinger, B. Antisense Oligonucleotide Mediated Splice Correction of a Deep Intronic Mutation in OPA1. Mol. Ther.-Nucleic Acids 2016, 5, e390. [Google Scholar] [CrossRef] [Green Version]
- Albert, S.; Garanto, A.; Sangermano, R.; Khan, M.; Bax, N.M.; Hoyng, C.B.; Zernant, J.; Lee, W.; Allikmets, R.; Collin, R.W.; et al. Identification and Rescue of Splice Defects Caused by Two Neighboring Deep-Intronic ABCA4 Mutations Underlying Stargardt Disease. Am. J. Hum. Genet. 2018, 102, 517–527. [Google Scholar] [CrossRef] [Green Version]
- Bauwens, M.; Garanto, A.; Sangermano, R.; Naessens, S.; Weisschuh, N.; De Zaeytijd, J.; Khan, M.; Sadler, F.; Balikova, I.; Van Cauwenbergh, C.; et al. ABCA4-associated disease as a model for missing heritability in autosomal recessive disorders: Novel noncoding splice, cis-regulatory, structural, and recurrent hypomorphic variants. Genet. Med. 2019, 21, 1761–1771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garanto, A.; Duijkers, L.; Tomkiewicz, T.Z.; Collin, R.W.J. Antisense Oligonucleotide Screening to Optimize the Rescue of the Splicing Defect Caused by the Recurrent Deep-Intronic ABCA4 Variant c.4539+2001G>A in Stargardt Disease. Genes 2019, 10, 452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khan, M.; Cornelis, S.S.; Khan, M.I.; Elmelik, D.; Manders, E.; Bakker, S.; Derks, R.; Neveling, K.; van de Vorst, M.; Gilissen, C.; et al. Cost-effective molecular inversion probe-based ABCA4 sequencing reveals deep-intronic variants in Stargardt disease. Hum. Mutat. 2019, 40, 1749–1759. [Google Scholar] [CrossRef] [PubMed]
- Zernant, J.; Collison, F.T.; Lee, W.; Fishman, G.A.; Noupuu, K.; Yuan, B.; Cai, C.; Lupski, J.R.; Yannuzzi, L.A.; Tsang, S.H.; et al. Genetic and Clinical Analysis ofABCA4-Associated Disease in African American Patients. Hum. Mutat. 2014, 35, 1187–1194. [Google Scholar] [CrossRef] [Green Version]
- Nassisi, M.; Mohand-Saïd, S.; Andrieu, C.; Antonio, A.; Condroyer, C.; Méjécase, C.; Varin, J.; Wohlschlegel, J.; Dhaenens, C.-M.; Sahel, J.-A.; et al. Prevalence of ABCA4 Deep-Intronic Variants and Related Phenotype in An Unsolved “One-Hit” Cohort with Stargardt Disease. Int. J. Mol. Sci. 2019, 20, 5053. [Google Scholar] [CrossRef] [Green Version]
- Garanto, A.; Collin, R.W.J. Design and In Vitro Use of Antisense Oligonucleotides to Correct Pre-mRNA Splicing Defects in Inherited Retinal Dystrophies. In Retinal Gene Therapy: Methods and Protocols; Boon, C.J.F., Wijnholds, J., Eds.; Springer: New York, NY, USA, 2018; pp. 61–78. [Google Scholar]
- Hammond, S.M.; Wood, M.J. Genetic therapies for RNA mis-splicing diseases. Trends Genet. 2011, 27, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Bennett, C.F.; Baker, B.F.; Pham, N.; Swayze, E.; Geary, R.S. Pharmacology of Antisense Drugs. Annu. Rev. Pharmacol. Toxicol. 2017, 57, 81–105. [Google Scholar] [CrossRef] [PubMed]
- Sheng, L.; Rigo, F.; Bennett, C.F.; Krainer, A.R.; Hua, Y. Comparison of the efficacy of MOE and PMO modifications of systemic antisense oligonucleotides in a severe SMA mouse model. Nucleic Acids Res. 2020, 48, 2853–2865. [Google Scholar] [CrossRef]
