
Islet Regeneration: Endogenous and Exogenous Approaches


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Beta Cell Mass Reduction in Diabetes
3. Intrinsic Beta Cell Mass Expansion
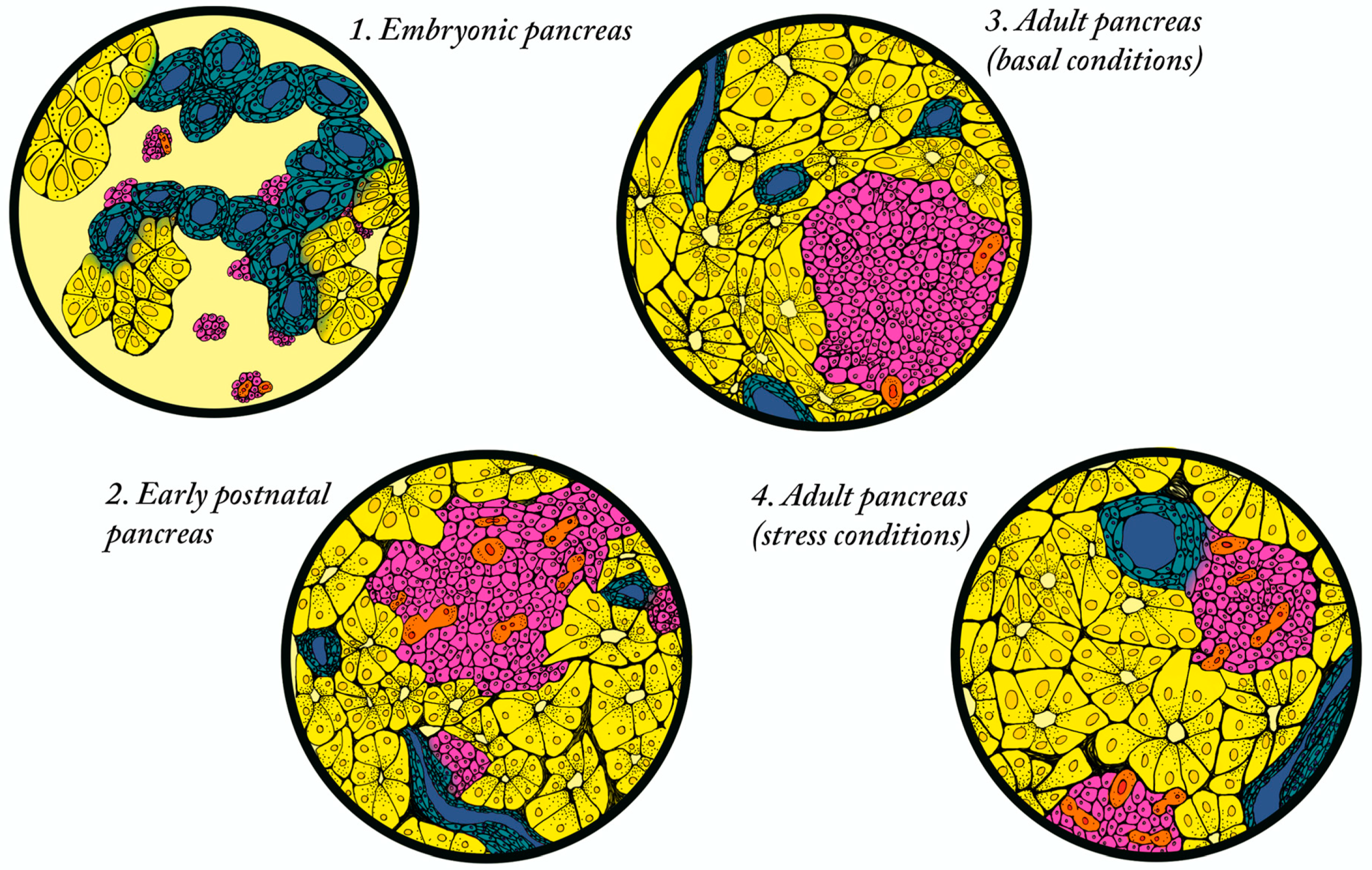
3.1. Establishment of Beta Cell Mass
3.2. Postnatal Expansion of Beta Cell Mass in Humans
4. Metabolic Stress Conditions Promote Increase in Beta Cell Mass
4.1. Beta Cell Mass in Metabolic Stress—Pregnancy
4.2. Beta Cell Mass in Metabolic Stress—Obesity
5. Models of Beta Cell Mass Expansion
5.1. Partial Duct Ligation: The Case for Neogenesis?
5.2. Partial Pancreatectomy: Dedifferentiation/Redifferentiation?
5.3. Beta Cell Ablation Models: Plasticity?
6. Mechanisms of Beta Cell Proliferation
6.1. Cell Cycle Regulators
6.2. Transcriptional Regulation of the Cell Cycle
6.3. Intracellular Signaling Pathways
6.4. Small Molecule Induction of Beta Cell Regeneration
7. Exogenous Sources of Beta Cells
7.1. Cadaveric Beta Cells
7.2. Mesenchymal Stem Cells
7.3. Human Pluripotent Cells: hESCs and iPSCs
8. Conclusions
Funding
Acknowledgments
Conflicts of Interest
References
- Deng, S.; Vatamaniuk, M.; Huang, X.; Doliba, N.; Lian, M.M.; Frank, A.; Velidedeoglu, E.; Desai, N.M.; Koeberlein, B.; Wolf, B.; et al. Structural and functional abnormalities in the islets isolated from type 2 diabetic subjects. Diabetes 2004, 53, 624–632. [Google Scholar] [CrossRef] [Green Version]
- Marselli, L.; Piron, A.; Suleiman, M.; Colli, M.L.; Yi, X.; Khamis, A.; Carrat, G.R.; Rutter, G.A.; Bugliani, M.; Giusti, L.; et al. Persistent or Transient Human beta Cell Dysfunction Induced by Metabolic Stress: Specific Signatures and Shared Gene Expression with Type 2 Diabetes. Cell Rep. 2020, 33, 108466. [Google Scholar] [CrossRef] [PubMed]
- Ritzel, R.A.; Butler, A.E.; Rizza, R.A.; Veldhuis, J.D.; Butler, P.C. Relationship between beta-cell mass and fasting blood glucose concentration in humans. Diabetes Care 2006, 29, 717–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linnemann, A.K.; Baan, M.; Davis, D.B. Pancreatic beta-cell proliferation in obesity. Adv. Nutr. 2014, 5, 278–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, J.J.; Saunders, D.C.; Dai, C.; Poffenberger, G.; Cairns, B.; Serreze, D.V.; Harlan, D.M.; Bottino, R.; Brissova, M.; Powers, A.C. Decreased pancreatic acinar cell number in type 1 diabetes. Diabetologia 2020, 63, 1418–1423. [Google Scholar] [CrossRef] [PubMed]
- Butler, A.E.; Janson, J.; Bonner-Weir, S.; Ritzel, R.; Rizza, R.A.; Butler, P.C. Beta-cell deficit and increased beta-cell apoptosis in humans with type 2 diabetes. Diabetes 2003, 52, 102–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, K.; Yaginuma, N.; Takahashi, T. Differential volumetry of A, B and D cells in the pancreatic islets of diabetic and nondiabetic subjects. Tohoku J. Exp. Med. 1979, 129, 273–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kloppel, G.; Lohr, M.; Habich, K.; Oberholzer, M.; Heitz, P.U. Islet pathology and the pathogenesis of type 1 and type 2 diabetes mellitus revisited. Surv. Synth. Pathol. Res. 1985, 4, 110–125. [Google Scholar] [CrossRef]
- Weir, G.C.; Gaglia, J.; Bonner-Weir, S. Inadequate beta-cell mass is essential for the pathogenesis of type 2 diabetes. Lancet Diabetes Endocrinol. 2020, 8, 249–256. [Google Scholar] [CrossRef]
- Lee, H.; Lee, Y.S.; Harenda, Q.; Pietrzak, S.; Oktay, H.Z.; Schreiber, S.; Liao, Y.; Sonthalia, S.; Ciecko, A.E.; Chen, Y.G.; et al. Beta Cell Dedifferentiation Induced by IRE1alpha Deletion Prevents Type 1 Diabetes. Cell Metab. 2020, 31, 822–836 e825. [Google Scholar] [CrossRef] [PubMed]
