
Loss of Fer Jeopardizes Metabolic Plasticity and Mitochondrial Homeostasis in Lung and Breast Carcinoma Cells



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
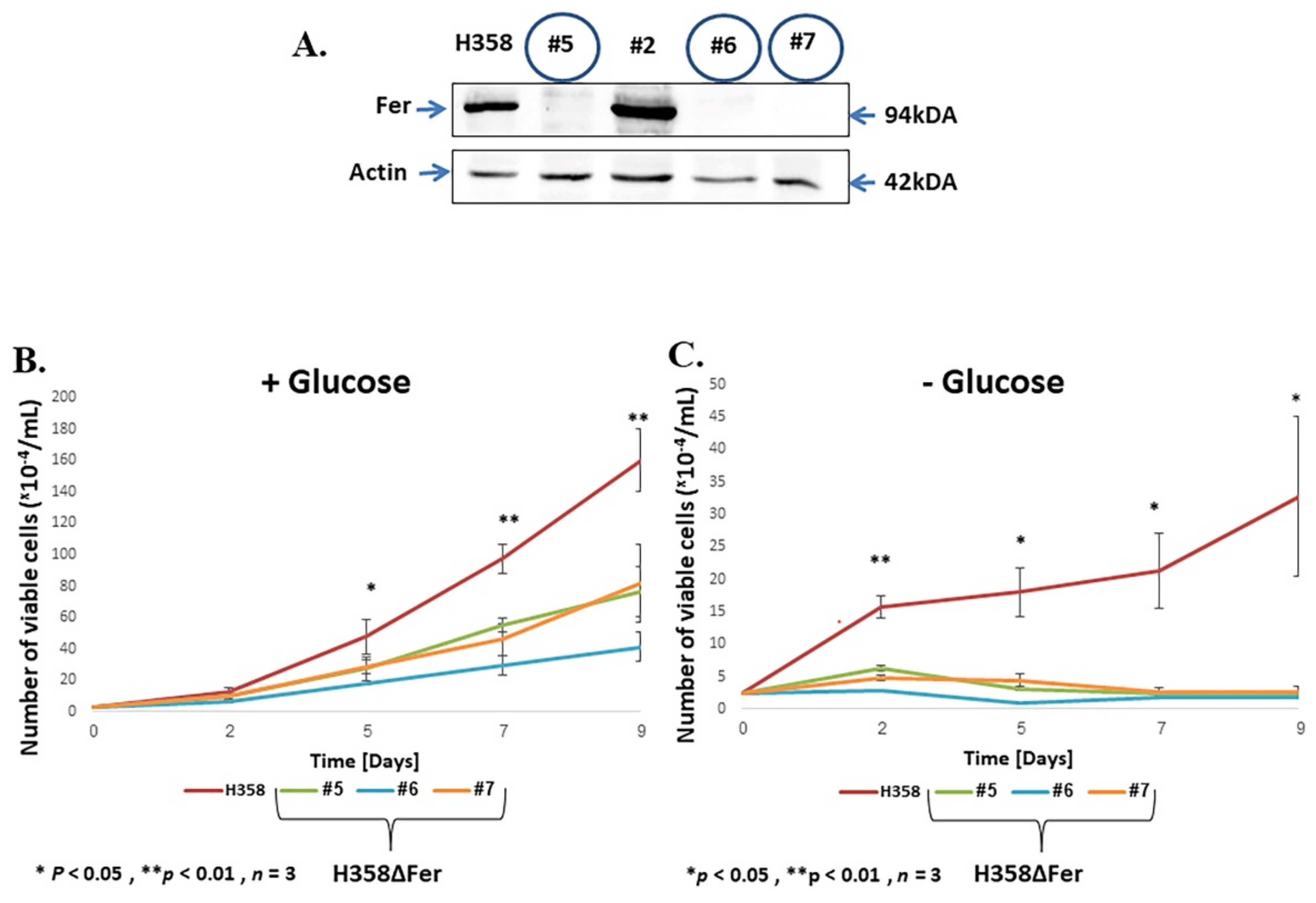
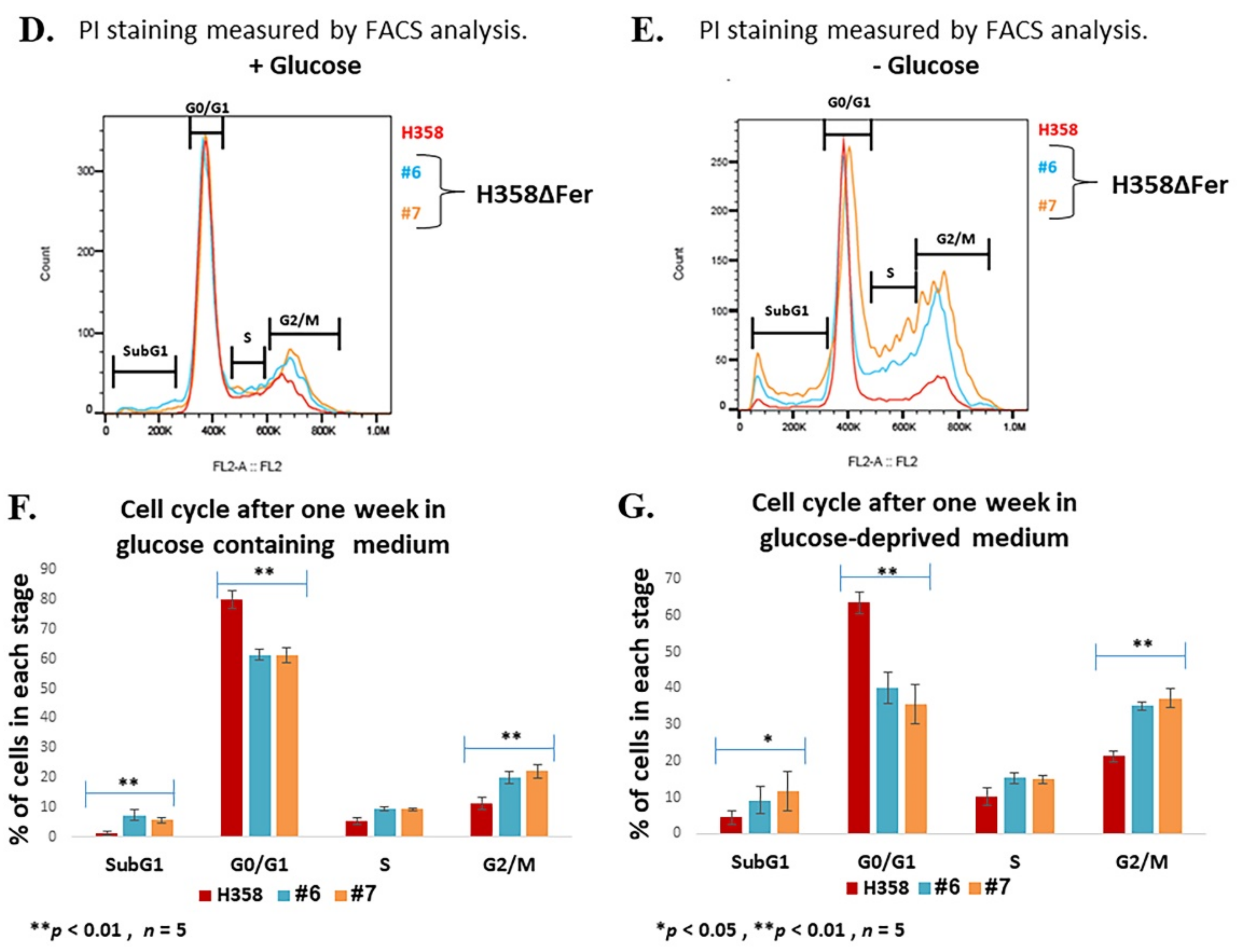
2.1. Impaired Metabolic Plasticity of Fer-Deficient H358 Cells
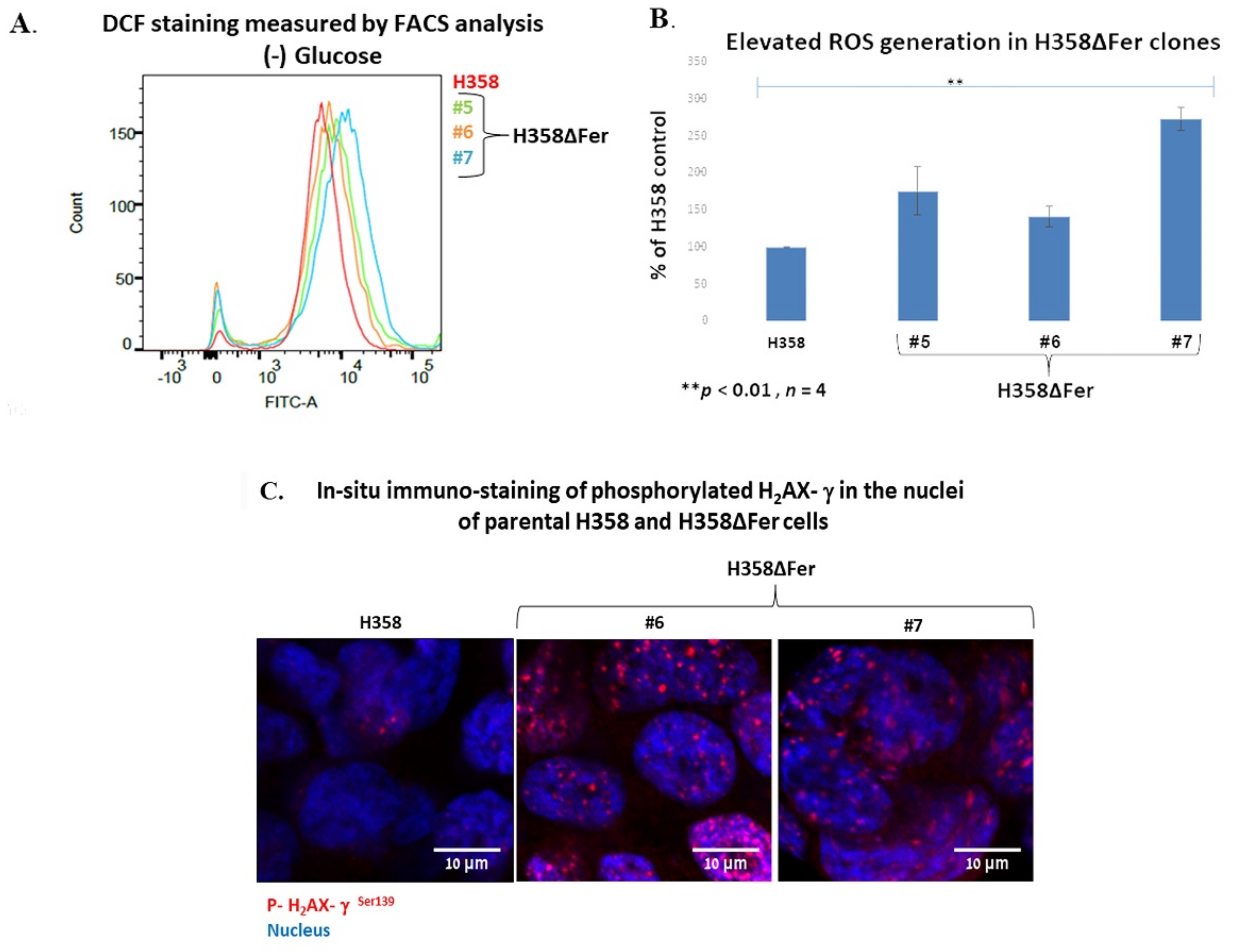
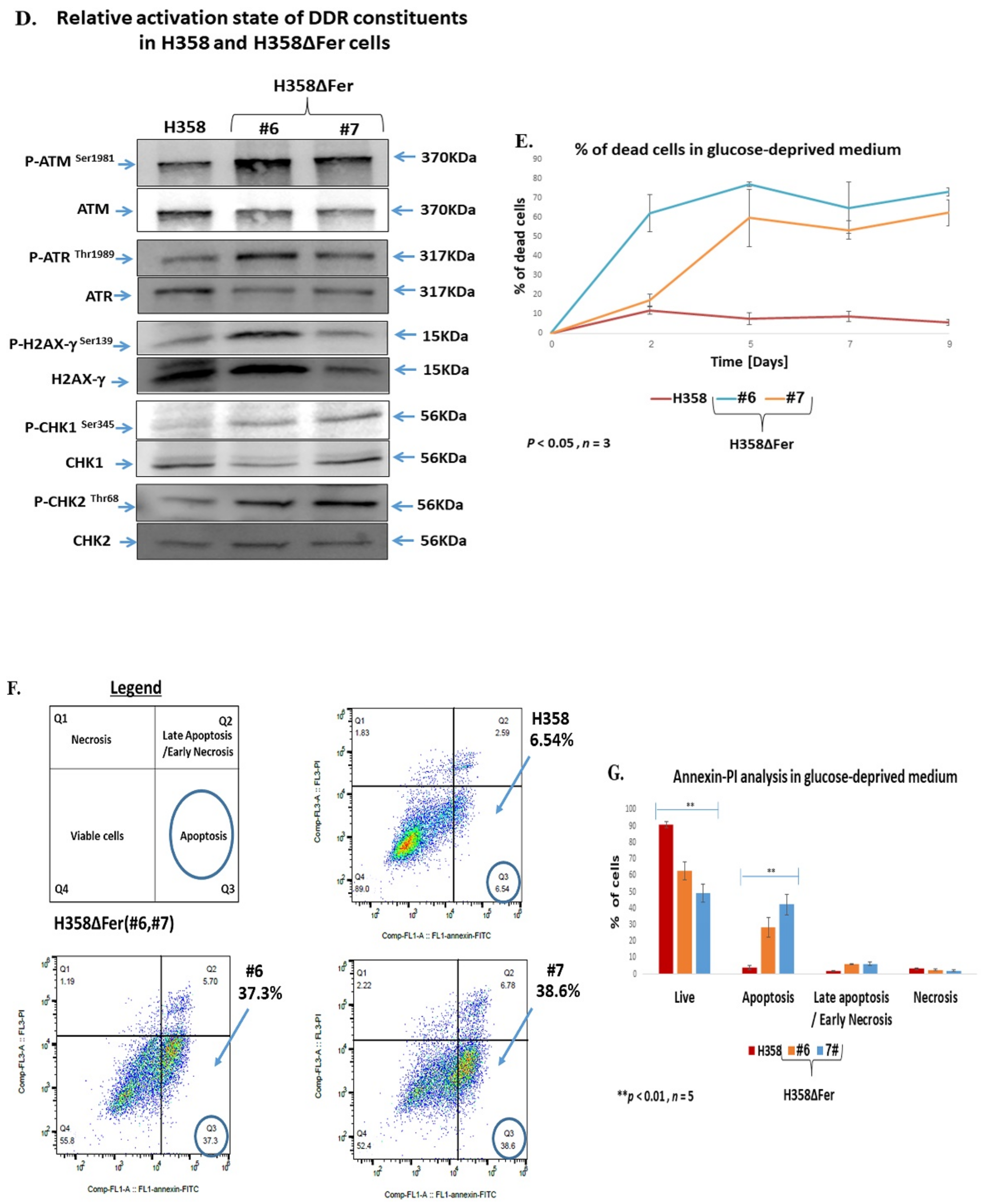
2.2. Elevated ROS Levels, Activation of the DNA Damage Response and Jeopardized Survival of Metabolically Challenged H358ΔFer Cells
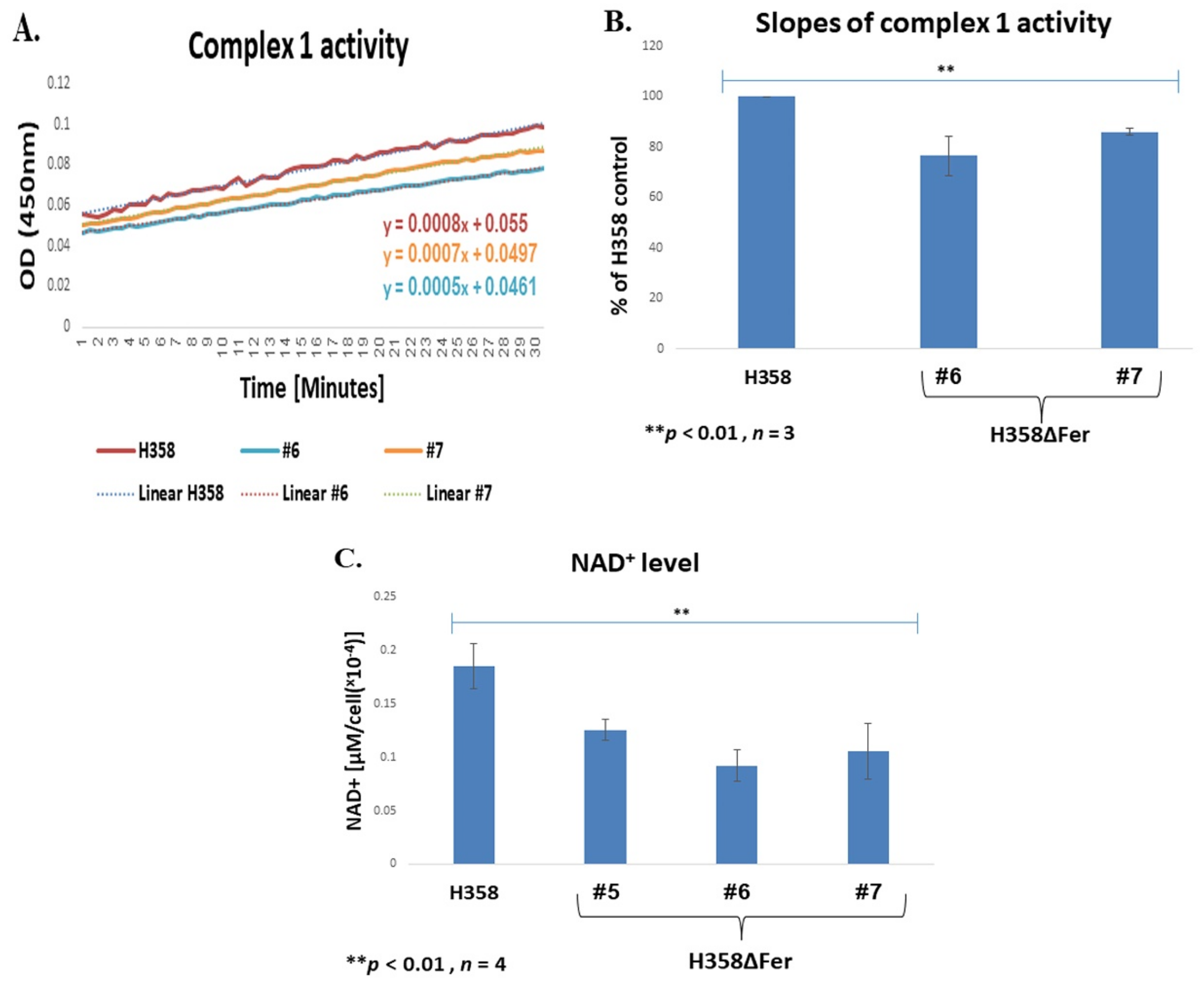
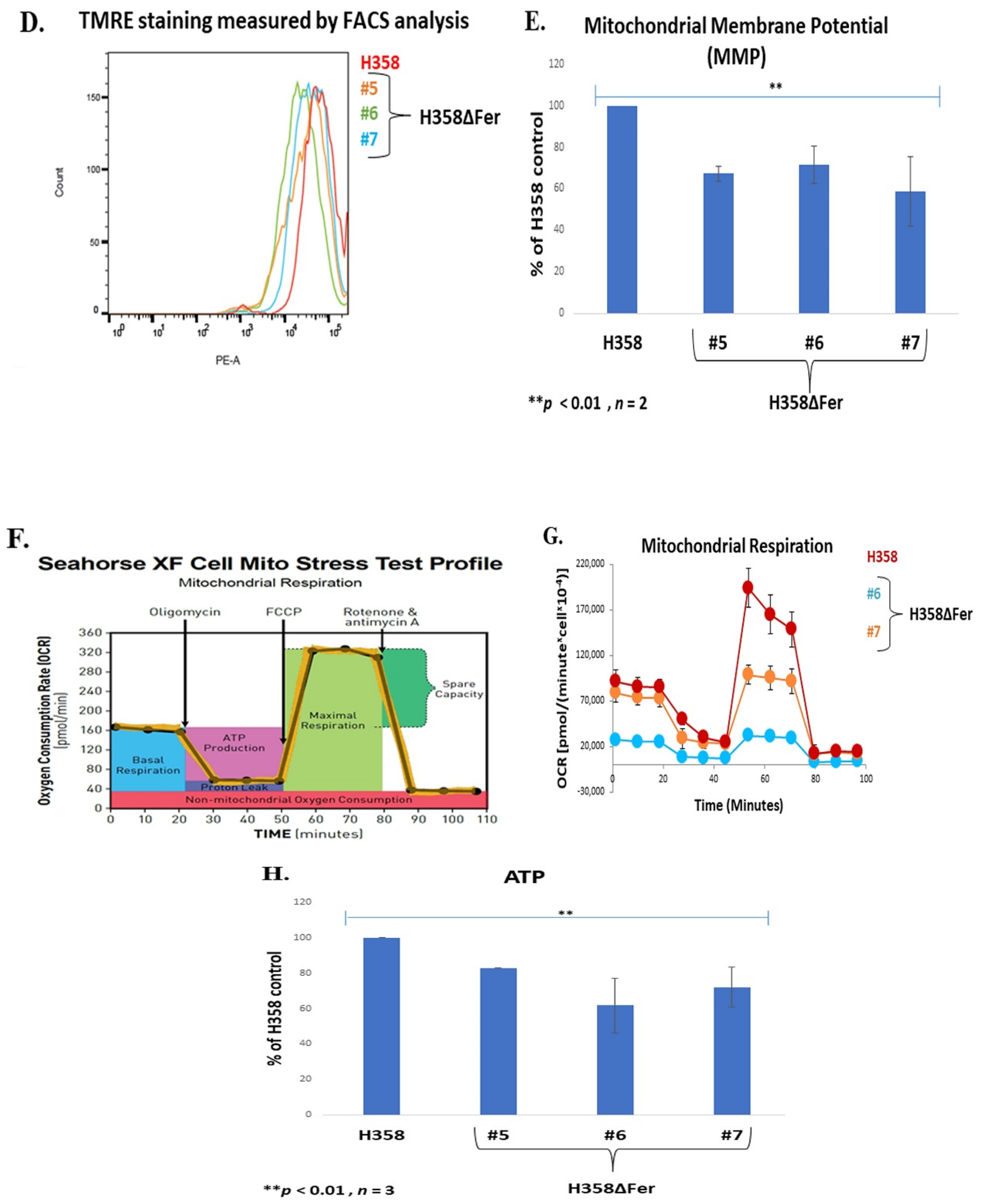
2.3. Impaired Mitochondrial Function in Fer-Deficient H358 Cells
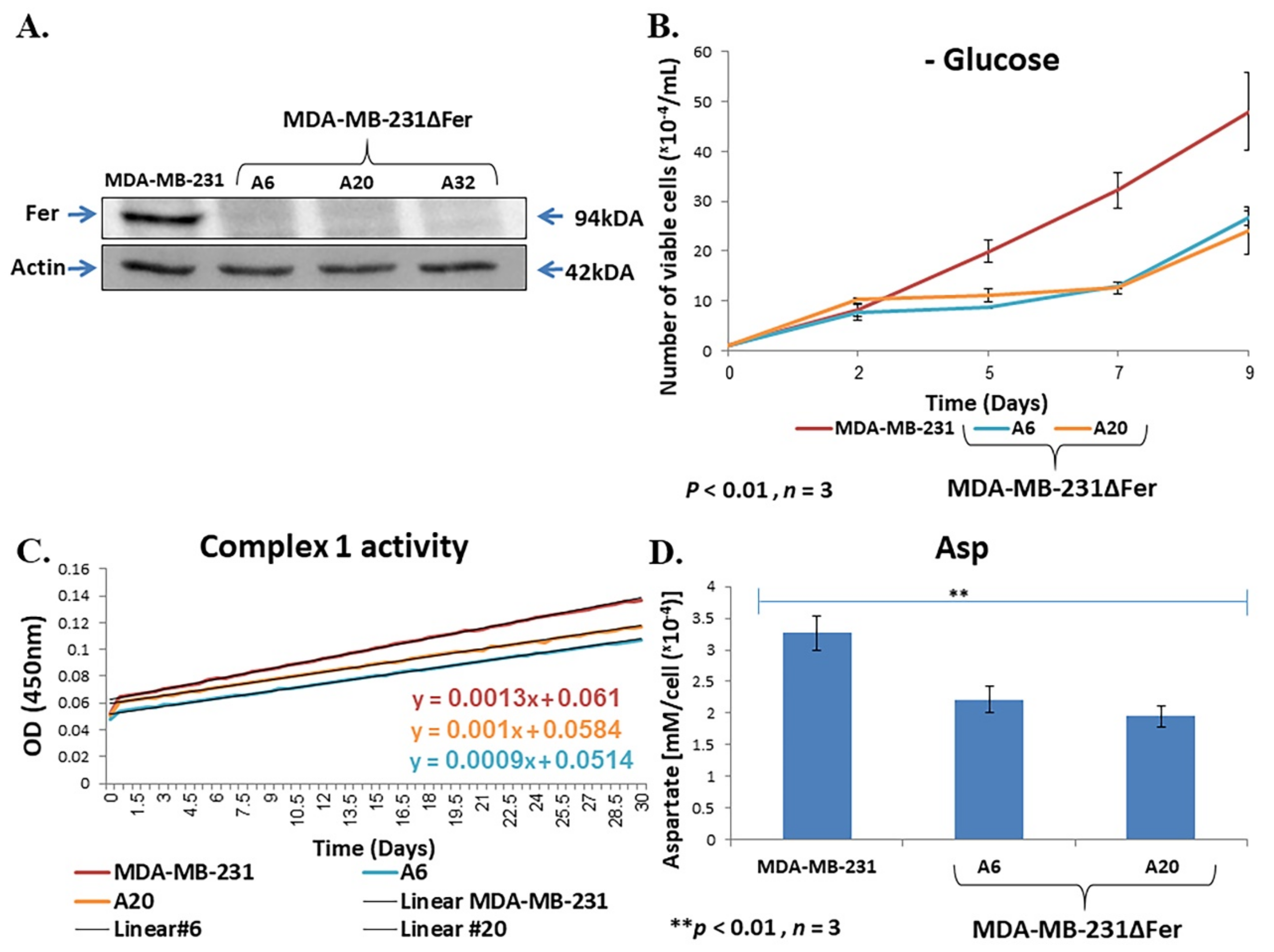
2.4. Fer Sustains Mitochondrial Metabolic Homeostasis in H358 and MDA-MB-231 Cells
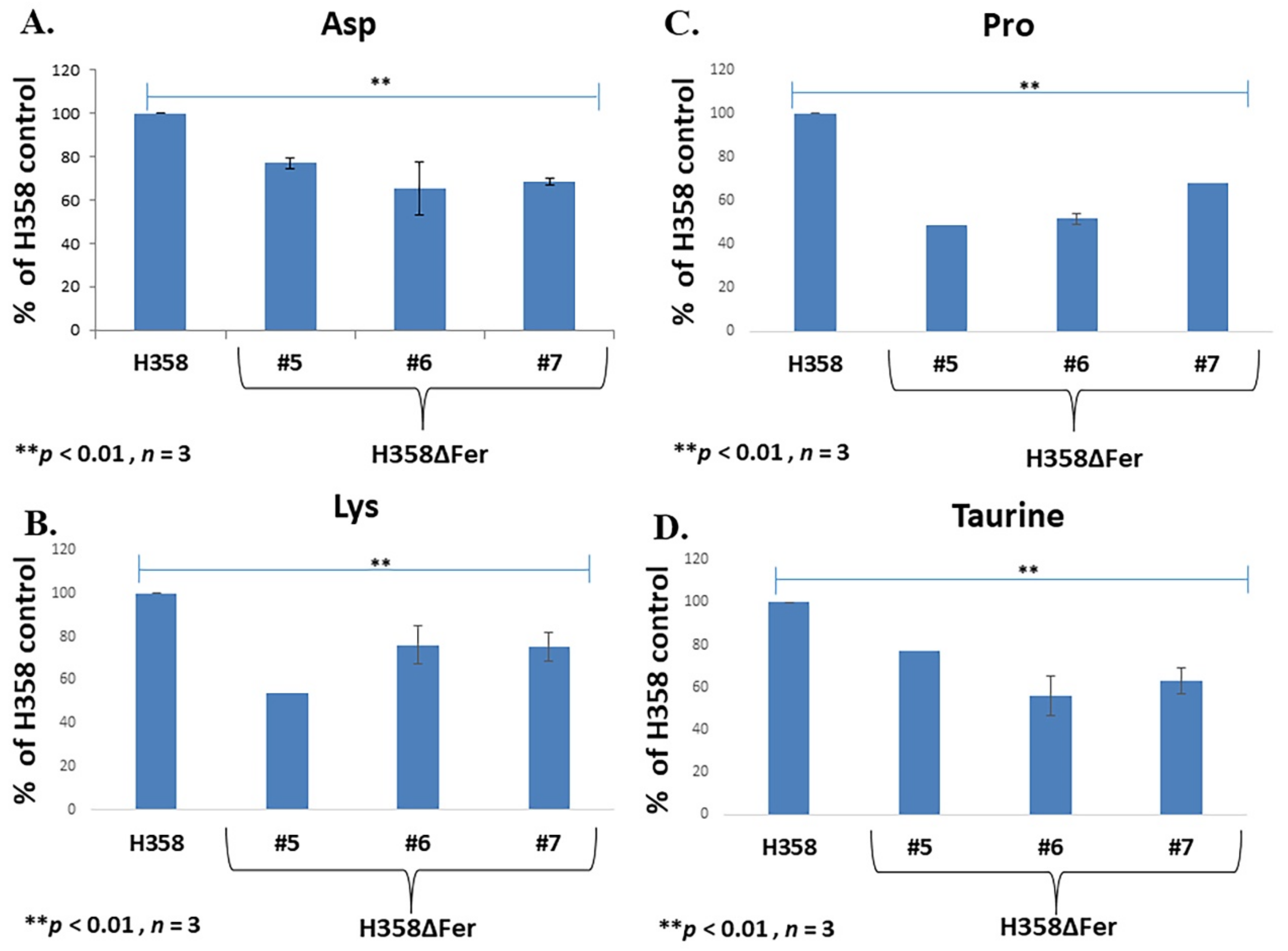
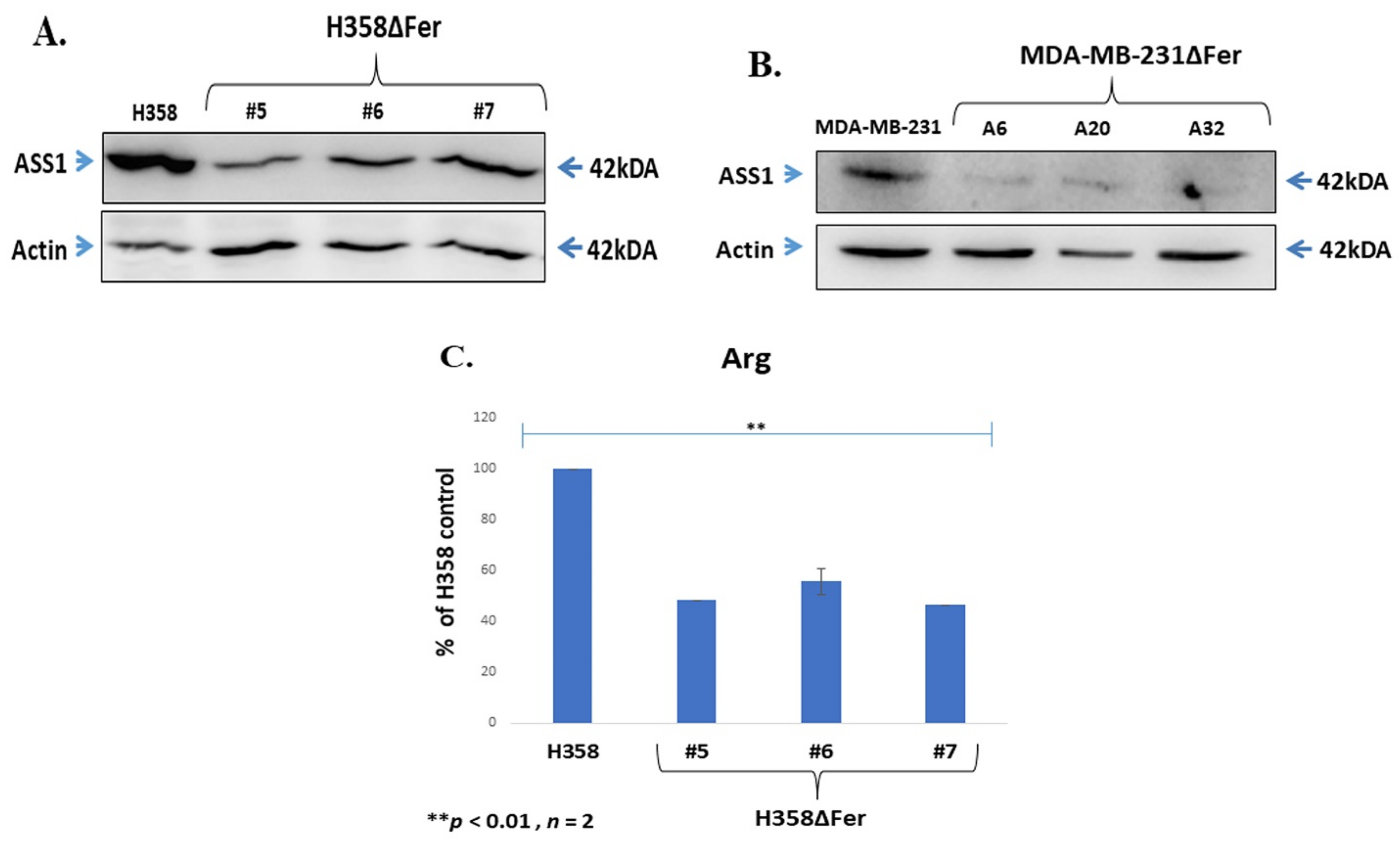
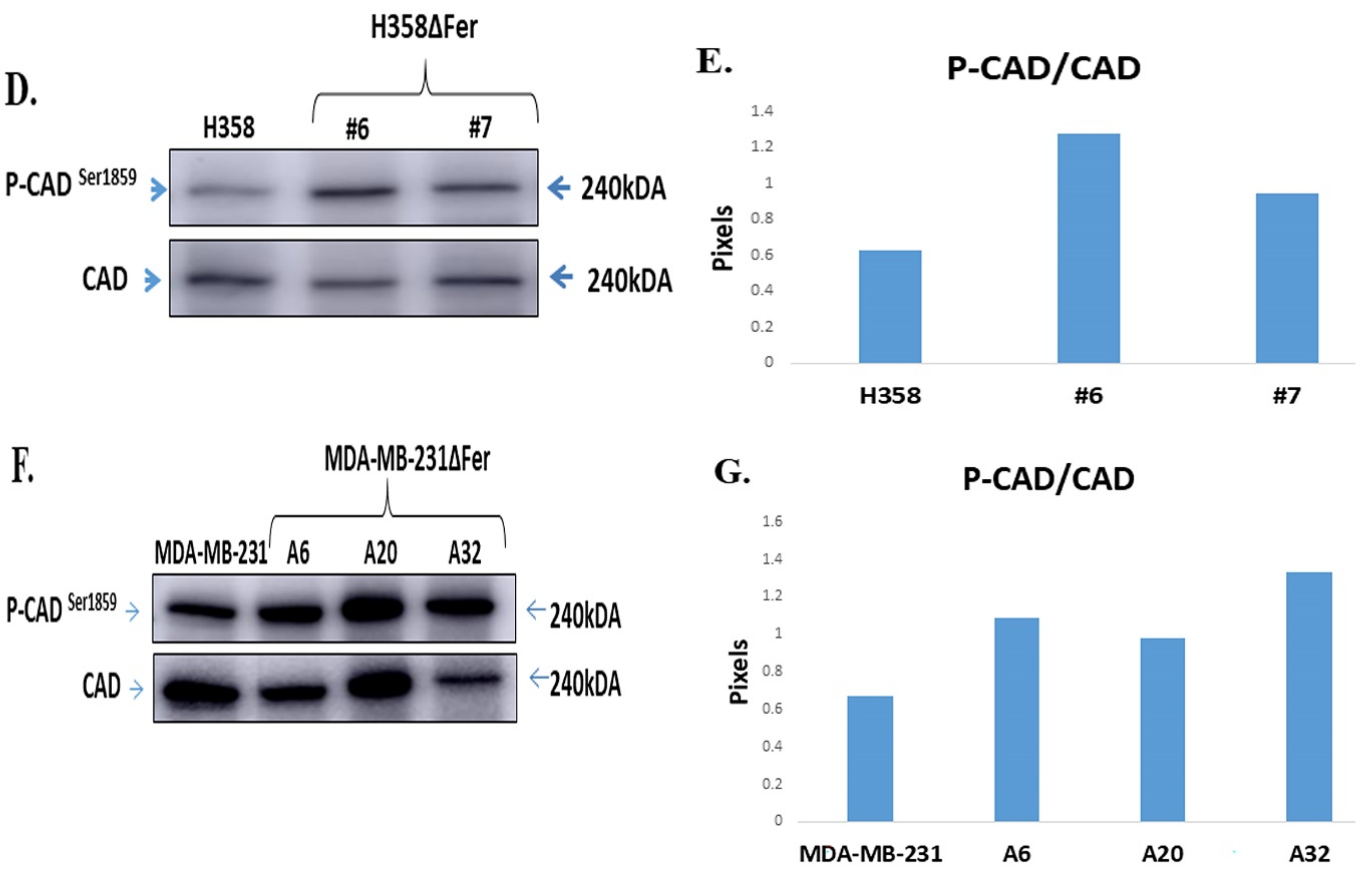
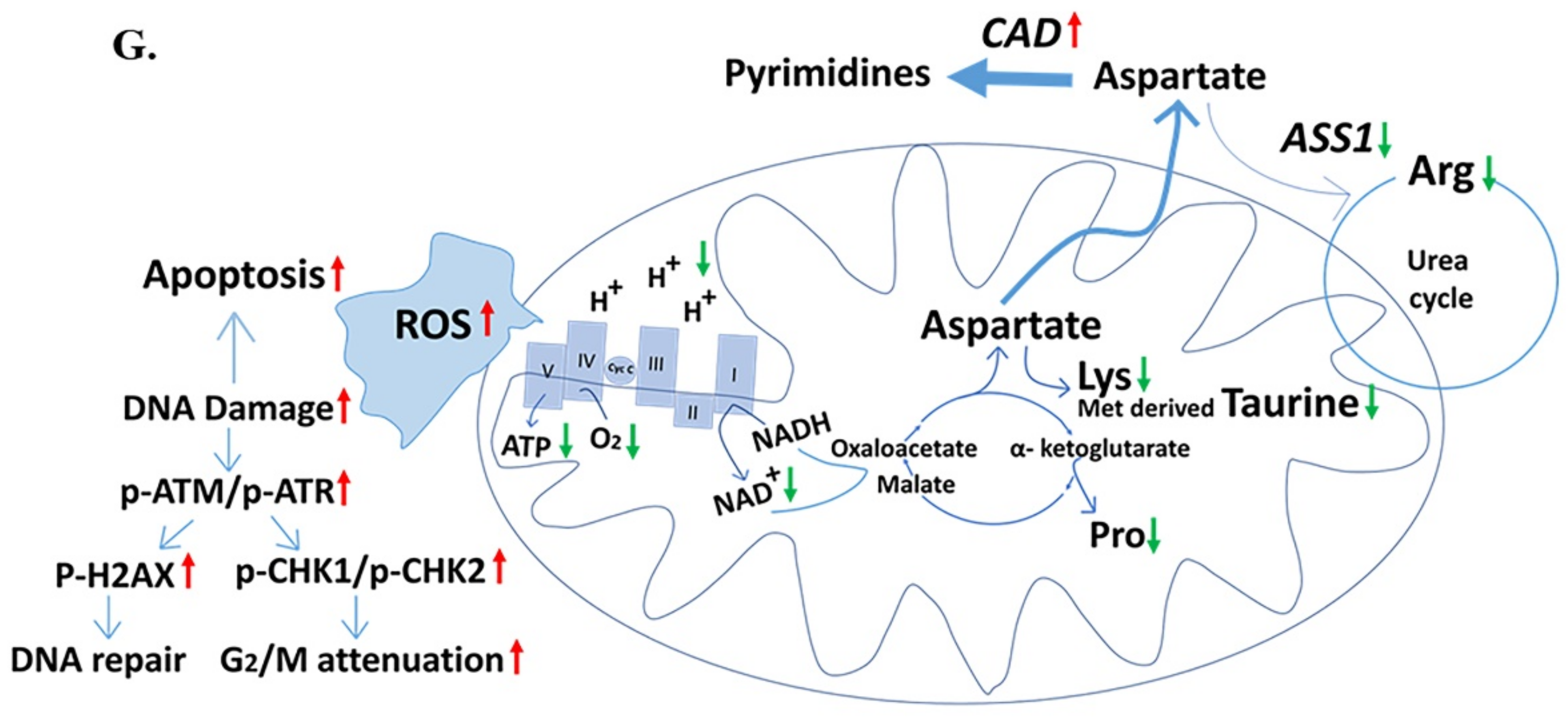
2.5. Down-Regulated Urea Cycle in Fer-Deficient H358ΔFer and MDA-MB-231ΔFer Cells