- Hammond, S.M.; Aartsma-Rus, A.; Alves, S.; E Borgos, S.; Buijsen, R.A.M.; Collin, R.W.J.; Covello, G.; A Denti, M.; Desviat, L.R.; Echevarría, L.; et al. Delivery of oligonucleotide-based therapeutics: Challenges and opportunities. EMBO Mol. Med. 2021, 13, e13243. [Google Scholar] [CrossRef]
- Aartsma-Rus, A.; Straub, V.; Hemmings, R.; Haas, M.; Schlosser-Weber, G.; Schlosser-Weber, G.; Stoyanova-Beninska, V.; Mercuri, E.; Sepodes, B.; Vroom, E.; et al. Development of Exon Skipping Therapies for Duchenne Muscular Dystrophy: A Critical Review and a Perspective on the Outstanding Issues. Nucleic Acids Ther. 2017, 27, 251–259. [Google Scholar] [CrossRef]
- Shi, Y. Mechanistic insights into precursor messenger RNA splicing by the spliceosome. Nat. Rev. Mol. Cell Biol. 2017, 18, 655–670. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, N.; Butler, D.C.D.; Svrzikapa, N.; Mohapatra, S.; Zlatev, I.; Sah, D.W.Y.; Meena; Standley, S.M.; Lu, G.; Apponi, L.H.; et al. Control of phosphorothioate stereochemistry substantially increases the efficacy of antisense oligonucleotides. Nat. Biotechnol. 2017, 35, 845–851. [Google Scholar] [CrossRef]
- Byrne, M.; Vathipadiekal, V.; Apponi, L.; Iwamoto, N.; Kandasamy, P.; Longo, K.; Liu, F.; Looby, R.; Norwood, L.; Shah, A.; et al. Stereochemistry Enhances Potency, Efficacy, and Durability of Malat1 Antisense Oligonucleotides In Vitro and In Vivo in Multiple Species. Transl. Vis. Sci. Technol. 2021, 10, 23. [Google Scholar] [CrossRef]
- Bartys, N.; Kierzek, R.; Lisowiec-Wachnicka, J. The regulation properties of RNA secondary structure in alternative splicing. Biochim. Biophys. Acta (BBA)—Gene Regul. Mech. 2019, 1862, 194401. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Cornelis, S.S.; Sangermano, R.; Post, I.J.; Groesbeek, A.J.; Amsu, J.; Gilissen, C.; Garanto, A.; Collin, R.W.; Cremers, F.P. In or Out? New Insights on Exon Recognition through Splice-Site Interdependency. Int. J. Mol. Sci. 2020, 21, 2300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Nostrand, E.L.; Pratt, G.A.; Yee, B.A.; Wheeler, E.C.; Blue, S.M.; Mueller, J.; Park, S.S.; Garcia, K.E.; Gelboin-Burkhart, C.; Nguyen, T.B.; et al. Principles of RNA processing from analysis of enhanced CLIP maps for 150 RNA binding proteins. Genome Biol. 2020, 21, 1–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmo-Fonseca, M.; Kirchhausen, T. The timing of pre-mRNA splicing visualized in real-time. Nucleus 2014, 5, 11–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pulyakhina, I.; Gazzoli, I.; ’t Hoen, P.A.; Verwey, N.; den Dunnen, J.; Aartsma-Rus, A.; Laros, J.F. SplicePie: A novel analytical approach for the detection of alternative, non-sequential and recursive splicing. Nucleic Acids Res. 2015, 43, e80. [Google Scholar] [CrossRef] [Green Version]
- Gazzoli, I.; Pulyakhina, I.; Verwey, N.E.; Ariyurek, Y.; Laros, J.F.J.; ’t Hoen, P.A.C.; Aartsma-Rus, A. Non-sequential and multi-step splicing of the dystrophin transcript. RNA Biol. 2016, 13, 290–305. [Google Scholar] [CrossRef] [Green Version]