- Talchai, C.; Xuan, S.; Lin, H.V.; Sussel, L.; Accili, D. Pancreatic beta cell dedifferentiation as a mechanism of diabetic beta cell failure. Cell 2012, 150, 1223–1234. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; York, N.W.; Nichols, C.G.; Remedi, M.S. Pancreatic beta cell dedifferentiation in diabetes and redifferentiation following insulin therapy. Cell Metab. 2014, 19, 872–882. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cinti, F.; Bouchi, R.; Kim-Muller, J.Y.; Ohmura, Y.; Sandoval, P.R.; Masini, M.; Marselli, L.; Suleiman, M.; Ratner, L.E.; Marchetti, P.; et al. Evidence of beta-Cell Dedifferentiation in Human Type 2 Diabetes. J. Clin. Endocrinol. Metab. 2016, 101, 1044–1054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.J.; Schug, J.; Won, K.J.; Liu, C.; Naji, A.; Avrahami, D.; Golson, M.L.; Kaestner, K.H. Single-Cell Transcriptomics of the Human Endocrine Pancreas. Diabetes 2016, 65, 3028–3038. [Google Scholar] [CrossRef] [Green Version]
- Moin, A.S.M.; Butler, A.E. Alterations in Beta Cell Identity in Type 1 and Type 2 Diabetes. Curr. Diab. Rep. 2019, 19, 83. [Google Scholar] [CrossRef] [Green Version]
- Finegood, D.T.; Scaglia, L.; Bonner-Weir, S. Dynamics of beta-cell mass in the growing rat pancreas. Estimation with a simple mathematical model. Diabetes 1995, 44, 249–256. [Google Scholar] [CrossRef]
- Georgia, S.; Bhushan, A. Beta cell replication is the primary mechanism for maintaining postnatal beta cell mass. J. Clin. Invest. 2004, 114, 963–968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gregg, B.E.; Moore, P.C.; Demozay, D.; Hall, B.A.; Li, M.; Husain, A.; Wright, A.J.; Atkinson, M.A.; Rhodes, C.J. Formation of a human beta-cell population within pancreatic islets is set early in life. J. Clin. Endocrinol. Metab. 2012, 97, 3197–3206. [Google Scholar] [CrossRef] [PubMed]
- Gradwohl, G.; Dierich, A.; LeMeur, M.; Guillemot, F. neurogenin3 is required for the development of the four endocrine cell lineages of the pancreas. Proc. Natl. Acad. Sci. USA 2000, 97, 1607–1611. [Google Scholar] [CrossRef] [Green Version]
- Gu, G.; Dubauskaite, J.; Melton, D.A. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development 2002, 129, 2447–2457. [Google Scholar] [PubMed]
- Romer, A.I.; Sussel, L. Pancreatic islet cell development and regeneration. Curr. Opin. Endocrinol. Diabetes Obes. 2015, 22, 255–264. [Google Scholar] [CrossRef] [Green Version]
- Bouwens, L.; Wang, R.N.; De Blay, E.; Pipeleers, D.G.; Kloppel, G. Cytokeratins as markers of ductal cell differentiation and islet neogenesis in the neonatal rat pancreas. Diabetes 1994, 43, 1279–1283. [Google Scholar] [CrossRef] [PubMed]
- Dhawan, S.; Georgia, S.; Bhushan, A. Formation and regeneration of the endocrine pancreas. Curr. Opin. Cell Biol. 2007, 19, 634–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seymour, P.A.; Sander, M. Historical perspective: Beginnings of the beta-cell: Current perspectives in beta-cell development. Diabetes 2011, 60, 364–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meier, J.J.; Butler, A.E.; Saisho, Y.; Monchamp, T.; Galasso, R.; Bhushan, A.; Rizza, R.A.; Butler, P.C. Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes 2008, 57, 1584–1594. [Google Scholar] [CrossRef] [Green Version]
- Nikolova, G.; Jabs, N.; Konstantinova, I.; Domogatskaya, A.; Tryggvason, K.; Sorokin, L.; Fassler, R.; Gu, G.; Gerber, H.P.; Ferrara, N.; et al. The vascular basement membrane: A niche for insulin gene expression and Beta cell proliferation. Dev. Cell 2006, 10, 397–405. [Google Scholar] [CrossRef] [Green Version]
- Diaferia, G.R.; Jimenez-Caliani, A.J.; Ranjitkar, P.; Yang, W.; Hardiman, G.; Rhodes, C.J.; Crisa, L.; Cirulli, V. beta1 integrin is a crucial regulator of pancreatic beta-cell expansion. Development 2013, 140, 3360–3372. [Google Scholar] [CrossRef] [Green Version]
- Epshtein, A.; Rachi, E.; Sakhneny, L.; Mizrachi, S.; Baer, D.; Landsman, L. Neonatal pancreatic pericytes support beta-cell proliferation. Mol. Metab. 2017, 6, 1330–1338. [Google Scholar] [CrossRef] [PubMed]
- Sakhneny, L.; Epshtein, A.; Landsman, L. Pericytes contribute to the islet basement membranes to promote beta-cell gene expression. Sci. Rep. 2021, 11, 2378. [Google Scholar] [CrossRef]
- Townsend, S.E.; Gannon, M. Extracellular Matrix-Associated Factors Play Critical Roles in Regulating Pancreatic beta-Cell Proliferation and Survival. Endocrinology 2019, 160, 1885–1894. [Google Scholar] [CrossRef] [Green Version]