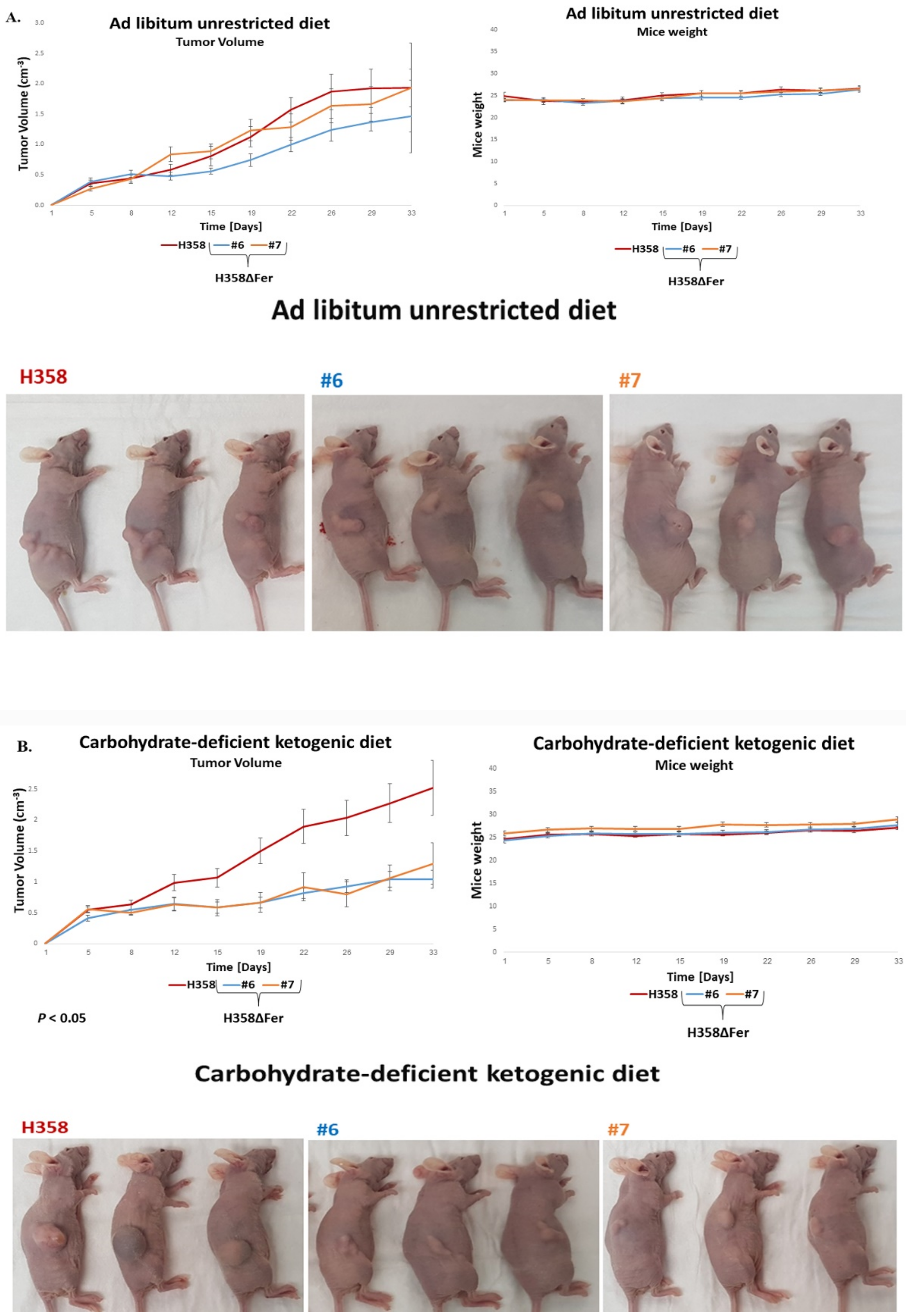
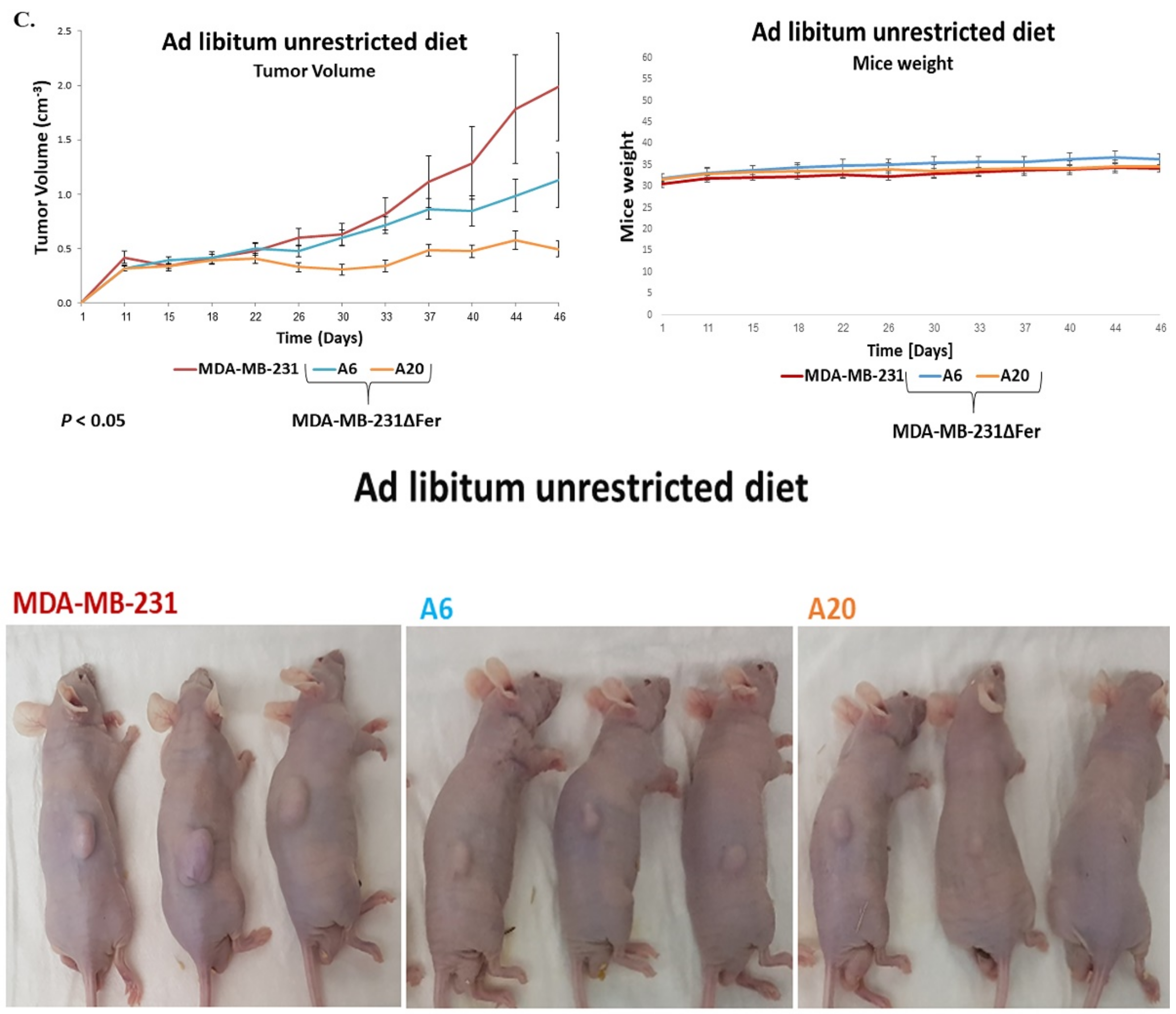
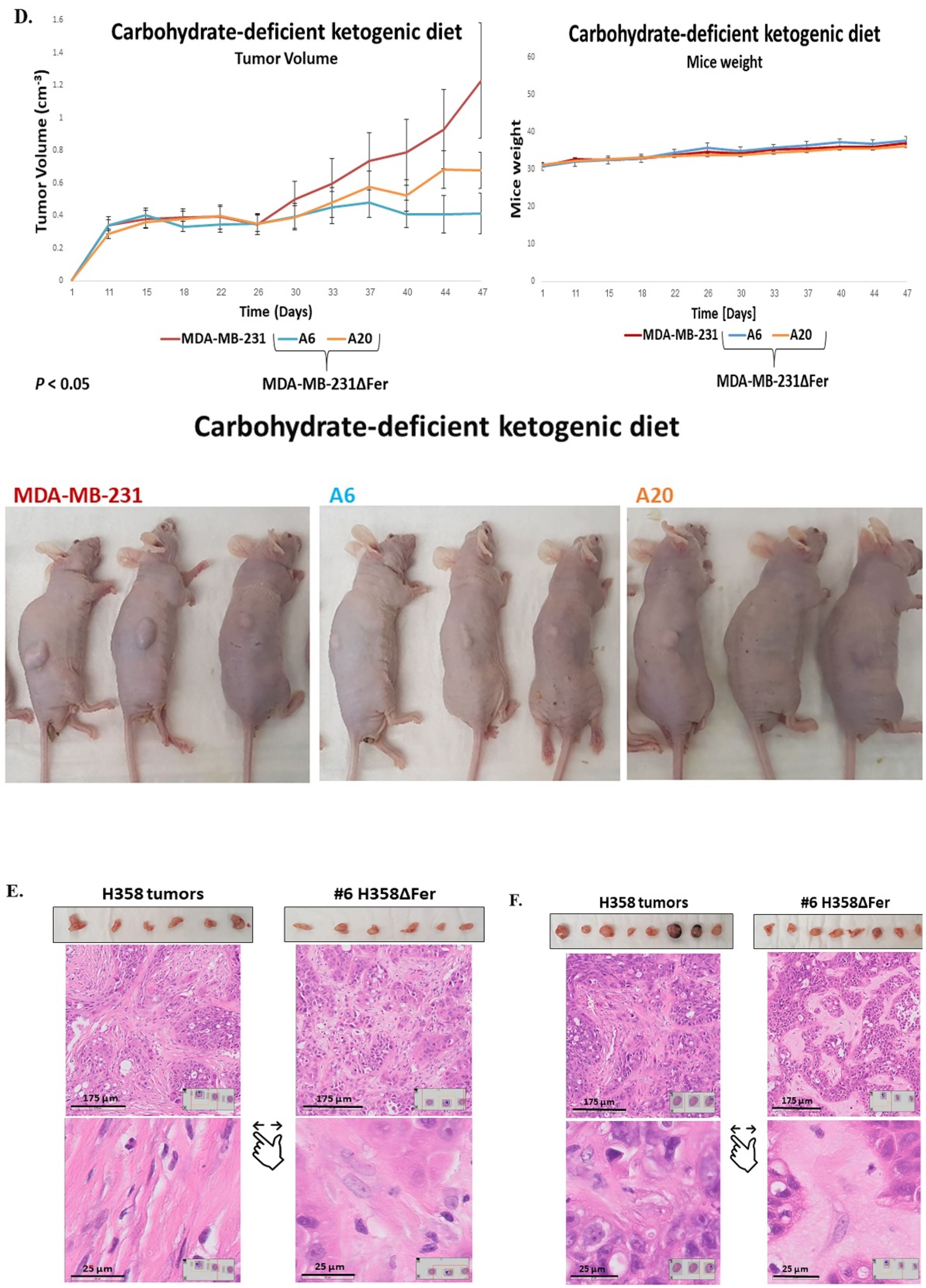
2.6. Absence of Fer Impedes the Development of H358 and MDA-MB-231 Xenografts in Mice
3. Discussion
4. Materials and Methods
4.1. Cell Cultures
4.2. Generation of Stable CRSPR-Cas9 Knockout Clones
4.3. Western Blot Analysis
4.4. Immuno-Cytostaining
4.5. Determination of Cell-Growth Curves
4.6. Flow-Cytometry Cell-Cycle Analysis
4.7. Cell Death Analysis using Annexin/PI Staining
4.8. Measurement of ROS Levels
4.9. Measurement of the Mitochondrial OXPHOS Complex I Activity
4.10. Determining Mitochondrial Respiration Profile using “Mito-Stress” Analysis
4.11. Measurement of the Mitochondrial Potential using TMRE Staining
4.12. Determining NAD+ and Aspartate Levels
4.13. Quantification of Cellular ATP, CTP, TTP, Lys, Pro and Taurine Levels
4.14. Animal Experiments