- Crooke, S.T.; Liang, X.-H.; Baker, B.F.; Crooke, R.M. Antisense technology: A review. J. Biol. Chem. 2021, 296, 100416. [Google Scholar] [CrossRef]
- Schwartz, S.D.; Hubschman, J.-P.; Heilwell, G.; Franco-Cardenas, V.; Pan, C.K.; Ostrick, R.M.; Mickunas, E.; Gay, R.; Klimanskaya, I.; Lanza, R. Embryonic stem cell trials for macular degeneration: A preliminary report. Lancet 2012, 379, 713–720. [Google Scholar] [CrossRef]
- Weiss, J.N.; Levy, S.; Benes, S.C. Stem Cell Ophthalmology Treatment Study (SCOTS): Bone marrow-derived stem cells in the treatment of Leber’s hereditary optic neuropathy. Neural Regen. Res. 2016, 11, 1685. [Google Scholar] [CrossRef]
- Saad, L.; Washington, I. Can Vitamin A be Improved to Prevent Blindness due to Age-Related Macular Degeneration, Stargardt Disease and Other Retinal Dystrophies? Adv. Exp. Med. Biol. 2015, 854, 355–361. [Google Scholar] [CrossRef]
- Piccardi, M.; Fadda, A.; Martelli, F.; Marangoni, D.; Magli, A.; Minnella, A.M.; Bertelli, M.; Di Marco, S.; Bisti, S.; Falsini, B. Antioxidant Saffron and Central Retinal Function in ABCA4-Related Stargardt Macular Dystrophy. Nutrients 2019, 11, 2461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, M.A.; Choi, D.; Erker, L.R.; Pennesi, M.E.; Yang, P.; Chegarnov, E.N.; Steinkamp, P.N.; Schlechter, C.L.; Dhaenens, C.-M.; Mohand-Said, S.; et al. Test–Retest Variability of Functional and Structural Parameters in Patients with Stargardt Disease Participating in the SAR422459 Gene Therapy Trial. Transl. Vis. Sci. Technol. 2016, 5, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vázquez-Domínguez, I.; Garanto, A.; Collin, R.W. Molecular Therapies for Inherited Retinal Diseases-Current Standing, Opportunities and Challenges. Genes 2019, 10, 654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cideciyan, A.V.; Jacobson, S.G.; Drack, A.V.; Ho, A.C.; Charng, J.; Garafalo, A.V.; Roman, A.J.; Sumaroka, A.; Han, I.C.; Hochstedler, M.D.; et al. Effect of an intravitreal antisense oligonucleotide on vision in Leber congenital amaurosis due to a photoreceptor cilium defect. Nat. Med. 2019, 25, 225–228. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Hu, C.; El Achkar, C.M.; Black, L.E.; Douville, J.; Larson, A.; Pendergast, M.K.; Goldkind, S.F.; Lee, E.A.; Kuniholm, A.; et al. Patient-Customized Oligonucleotide Therapy for a Rare Genetic Disease. N. Engl. J. Med. 2019, 381, 1644–1652. [Google Scholar] [CrossRef] [PubMed]
- Kibbe, W.A. OligoCalc: An online oligonucleotide properties calculator. Nucleic Acids Res. 2007, 35, W43–W46. [Google Scholar] [CrossRef]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Variant | AON# | Sequence (5′ to 3′) | L | Tm | GC | % Correction | RNA Defect |
|---|---|---|---|---|---|---|---|
| c.1937+37C>G | AON1 | CCGUGUCAUGGAGGAGGAUC | 20 | 55.9/64.0 | 60 | 100.00 | 36-nt exon elongation |
| AON2 | CCAUUACCGUGUCAUGGAGGA | 21 | 54.4/49.2 | 52 | 84.64 | ||
| c.3191-11T>A | AON1 | GCACUAGAAGGACGGGAG | 18 | 52.6/58.0 | 61 | 0.00 | 9-nt exon elongation |
| c.4352+61G>A | AON1 | GAACUCACCGUUGGGUCCU | 19 | 53.2/60.0 | 58 | 28.40 | 57-nt exon elongation |
| AON2 | UCUUGAACUCACCGUUGG | 18 | 48/54.0 | 50 | 67.07 | ||