- Aamodt, K.I.; Powers, A.C. Signals in the pancreatic islet microenvironment influence beta-cell proliferation. Diabetes Obes. Metab. 2017, 19 (Suppl. 1), 124–136. [Google Scholar] [CrossRef] [Green Version]
- Mussar, K.; Pardike, S.; Hohl, T.M.; Hardiman, G.; Cirulli, V.; Crisa, L. A CCR2+ myeloid cell niche required for pancreatic beta cell growth. JCI Insight 2017, 2. [Google Scholar] [CrossRef] [Green Version]
- Brissova, M.; Aamodt, K.; Brahmachary, P.; Prasad, N.; Hong, J.Y.; Dai, C.; Mellati, M.; Shostak, A.; Poffenberger, G.; Aramandla, R.; et al. Islet microenvironment, modulated by vascular endothelial growth factor-A signaling, promotes beta cell regeneration. Cell Metab. 2014, 19, 498–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teta, M.; Rankin, M.M.; Long, S.Y.; Stein, G.M.; Kushner, J.A. Growth and regeneration of adult beta cells does not involve specialized progenitors. Dev. Cell 2007, 12, 817–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dor, Y.; Brown, J.; Martinez, O.I.; Melton, D.A. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 2004, 429, 41–46. [Google Scholar] [CrossRef] [Green Version]
- Montanya, E.; Nacher, V.; Biarnes, M.; Soler, J. Linear correlation between beta-cell mass and body weight throughout the lifespan in Lewis rats: Role of beta-cell hyperplasia and hypertrophy. Diabetes 2000, 49, 1341–1346. [Google Scholar] [CrossRef] [Green Version]
- Teta, M.; Long, S.Y.; Wartschow, L.M.; Rankin, M.M.; Kushner, J.A. Very slow turnover of beta-cells in aged adult mice. Diabetes 2005, 54, 2557–2567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dominguez-Bendala, J.; Qadir, M.M.F.; Pastori, R.L. Pancreatic Progenitors: There and Back Again. Trends Endocrinol. Metab. 2019, 30, 4–11. [Google Scholar] [CrossRef] [PubMed]
- van der Meulen, T.; Mawla, A.M.; DiGruccio, M.R.; Adams, M.W.; Nies, V.; Dolleman, S.; Liu, S.; Ackermann, A.M.; Caceres, E.; Hunter, A.E.; et al. Virgin Beta Cells Persist throughout Life at a Neogenic Niche within Pancreatic Islets. Cell Metab. 2017, 25, 911–926 e916. [Google Scholar] [CrossRef] [Green Version]
- Laurent, D.; Vinet, L.; Lamprianou, S.; Daval, M.; Filhoulaud, G.; Ktorza, A.; Wang, H.; Sewing, S.; Juretschke, H.P.; Glombik, H.; et al. Pancreatic beta-cell imaging in humans: Fiction or option? Diabetes Obes. Metab. 2016, 18, 6–15. [Google Scholar] [CrossRef]
- Wei, W.; Ehlerding, E.B.; Lan, X.; Luo, Q.Y.; Cai, W. Molecular imaging of beta-cells: Diabetes and beyond. Adv. Drug Deliv. Rev. 2019, 139, 16–31. [Google Scholar] [CrossRef] [PubMed]
- Aguayo-Mazzucato, C.; Bonner-Weir, S. Pancreatic beta Cell Regeneration as a Possible Therapy for Diabetes. Cell Metab. 2018, 27, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Swartz, F.J.; Carstens, P.H.B. An Islet of Langerhans Located within the Epithelium of a Human Pancreatic Duct. Histol. Histopathol. 1986, 1, 111–117. [Google Scholar] [PubMed]
- Loomans, C.J.M.; Williams Giuliani, N.; Balak, J.; Ringnalda, F.; van Gurp, L.; Huch, M.; Boj, S.F.; Sato, T.; Kester, L.; de Sousa Lopes, S.M.C.; et al. Expansion of Adult Human Pancreatic Tissue Yields Organoids Harboring Progenitor Cells with Endocrine Differentiation Potential. Stem Cell Rep. 2018, 10, 712–724. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.; Newton, K.M.; Knopp, R.H. Gestational diabetes and the incidence of type 2 diabetes: A systematic review. Diabetes Care 2002, 25, 1862–1868. [Google Scholar] [CrossRef] [Green Version]
- Parsons, J.A.; Bartke, A.; Sorenson, R.L. Number and size of islets of Langerhans in pregnant, human growth hormone-expressing transgenic, and pituitary dwarf mice: Effect of lactogenic hormones. Endocrinology 1995, 136, 2013–2021. [Google Scholar] [CrossRef]
- Parsons, J.A.; Brelje, T.C.; Sorenson, R.L. Adaptation of islets of Langerhans to pregnancy: Increased islet cell proliferation and insulin secretion correlates with the onset of placental lactogen secretion. Endocrinology 1992, 130, 1459–1466. [Google Scholar] [CrossRef]