4.15. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Luzzi, K.J.; MacDonald, I.C.; Schmidt, E.E.; Kerkvliet, N.; Morris, V.L.; Chambers, A.F.; Groom, A.C. Multistep nature of metastatic inefficiency: Dormancy of solitary cells after successful extravasation and limited survival of early micrometastases. Am. J. Pathol. 1998, 153, 865–873. [Google Scholar] [CrossRef]
- Mason, J.A.; Hagel, K.R.; Hawk, M.A.; Schafer, Z.T. Metabolism during ECM Detachment: Achilles Heel of cancer cells? Trends Cancer 2017, 3, 475–481. [Google Scholar] [CrossRef]
- LeBleu, V.S.; O’Connell, J.T.; Gonzalez Herrera, K.N.; Wikman, H.; Pantel, K.; Haigis, M.C.; de Carvalho, F.M.; Damascena, A.; Domingos Chinen, L.T.; Rocha, R.M.; et al. PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nat. Cell Biol. 2014, 16, 992–1003. [Google Scholar] [CrossRef]
- Viale, A.; Corti, D.; Draetta, G.F. Tumors and mitochondrial respiration: A neglected connection. Cancer Res. 2015, 75, 3687–3691. [Google Scholar] [CrossRef]
- Aguilar, E.; Marin de Mas, I.; Zodda, E.; Marín, S.; Morrish, F.; Selivanov, V.; Meca-Cortés, Ó.; Delowar, H.; Pons, M.; Izquierdo, I.; et al. Metabolic reprogramming and dependencies associated with epithelial cancer stem cells independent of the epithelial-mesenchymal transition program. Stem Cells 2016, 34, 1163–1176. [Google Scholar] [CrossRef] [PubMed]
- Caino, M.C.; Ghosh, J.C.; Chae, Y.C.; Vaira, V.; Rivadeneira, D.B.; Faversani, A.; Rampini, P.; Kossenkov, A.V.; Aird, K.M.; Zhang, R.; et al. PI3K therapy reprograms mitochondrial trafficking to fuel tumor cell invasion. Proc. Natl. Acad. Sci. USA 2015, 112, 8638–8643. [Google Scholar] [CrossRef]
- Weber, G.F. Metabolism in cancer metastasis. Int. J. Cancer 2016, 138, 2061–2066. [Google Scholar] [CrossRef] [PubMed]
- Celià-Terrassa, T.; Kang, Y. Distinctive properties of metastasis-initiating cells. Genes Dev. 2016, 30, 892–908. [Google Scholar] [CrossRef] [PubMed]
- Lupas, A. Coiled coils: New structures and new functions. Trends Biochem. Sci. 1996, 21, 375–382. [Google Scholar] [CrossRef]