| c.67-2023T>G | AON1 | UGCGGCAACAUCUAUCUGG | 19 | 51.1/58.8 | 53 | 11.47 | 243-nt PE inclusion |
| AON2 | CAUCAGUGGGUAAGGCUG | 18 | 50.3/56.0 | 56 | 87.54 | ||
| c.570+1798A>G | AON1 | CUGGAAGUCAUCAAGGCAUUG | 21 | 52.4/47.3 | 48 | 100.00 | 65-nt PE inclusion |
| AON2 | GACUUGAGUUUUACGAGCUG | 20 | 49.7/58.0 | 45 | 100.00 | ||
| c.769-788A>T | AON1 | GGAAUCACUGAUCCUAGAGG | 20 | 51.8/60.0 | 50 | 92.72 | 162-nt PE inclusion |
| AON2 | GGAUGUGGAAGUCCCCAGG | 19 | 55.4/62.0 | 63 | 93.11 | ||
| c.859-640A>G | AON1 | CCAGUUCUUGGGUUCUGUUG | 20 | 51.8/60.0 | 50 | 100.00 | 46-nt PE inclusion |
| AON2 | CACCAAGAUGGGGAUACUGG | 20 | 53.8/62.0 | 55 | 100.00 | ||
| AON3 | CCUCUCUUCUUCUAGUCUCC | 20 | 51.8/60.0 | 50 | 24.34 | ||
| c.859-546G>A | AON1 | CCAGUUCUUGGGUUCUGUUG | 20 | 51.8/60.0 | 50 | 100.00 | 141-nt PE inclusion |
| AON2 | CACCAAGAUGGGGAUACUGG | 20 | 53.8/62.0 | 55 | 100.00 | ||
| AON3 | CCUCUCUUCUUCUAGUCUCC | 20 | 51.8/60.0 | 50 | 100.00 | ||
| c.2588-706C>T | AON1 | ACUGGACUGUCUAUUCCUCG | 20 | 51.8/60.0 | 50 | 93.83 | 134-nt PE inclusion |
| AON2 | UCUUUAUCUCCACCGCUCUG | 20 | 51.8/60.0 | 50 | 100.00 | ||
| c.2919-826T>A | AON1 | CAGCUCUCUGACCUUAUCAGU | 21 | 52.4/47.3 | 48 | 47.33 | 133-nt PE inclusion |
| AON2 | GCUCUGUCCCUGAGUUCUG | 19 | 53.8/60.0 | 58 | 100.00 | ||
| c.3050+370C>T | AON1 | CAGAGUCCCAUAUUCUCAGG | 20 | 51.8/60.0 | 50 | 61.28 | 205-nt PE inclusion |
| AON2 | GGAUCGAUCAGCUGCUCUG | 19 | 53.2/60.0 | 58 | 71.07 | ||
| c.4539+2064C>T | AON4 | GGGGCACAGAGGACUGAGA | 19 | 55.4/62.0 | 63 | 100.00 | 345-nt PE inclusion |
| AON17 | GCCAAGAGCUCAGGGUACAG | 20 | 60/64.0 | 55.9 | 72.35 | ||
| c.4539+2065C>G | AON4 | GGGGCACAGAGGACUGAGA | 19 | 55.4/62.0 | 63 | 100.00 | 170-nt PE inclusion |
| AON17 | GCCAAGAGCUCAGGGUACAG | 20 | 60/64.0 | 55.9 | 55.77 | ||
| c.4634+741A>G | AON1 | UCUGAUACGGGCUGCCAAAG | 20 | 53.8/62.0 | 55 | 100.00 | 127-nt PE inclusion |
| AON2 | UCCUUAGGAUCCUCUCUCCU | 20 | 51.8/60.0 | 50 | 100.00 | ||
| c.6283-78G>T | AON1 | GGCAAUGACAGAAUUCUCCUC | 21 | 52.4/47.3 | 48 | 100.00 | 203-nt PE inclusion |
| AON2 | GCUGACAGAAGGCGCACAC | 19 | 55.4/62.0 | 63 | 100.00 | ||
| N/A | SON | CGCCAAUUGCAAGGUGAUUCC | 21 | 54.4/49.2 | 52 | N/A | N/A |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tomkiewicz, T.Z.; Suárez-Herrera, N.; Cremers, F.P.M.; Collin, R.W.J.; Garanto, A. Antisense Oligonucleotide-Based Rescue of Aberrant Splicing Defects Caused by 15 Pathogenic Variants in ABCA4. Int. J. Mol. Sci. 2021, 22, 4621. https://doi.org/10.3390/ijms22094621
Tomkiewicz TZ, Suárez-Herrera N, Cremers FPM, Collin RWJ, Garanto A. Antisense Oligonucleotide-Based Rescue of Aberrant Splicing Defects Caused by 15 Pathogenic Variants in ABCA4. International Journal of Molecular Sciences. 2021; 22(9):4621. https://doi.org/10.3390/ijms22094621
Chicago/Turabian StyleTomkiewicz, Tomasz Z., Nuria Suárez-Herrera, Frans P. M. Cremers, Rob W. J. Collin, and Alejandro Garanto. 2021. "Antisense Oligonucleotide-Based Rescue of Aberrant Splicing Defects Caused by 15 Pathogenic Variants in ABCA4" International Journal of Molecular Sciences 22, no. 9: 4621. https://doi.org/10.3390/ijms22094621