- Rieck, S.; White, P.; Schug, J.; Fox, A.J.; Smirnova, O.; Gao, N.; Gupta, R.K.; Wang, Z.V.; Scherer, P.E.; Keller, M.P.; et al. The transcriptional response of the islet to pregnancy in mice. Mol. Endocrinol. 2009, 23, 1702–1712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scaglia, L.; Smith, F.E.; Bonner-Weir, S. Apoptosis contributes to the involution of beta cell mass in the post partum rat pancreas. Endocrinology 1995, 136, 5461–5468. [Google Scholar] [CrossRef] [PubMed]
- Sorenson, R.L.; Brelje, T.C. Adaptation of islets of Langerhans to pregnancy: Beta-cell growth, enhanced insulin secretion and the role of lactogenic hormones. Horm. Metab. Res. 1997, 29, 301–307. [Google Scholar] [CrossRef]
- Baeyens, L.; Hindi, S.; Sorenson, R.L.; German, M.S. beta-Cell adaptation in pregnancy. Diabetes Obes. Metab. 2016, 18 (Suppl. 1), 63–70. [Google Scholar] [CrossRef] [Green Version]
- Brelje, T.C.; Bhagroo, N.V.; Stout, L.E.; Sorenson, R.L. Beneficial effects of lipids and prolactin on insulin secretion and beta-cell proliferation: A role for lipids in the adaptation of islets to pregnancy. J. Endocrinol. 2008, 197, 265–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorenson, R.L.; Brelje, T.C. Prolactin receptors are critical to the adaptation of islets to pregnancy. Endocrinology 2009, 150, 1566–1569. [Google Scholar] [CrossRef] [Green Version]
- Freemark, M.; Avril, I.; Fleenor, D.; Driscoll, P.; Petro, A.; Opara, E.; Kendall, W.; Oden, J.; Bridges, S.; Binart, N.; et al. Targeted deletion of the PRL receptor: Effects on islet development, insulin production, and glucose tolerance. Endocrinology 2002, 143, 1378–1385. [Google Scholar] [CrossRef]
- Goyvaerts, L.; Lemaire, K.; Arijs, I.; Auffret, J.; Granvik, M.; Van Lommel, L.; Binart, N.; in’t Veld, P.; Schuit, F.; Schraenen, A. Prolactin receptors and placental lactogen drive male mouse pancreatic islets to pregnancy-related mRNA changes. PLoS ONE 2015, 10, e0121868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, C.; Snider, F.; Cross, J.C. Prolactin receptor is required for normal glucose homeostasis and modulation of beta-cell mass during pregnancy. Endocrinology 2009, 150, 1618–1626. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Zhang, J.; Pope, C.F.; Crawford, L.A.; Vasavada, R.C.; Jagasia, S.M.; Gannon, M. Gestational diabetes mellitus resulting from impaired beta-cell compensation in the absence of FoxM1, a novel downstream effector of placental lactogen. Diabetes 2010, 59, 143–152. [Google Scholar] [CrossRef] [Green Version]
- Hanley, S.C.; Austin, E.; Assouline-Thomas, B.; Kapeluto, J.; Blaichman, J.; Moosavi, M.; Petropavlovskaia, M.; Rosenberg, L. {beta}-Cell mass dynamics and islet cell plasticity in human type 2 diabetes. Endocrinology 2010, 151, 1462–1472. [Google Scholar] [CrossRef] [Green Version]
- Rahier, J.; Guiot, Y.; Goebbels, R.M.; Sempoux, C.; Henquin, J.C. Pancreatic beta-cell mass in European subjects with type 2 diabetes. Diabetes Obes. Metab. 2008, 10 (Suppl. 4), 32–42. [Google Scholar] [CrossRef]
- Saisho, Y.; Butler, A.E.; Manesso, E.; Elashoff, D.; Rizza, R.A.; Butler, P.C. beta-cell mass and turnover in humans: Effects of obesity and aging. Diabetes Care 2013, 36, 111–117. [Google Scholar] [CrossRef] [Green Version]
- Flier, S.N.; Kulkarni, R.N.; Kahn, C.R. Evidence for a circulating islet cell growth factor in insulin-resistant states. Proc. Natl. Acad. Sci. USA 2001, 98, 7475–7480. [Google Scholar] [CrossRef] [Green Version]
- Bock, T.; Pakkenberg, B.; Buschard, K. Increased islet volume but unchanged islet number in ob/ob mice. Diabetes 2003, 52, 1716–1722. [Google Scholar] [CrossRef] [Green Version]
- Hull, R.L.; Kodama, K.; Utzschneider, K.M.; Carr, D.B.; Prigeon, R.L.; Kahn, S.E. Dietary-fat-induced obesity in mice results in beta cell hyperplasia but not increased insulin release: Evidence for specificity of impaired beta cell adaptation. Diabetologia 2005, 48, 1350–1358. [Google Scholar] [CrossRef] [Green Version]