- Kim, L.; Wong, T.W. The cytoplasmic tyrosine kinase FER is associated with the catenin-like substrate pp120 and is activated by growth factors. Mol. Cell. Biol. 1995, 15, 4553–4561. [Google Scholar] [CrossRef]
- Craig, A.W.B.; Zirngibl, R.; Greer, P. Disruption of coiled-coil domains in fer protein-tyrosine kinase abolishes trimerization but not kinase activation. J. Biol. Chem. 1999, 274, 19934–19942. [Google Scholar] [CrossRef] [PubMed]
- Hazan, B.; Bern, O.; Carmel, M.; Lejbkowicz, F.; Goldstein, R.S.; Nir, U. ferT encodes a meiosis-specific nuclear tyrosine kinase. Cell Growth Differ. 1993, 4, 443–449. [Google Scholar] [PubMed]
- Letwin, K.; Yee, S.P.; Pawson, T. Novel protein-tyrosine kinase cDNAs related to fps/fes and eph cloned using anti-phosphotyrosine antibody. Oncogene 1988, 3, 621–627. [Google Scholar] [PubMed]
- Halachmy, S.; Bern, O.; Schreiber, L.; Carmel, M.; Sharabi, Y.; Shoham, J.; Nir, U. p94fer facilitates cellular recovery of gamma irradiated pre-T cells. Oncogene 1997, 14, 2871–2880. [Google Scholar] [CrossRef] [PubMed]