- Keller, M.P.; Choi, Y.; Wang, P.; Davis, D.B.; Rabaglia, M.E.; Oler, A.T.; Stapleton, D.S.; Argmann, C.; Schueler, K.L.; Edwards, S.; et al. A gene expression network model of type 2 diabetes links cell cycle regulation in islets with diabetes susceptibility. Genome Res. 2008, 18, 706–716. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasavada, R.C.; Gonzalez-Pertusa, J.A.; Fujinaka, Y.; Fiaschi-Taesch, N.; Cozar-Castellano, I.; Garcia-Ocana, A. Growth factors and beta cell replication. Int. J. Biochem. Cell Biol. 2006, 38, 931–950. [Google Scholar] [CrossRef] [PubMed]
- El Ouaamari, A.; Kawamori, D.; Dirice, E.; Liew, C.W.; Shadrach, J.L.; Hu, J.; Katsuta, H.; Hollister-Lock, J.; Qian, W.J.; Wagers, A.J.; et al. Liver-derived systemic factors drive beta cell hyperplasia in insulin-resistant states. Cell Rep. 2013, 3, 401–410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabir, S.; Saleem, A.; Akhtar, M.F.; Saleem, M.; Raza, M. Increasing beta cell mass to treat diabetes mellitus. Adv. Clin. Exp. Med. 2018, 27, 1309–1315. [Google Scholar] [CrossRef] [PubMed]
- Ogilvie, R.F. The islands of langerhans in 19 cases of obesity. J. Pathol. Bacteriol. 1933, 37, 473–481. [Google Scholar] [CrossRef]
- Folli, F.; Okada, T.; Perego, C.; Gunton, J.; Liew, C.W.; Akiyama, M.; D’Amico, A.; La Rosa, S.; Placidi, C.; Lupi, R.; et al. Altered insulin receptor signalling and beta-cell cycle dynamics in type 2 diabetes mellitus. PLoS ONE 2011, 6, e28050. [Google Scholar] [CrossRef] [Green Version]
- Davis, D.B.; Lavine, J.A.; Suhonen, J.I.; Krautkramer, K.A.; Rabaglia, M.E.; Sperger, J.M.; Fernandez, L.A.; Yandell, B.S.; Keller, M.P.; Wang, I.M.; et al. FoxM1 is up-regulated by obesity and stimulates beta-cell proliferation. Mol. Endocrinol. 2010, 24, 1822–1834. [Google Scholar] [CrossRef] [Green Version]
- Fiaschi-Taesch, N.M.; Kleinberger, J.W.; Salim, F.G.; Troxell, R.; Wills, R.; Tanwir, M.; Casinelli, G.; Cox, A.E.; Takane, K.K.; Scott, D.K.; et al. Human pancreatic beta-cell G1/S molecule cell cycle atlas. Diabetes 2013, 62, 2450–2459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fiaschi-Taesch, N.M.; Kleinberger, J.W.; Salim, F.G.; Troxell, R.; Wills, R.; Tanwir, M.; Casinelli, G.; Cox, A.E.; Takane, K.K.; Srinivas, H.; et al. Cytoplasmic-nuclear trafficking of G1/S cell cycle molecules and adult human beta-cell replication: A revised model of human beta-cell G1/S control. Diabetes 2013, 62, 2460–2470. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.N.; Kloppel, G.; Bouwens, L. Duct- to islet-cell differentiation and islet growth in the pancreas of duct-ligated adult rats. Diabetologia 1995, 38, 1405–1411. [Google Scholar] [CrossRef]
- Xu, X.; D’Hoker, J.; Stange, G.; Bonne, S.; De Leu, N.; Xiao, X.; Van de Casteele, M.; Mellitzer, G.; Ling, Z.; Pipeleers, D.; et al. Beta cells can be generated from endogenous progenitors in injured adult mouse pancreas. Cell 2008, 132, 197–207. [Google Scholar] [CrossRef] [Green Version]
- Kopp, J.L.; Dubois, C.L.; Schaffer, A.E.; Hao, E.; Shih, H.P.; Seymour, P.A.; Ma, J.; Sander, M. Sox9+ ductal cells are multipotent progenitors throughout development but do not produce new endocrine cells in the normal or injured adult pancreas. Development 2011, 138, 653–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Solar, M.; Cardalda, C.; Houbracken, I.; Martin, M.; Maestro, M.A.; De Medts, N.; Xu, X.; Grau, V.; Heimberg, H.; Bouwens, L.; et al. Pancreatic exocrine duct cells give rise to insulin-producing beta cells during embryogenesis but not after birth. Dev. Cell 2009, 17, 849–860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van de Casteele, M.; Leuckx, G.; Baeyens, L.; Cai, Y.; Yuchi, Y.; Coppens, V.; De Groef, S.; Eriksson, M.; Svensson, C.; Ahlgren, U.; et al. Neurogenin 3+ cells contribute to beta-cell neogenesis and proliferation in injured adult mouse pancreas. Cell Death Dis 2013, 4, e523. [Google Scholar] [CrossRef] [Green Version]
- Xiao, X.; Guo, P.; Shiota, C.; Prasadan, K.; El-Gohary, Y.; Wiersch, J.; Gaffar, I.; Gittes, G.K. Neurogenin3 activation is not sufficient to direct duct-to-beta cell transdifferentiation in the adult pancreas. J. Biol. Chem. 2013, 288, 25297–25308. [Google Scholar] [CrossRef] [Green Version]
- Baron, M.; Veres, A.; Wolock, S.L.; Faust, A.L.; Gaujoux, R.; Vetere, A.; Ryu, J.H.; Wagner, B.K.; Shen-Orr, S.S.; Klein, A.M.; et al. A Single-Cell Transcriptomic Map of the Human and Mouse Pancreas Reveals Inter- and Intra-cell Population Structure. Cell Syst. 2016, 3, 346–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qadir, M.M.F.; Alvarez-Cubela, S.; Klein, D.; van Dijk, J.; Muniz-Anquela, R.; Moreno-Hernandez, Y.B.; Lanzoni, G.; Sadiq, S.; Navarro-Rubio, B.; Garcia, M.T.; et al. Single-cell resolution analysis of the human pancreatic ductal progenitor cell niche. Proc. Natl. Acad. Sci. USA 2020, 117, 10876–10887. [Google Scholar] [CrossRef]
- Tritschler, S.; Theis, F.J.; Lickert, H.; Bottcher, A. Systematic single-cell analysis provides new insights into heterogeneity and plasticity of the pancreas. Mol. Metab. 2017, 6, 974–990. [Google Scholar] [CrossRef]
- Jonas, J.C.; Sharma, A.; Hasenkamp, W.; Ilkova, H.; Patane, G.; Laybutt, R.; Bonner-Weir, S.; Weir, G.C. Chronic hyperglycemia triggers loss of pancreatic beta cell differentiation in an animal model of diabetes. J. Biol. Chem. 1999, 274, 14112–14121. [Google Scholar] [CrossRef] [Green Version]
- Quintens, R.; Hendrickx, N.; Lemaire, K.; Schuit, F. Why expression of some genes is disallowed in beta-cells. Biochem. Soc. Trans. 2008, 36, 300–305. [Google Scholar] [CrossRef]
- Sekine, N.; Cirulli, V.; Regazzi, R.; Brown, L.J.; Gine, E.; Tamarit-Rodriguez, J.; Girotti, M.; Marie, S.; MacDonald, M.J.; Wollheim, C.B.; et al. Low lactate dehydrogenase and high mitochondrial glycerol phosphate dehydrogenase in pancreatic beta-cells. Potential role in nutrient sensing. J. Biol. Chem. 1994, 269, 4895–4902. [Google Scholar] [CrossRef]
- Gao, T.; McKenna, B.; Li, C.; Reichert, M.; Nguyen, J.; Singh, T.; Yang, C.; Pannikar, A.; Doliba, N.; Zhang, T.; et al. Pdx1 maintains beta cell identity and function by repressing an alpha cell program. Cell Metab. 2014, 19, 259–271. [Google Scholar] [CrossRef] [Green Version]
- Gu, C.; Stein, G.H.; Pan, N.; Goebbels, S.; Hornberg, H.; Nave, K.A.; Herrera, P.; White, P.; Kaestner, K.H.; Sussel, L.; et al. Pancreatic beta cells require NeuroD to achieve and maintain functional maturity. Cell Metab. 2010, 11, 298–310. [Google Scholar] [CrossRef] [Green Version]
- Segerstolpe, A.; Palasantza, A.; Eliasson, P.; Andersson, E.M.; Andreasson, A.C.; Sun, X.; Picelli, S.; Sabirsh, A.; Clausen, M.; Bjursell, M.K.; et al. Single-Cell Transcriptome Profiling of Human Pancreatic Islets in Health and Type 2 Diabetes. Cell Metab. 2016, 24, 593–607. [Google Scholar] [CrossRef] [Green Version]
- Xin, Y.; Kim, J.; Okamoto, H.; Ni, M.; Wei, Y.; Adler, C.; Murphy, A.J.; Yancopoulos, G.D.; Lin, C.; Gromada, J. RNA Sequencing of Single Human Islet Cells Reveals Type 2 Diabetes Genes. Cell Metab. 2016, 24, 608–615. [Google Scholar] [CrossRef] [Green Version]
- Collombat, P.; Mansouri, A.; Hecksher-Sorensen, J.; Serup, P.; Krull, J.; Gradwohl, G.; Gruss, P. Opposing actions of Arx and Pax4 in endocrine pancreas development. Genes Dev. 2003, 17, 2591–2603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Courtney, M.; Gjernes, E.; Druelle, N.; Ravaud, C.; Vieira, A.; Ben-Othman, N.; Pfeifer, A.; Avolio, F.; Leuckx, G.; Lacas-Gervais, S.; et al. The inactivation of Arx in pancreatic alpha-cells triggers their neogenesis and conversion into functional beta-like cells. PLoS Genet. 2013, 9, e1003934. [Google Scholar] [CrossRef]
- Lu, J.; Herrera, P.L.; Carreira, C.; Bonnavion, R.; Seigne, C.; Calender, A.; Bertolino, P.; Zhang, C.X. Alpha cell-specific Men1 ablation triggers the transdifferentiation of glucagon-expressing cells and insulinoma development. Gastroenterology 2010, 138, 1954–1965. [Google Scholar] [CrossRef]