- Elkis, Y.; Cohen, M.; Yaffe, E.; Satmary-Tusk, S.; Feldman, T.; Hikri, E.; Nyska, A.; Feiglin, A.; Ofran, Y.; Shpungin, S.; et al. A novel Fer/FerT targeting compound selectively evokes metabolic stress and necrotic death in malignant cells. Nat. Commun. 2017, 8, 940. [Google Scholar] [CrossRef]
- Yaffe, E.; Hikri, E.; Elkis, Y.; Cohen, O.; Segal, A.; Makovski, A.; Varvak, A.; Shpungin, S.; Nir, U. Oncogenic properties of a spermatogenic meiotic variant of fer kinase expressed in somatic cells. Cancer Res. 2014, 74, 6474–6485. [Google Scholar] [CrossRef]
- Ivanova, I.A.; Vermeulen, J.F.; Ercan, C.; Houthuijzen, J.M.; Saig, F.A.; Vlug, E.J.; van der Wall, E.; van Diest, P.J.; Vooijs, M.; Derksen, P.W. FER kinase promotes breast cancer metastasis by regulating alpha- and beta-integrin-dependent cell adhesion and anoikis resistance. Oncogene 2013, 32, 5582–5592. [Google Scholar] [CrossRef] [PubMed]
- Fan, G.; Zhang, S.; Gao, Y.; Greer, P.A.; Tonks, N.K. HGF-independent regulation of MET and GAB1 by nonreceptor tyrosine kinase FER potentiates metastasis in ovarian cancer. Genes Dev. 2016, 30, 1542–1557. [Google Scholar] [CrossRef]
- Ahn, J.; Truesdell, P.; Meens, J.; Kadish, C.; Yang, X.; Boag, A.H.; Craig, A.W. Fer protein-tyrosine kinase promotes lung adenocarcinoma cell invasion and tumor metastasis. Mol. Cancer Res. 2013, 11, 952–963. [Google Scholar] [CrossRef] [PubMed]
- Rikova, K.; Guo, A.; Zeng, Q.; Possemato, A.; Yu, J.; Haack, H.; Nardone, J.; Lee, K.; Reeves, C.; Li, Y.; et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 2007, 131, 1190–1203. [Google Scholar] [CrossRef]
- Kawakami, M.; Morita, S.; Sunohara, M.; Amano, Y.; Ishikawa, R.; Watanabe, K.; Hamano, E.; Ohishi, N.; Nakajima, J.; Yatomi, Y.; et al. FER overexpression is associated with poor postoperative prognosis and cancer-cell survival in non-small cell lung cancer. Int. J. Clin. Exp. Pathol. 2013, 6, 598–612. [Google Scholar] [PubMed]
- International Agency for Research on Cancer. Globocan. 2018. Available online: http://gco.iarc.fr/today (accessed on 20 February 2021).