- Papizan, J.B.; Singer, R.A.; Tschen, S.I.; Dhawan, S.; Friel, J.M.; Hipkens, S.B.; Magnuson, M.A.; Bhushan, A.; Sussel, L. Nkx2.2 repressor complex regulates islet beta-cell specification and prevents beta-to-alpha-cell reprogramming. Genes Dev. 2011, 25, 2291–2305. [Google Scholar] [CrossRef] [Green Version]
- Thorel, F.; Nepote, V.; Avril, I.; Kohno, K.; Desgraz, R.; Chera, S.; Herrera, P.L. Conversion of adult pancreatic alpha-cells to beta-cells after extreme beta-cell loss. Nature 2010, 464, 1149–1154. [Google Scholar] [CrossRef] [Green Version]
- Chera, S.; Baronnier, D.; Ghila, L.; Cigliola, V.; Jensen, J.N.; Gu, G.; Furuyama, K.; Thorel, F.; Gribble, F.M.; Reimann, F.; et al. Diabetes recovery by age-dependent conversion of pancreatic delta-cells into insulin producers. Nature 2014, 514, 503–507. [Google Scholar] [CrossRef] [PubMed]
- Spijker, H.S.; Song, H.; Ellenbroek, J.H.; Roefs, M.M.; Engelse, M.A.; Bos, E.; Koster, A.J.; Rabelink, T.J.; Hansen, B.C.; Clark, A.; et al. Loss of beta-Cell Identity Occurs in Type 2 Diabetes and Is Associated With Islet Amyloid Deposits. Diabetes 2015, 64, 2928–2938. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, M.G.; Marshall, H.L.; Rigby, R.; Huang, G.C.; Amer, A.; Booth, T.; White, S.; Shaw, J.A. Expression of mesenchymal and alpha-cell phenotypic markers in islet beta-cells in recently diagnosed diabetes. Diabetes Care 2013, 36, 3818–3820. [Google Scholar] [CrossRef] [Green Version]
- Kushner, J.A.; Ciemerych, M.A.; Sicinska, E.; Wartschow, L.M.; Teta, M.; Long, S.Y.; Sicinski, P.; White, M.F. Cyclins D2 and D1 are essential for postnatal pancreatic beta-cell growth. Mol. Cell Biol. 2005, 25, 3752–3762. [Google Scholar] [CrossRef] [Green Version]
- Rane, S.G.; Dubus, P.; Mettus, R.V.; Galbreath, E.J.; Boden, G.; Reddy, E.P.; Barbacid, M. Loss of Cdk4 expression causes insulin-deficient diabetes and Cdk4 activation results in beta-islet cell hyperplasia. Nat. Genet. 1999, 22, 44–52. [Google Scholar] [CrossRef]
- Hino, S.; Yamaoka, T.; Yamashita, Y.; Yamada, T.; Hata, J.; Itakura, M. In vivo proliferation of differentiated pancreatic islet beta cells in transgenic mice expressing mutated cyclin-dependent kinase 4. Diabetologia 2004, 47, 1819–1830. [Google Scholar] [CrossRef] [PubMed]
- Marzo, N.; Mora, C.; Fabregat, M.E.; Martin, J.; Usac, E.F.; Franco, C.; Barbacid, M.; Gomis, R. Pancreatic islets from cyclin-dependent kinase 4/R24C (Cdk4) knockin mice have significantly increased beta cell mass and are physiologically functional, indicating that Cdk4 is a potential target for pancreatic beta cell mass regeneration in Type 1 diabetes. Diabetologia 2004, 47, 686–694. [Google Scholar] [CrossRef] [Green Version]
- Dhawan, S.; Tschen, S.I.; Bhushan, A. Bmi-1 regulates the Ink4a/Arf locus to control pancreatic beta-cell proliferation. Genes Dev. 2009, 23, 906–911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnamurthy, J.; Ramsey, M.R.; Ligon, K.L.; Torrice, C.; Koh, A.; Bonner-Weir, S.; Sharpless, N.E. p16INK4a induces an age-dependent decline in islet regenerative potential. Nature 2006, 443, 453–457. [Google Scholar] [CrossRef] [PubMed]
- Salpeter, S.J.; Khalaileh, A.; Weinberg-Corem, N.; Ziv, O.; Glaser, B.; Dor, Y. Systemic regulation of the age-related decline of pancreatic beta-cell replication. Diabetes 2013, 62, 2843–2848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puri, S.; Roy, N.; Russ, H.A.; Leonhardt, L.; French, E.K.; Roy, R.; Bengtsson, H.; Scott, D.K.; Stewart, A.F.; Hebrok, M. Replication confers beta cell immaturity. Nat. Commun. 2018, 9, 485. [Google Scholar] [CrossRef]
- Bader, E.; Migliorini, A.; Gegg, M.; Moruzzi, N.; Gerdes, J.; Roscioni, S.S.; Bakhti, M.; Brandl, E.; Irmler, M.; Beckers, J.; et al. Identification of proliferative and mature beta-cells in the islets of Langerhans. Nature 2016, 535, 430–434. [Google Scholar] [CrossRef]