- Reno, T.A.; Kim, J.Y.; Raz, D.J. Triptolide inhibits lung cancer cell migration, invasion, and metastasis. Ann. Thorac. Surg. 2015, 100, 1817–1825. [Google Scholar] [CrossRef]
- Raz, D.J.; He, B.; Rosell, R.; Jablons, D.M. Bronchioloalveolar carcinoma: A review. Clin. Lung Cancer 2006, 7, 313–322. [Google Scholar] [CrossRef] [PubMed]
- Ran, F.A.; Hsu, P.D.; Lin, C.Y.; Gootenberg, J.S.; Konermann, S.; Trevino, A.E.; Scott, D.A.; Inoue, A.; Matoba, S.; Zhang, Y.; et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell 2013, 154, 1380–1389. [Google Scholar] [CrossRef] [PubMed]
- Gasiunas, G.; Barrangou, R.; Horvath, P.; Siksnys, V. Cas9-crRNA ribonucleoprotein complex mediates specific DNA cleavage for adaptive immunity in bacteria. Proc. Natl. Acad. Sci. USA 2012, 109, E2579–E2586. [Google Scholar] [CrossRef]
- Budczies, J.; Brockmöller, S.F.; Müller, B.M.; Barupal, D.K.; Richter-Ehrenstein, C.; Kleine-Tebbe, A.; Griffin, J.L.; Orešič, M.; Dietel, M.; Denkert, C.; et al. Comparative metabolomics of estrogen receptor positive and estrogen receptor negative breast cancer: Alterations in glutamine and beta-alanine metabolism. J. Proteom. 2013, 94, 279–288. [Google Scholar] [CrossRef] [PubMed]
- Jin, L.; Alesi, G.N.; Kang, S. Glutaminolysis as a target for cancer therapy. Oncogene 2016, 35, 3619–3625. [Google Scholar] [CrossRef] [PubMed]
- Boonstra, J.; Post, J.A. Molecular events associated with reactive oxygen species and cell cycle progression in mammalian cells. Gene 2004, 337, 1–13. [Google Scholar] [CrossRef]
- Kinner, A.; Wu, W.; Staudt, C.; Iliakis, G. Gamma-H2AX in recognition and signaling of DNA double-strand breaks in the context of chromatin. Nucleic Acids Res. 2008, 36, 5678–5694. [Google Scholar] [CrossRef] [PubMed]
- Krempler, A.; Deckbar, D.; Jeggo, P.A.; Lobrich, M. An imperfect G2M checkpoint contributes to chromosome instability following irradiation of S and G2 phase cells. Cell Cycle 2007, 6, 1682–1686. [Google Scholar] [CrossRef]
- Pandita, T.K.; Lieberman, H.B.; Lim, D.-S.; Dhar, S.; Zheng, W.; Taya, Y.; Kastan, M.B. Ionizing radiation activates the ATM kinase throughout the cell cycle. Oncogene 2000, 19, 1386–1391. [Google Scholar] [CrossRef]
- Yan, T.; Desai, A.B.; Jacobberger, J.W.; Sramkoski, R.M.; Loh, T.; Kinsella, T.J. CHK1 and CHK2 are differentially involved in mismatch repair-mediated 6-thioguanine-induced cell cycle checkpoint responses. Mol. Cancer Ther. 2004, 3, 1147–1157. [Google Scholar]
- Ashwell, S.; Zabludoff, S. DNA damage detection and repair pathways—Recent advances with inhibitors of checkpoint kinases in cancer therapy. Clin. Cancer Res. 2008, 14, 4032–4037. [Google Scholar] [CrossRef] [PubMed]
- Redza-Dutordoir, M.; Averill-Bates, D.A. Activation of apoptosis signalling pathways by reactive oxygen species. Biochim. Biophys. Acta 2016, 1863, 2977–2992. [Google Scholar] [CrossRef]
- Collins, J.A.; Schandl, C.A.; Young, K.K.; Vesely, J.; Willingham, M.C. Major DNA fragmentation is a late event in apoptosis. J. Histochem. Cytochem. 1997, 45, 923–934. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552 Pt 2, 335–344. [Google Scholar] [CrossRef]
- Inoue, M.; Sato, E.F.; Nishikawa, M.; Park, A.-M.; Kira, Y.; Imada, I.; Utsumi, K. Cross talk of nitric oxide, oxygen radicals, and superoxide dismutase regulates the energy metabolism and cell death and determines the fates of aerobic life. Antioxid. Redox Signal. 2003, 5, 475–484. [Google Scholar] [CrossRef]
- Sharma, L.K.; Lu, J.; Bai, Y. Mitochondrial respiratory complex i: Structure, function and implication in human diseases. Curr. Med. Chem. 2009, 16, 1266–1277. [Google Scholar] [CrossRef] [PubMed]
- Birsoy, K.; Wang, T.; Chen, W.W.; Freinkman, E.; Abu-Remaileh, M.; Sabatini, D.M. An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell 2015, 162, 540–551. [Google Scholar] [CrossRef] [PubMed]
- Park, J.H.; Vithayathil, S.; Kumar, S.; Sung, P.-L.; Dobrolecki, L.E.; Putluri, V.; Bhat, V.B.; Bhowmik, S.K.; Gupta, V.; Arora, K.; et al. Fatty acid oxidation-driven Src links mitochondrial energy reprogramming and oncogenic properties in triple-negative breast cancer. Cell Rep. 2016, 14, 2154–2165. [Google Scholar] [CrossRef]
- Evans, D.R.; Guy, H.I. Mammalian pyrimidine biosynthesis: Fresh insights into an ancient pathway. J. Biol. Chem. 2004, 279, 33035–33038. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.; Menon, D.; Bernfeld, E.; Mroz, V.; Kalan, S.; Loayza, D.; Foster, D.A. Aspartate rescues S-phase arrest caused by suppression of glutamine utilization in KRas-driven cancer cells. J. Biol. Chem. 2016, 291, 9322–9329. [Google Scholar] [CrossRef] [PubMed]
- Rabinovich, S.; Adler, L.; Yizhak, K.; Sarver, A.; Silberman, A.; Agron, S.; Stettner, N.; Sun, Q.; Brandis, A.; Helbling, D.; et al. Diversion of aspartate in ASS1-deficient tumours fosters de novo pyrimidine synthesis. Nature 2015, 527, 379–383. [Google Scholar] [CrossRef] [PubMed]
- Husson, A.; Brasse-Lagnel, C.; Fairand, A.; Renouf, S.; Lavoinne, A. Argininosuccinate synthetase from the urea cycle to the citrulline-NO cycle. Eur. J. Biochem. 2003, 270, 1887–1899. [Google Scholar] [CrossRef]
- Keshet, R.; Szlosarek, P.; Carracedo, A.; Erez, A. Rewiring urea cycle metabolism in cancer to support anabolism. Nat. Rev. Cancer 2018, 18, 634–645. [Google Scholar] [CrossRef]
- Ng, B.G.; Wolfe, L.A.; Ichikawa, M.; Markello, T.; He, M.; Tifft, C.J.; Gahl, W.A.; Freeze, H.H. Biallelic mutations in CAD, impair de novo pyrimidine biosynthesis and decrease glycosylation precursors. Hum. Mol. Genet. 2015, 24, 3050–3057. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Birsoy, K.; Possemato, R.; Lorbeer, F.K.; Bayraktar, E.C.; Thiru, P.; Yucel, B.; Wang, T.; Chen, W.W.; Clish, C.B.; Sabatini, D.M. Metabolic determinants of cancer cell sensitivity to glucose limitation and biguanides. Nature 2014, 508, 108–112. [Google Scholar] [CrossRef] [PubMed]
- Votyakova, T.V.; Reynolds, I.J. DeltaPsi(m)-Dependent and -independent production of reactive oxygen species by rat brain mitochondria. J. Neurochem. 2001, 79, 266–277. [Google Scholar] [CrossRef] [PubMed]
- Martínez-Reyes, I.; Cardona, L.R.; Kong, H.; Vasan, K.; McElroy, G.S.; Werner, M.; Kihshen, H.; Reczek, C.R.; Weinberg, S.E.; Gao, P.; et al. Mitochondrial ubiquinol oxidation is necessary for tumour growth. Nature 2020, 585, 288–292. [Google Scholar] [CrossRef]
- Sullivan, L.B.; Gui, D.Y.; Hosios, A.M.; Bush, L.N.; Freinkman, E.; Heiden, M.G.V. Supporting aspartate biosynthesis is an essential function of respiration in proliferating cells. Cell 2015, 162, 552–563. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Reid, M.A.; Kong, M.; Locasale, J.W. Metabolic interactions with cancer epigenetics. Mol. Asp. Med. 2017, 54, 50–57. [Google Scholar] [CrossRef]
- Muz, B.; De La Puente, P.; Azab, F.; Azab, A.K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia 2015, 3, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Vaupel, P.; Kallinowski, F.; Okunieff, P. Blood flow, oxygen and nutrient supply, and metabolic microenvironment of human tumors: A review. Cancer Res. 1989, 49, 6449–6465. [Google Scholar]
- Makovski, A.; Yaffe, E.; Shpungin, S.; Nir, U. Down-regulation of Fer induces ROS levels accompanied by ATM and p53 activation in colon carcinoma cells. Cell. Signal. 2012, 24, 1369–1374. [Google Scholar] [CrossRef] [PubMed]














Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mehazri, L.; Shpungin, S.; Bel, S.; Nir, U. Loss of Fer Jeopardizes Metabolic Plasticity and Mitochondrial Homeostasis in Lung and Breast Carcinoma Cells. Int. J. Mol. Sci. 2021, 22, 3387. https://doi.org/10.3390/ijms22073387
Mehazri L, Shpungin S, Bel S, Nir U. Loss of Fer Jeopardizes Metabolic Plasticity and Mitochondrial Homeostasis in Lung and Breast Carcinoma Cells. International Journal of Molecular Sciences. 2021; 22(7):3387. https://doi.org/10.3390/ijms22073387
Chicago/Turabian StyleMehazri, Linoy, Sally Shpungin, Shai Bel, and Uri Nir. 2021. "Loss of Fer Jeopardizes Metabolic Plasticity and Mitochondrial Homeostasis in Lung and Breast Carcinoma Cells" International Journal of Molecular Sciences 22, no. 7: 3387. https://doi.org/10.3390/ijms22073387