- Arda, H.E.; Li, L.; Tsai, J.; Torre, E.A.; Rosli, Y.; Peiris, H.; Spitale, R.C.; Dai, C.; Gu, X.; Qu, K.; et al. Age-Dependent Pancreatic Gene Regulation Reveals Mechanisms Governing Human beta Cell Function. Cell Metab. 2016, 23, 909–920. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Bender, A.; Wang, P.; Karakose, E.; Inabnet, W.B.; Libutti, S.K.; Arnold, A.; Lambertini, L.; Stang, M.; Chen, H.; et al. Insights into beta cell regeneration for diabetes via integration of molecular landscapes in human insulinomas. Nat. Commun. 2017, 8, 767. [Google Scholar] [CrossRef] [Green Version]
- Ackermann Misfeldt, A.; Costa, R.H.; Gannon, M. Beta-cell proliferation, but not neogenesis, following 60% partial pancreatectomy is impaired in the absence of FoxM1. Diabetes 2008, 57, 3069–3077. [Google Scholar] [CrossRef] [Green Version]
- Karslioglu, E.; Kleinberger, J.W.; Salim, F.G.; Cox, A.E.; Takane, K.K.; Scott, D.K.; Stewart, A.F. cMyc is a principal upstream driver of beta-cell proliferation in rat insulinoma cell lines and is an effective mediator of human beta-cell replication. Mol. Endocrinol. 2011, 25, 1760–1772. [Google Scholar] [CrossRef] [Green Version]
- Rosselot, C.; Baumel-Alterzon, S.; Li, Y.; Brill, G.; Lambertini, L.; Katz, L.S.; Lu, G.; Garcia-Ocana, A.; Scott, D.K. The Many Lives of Myc in the Pancreatic beta-Cell. J. Biol. Chem. 2020. [Google Scholar] [CrossRef]
- Rosselot, C.; Kumar, A.; Lakshmipathi, J.; Zhang, P.; Lu, G.; Katz, L.S.; Prochownik, E.V.; Stewart, A.F.; Lambertini, L.; Scott, D.K.; et al. Myc Is Required for Adaptive beta-Cell Replication in Young Mice but Is Not Sufficient in One-Year-Old Mice Fed With a High-Fat Diet. Diabetes 2019, 68, 1934–1949. [Google Scholar] [CrossRef]
- Sander, M.; Sussel, L.; Conners, J.; Scheel, D.; Kalamaras, J.; Dela Cruz, F.; Schwitzgebel, V.; Hayes-Jordan, A.; German, M. Homeobox gene Nkx6.1 lies downstream of Nkx2.2 in the major pathway of beta-cell formation in the pancreas. Development 2000, 127, 5533–5540. [Google Scholar]
- Schisler, J.C.; Fueger, P.T.; Babu, D.A.; Hohmeier, H.E.; Tessem, J.S.; Lu, D.; Becker, T.C.; Naziruddin, B.; Levy, M.; Mirmira, R.G.; et al. Stimulation of human and rat islet beta-cell proliferation with retention of function by the homeodomain transcription factor Nkx6.1. Mol. Cell Biol. 2008, 28, 3465–3476. [Google Scholar] [CrossRef] [Green Version]
- Schisler, J.C.; Jensen, P.B.; Taylor, D.G.; Becker, T.C.; Knop, F.K.; Takekawa, S.; German, M.; Weir, G.C.; Lu, D.; Mirmira, R.G.; et al. The Nkx6.1 homeodomain transcription factor suppresses glucagon expression and regulates glucose-stimulated insulin secretion in islet beta cells. Proc. Natl. Acad. Sci. USA 2005, 102, 7297–7302. [Google Scholar] [CrossRef] [Green Version]
- Stephens, S.B.; Schisler, J.C.; Hohmeier, H.E.; An, J.; Sun, A.Y.; Pitt, G.S.; Newgard, C.B. A VGF-derived peptide attenuates development of type 2 diabetes via enhancement of islet beta-cell survival and function. Cell Metab. 2012, 16, 33–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernal-Mizrachi, E.; Kulkarni, R.N.; Scott, D.K.; Mauvais-Jarvis, F.; Stewart, A.F.; Garcia-Ocana, A. Human beta-cell proliferation and intracellular signaling part 2: Still driving in the dark without a road map. Diabetes 2014, 63, 819–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulkarni, R.N.; Mizrachi, E.B.; Ocana, A.G.; Stewart, A.F. Human beta-cell proliferation and intracellular signaling: Driving in the dark without a road map. Diabetes 2012, 61, 2205–2213. [Google Scholar] [CrossRef] [Green Version]
- Stewart, A.F.; Hussain, M.A.; Garcia-Ocana, A.; Vasavada, R.C.; Bhushan, A.; Bernal-Mizrachi, E.; Kulkarni, R.N. Human beta-cell proliferation and intracellular signaling: Part 3. Diabetes 2015, 64, 1872–1885. [Google Scholar] [CrossRef] [Green Version]
- Dirice, E.; Walpita, D.; Vetere, A.; Meier, B.C.; Kahraman, S.; Hu, J.; Dancik, V.; Burns, S.M.; Gilbert, T.J.; Olson, D.E.; et al. Inhibition of DYRK1A Stimulates Human beta-Cell Proliferation. Diabetes 2016, 65, 1660–1671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, W.; Taylor, B.; Jin, Q.; Nguyen-Tran, V.; Meeusen, S.; Zhang, Y.Q.; Kamireddy, A.; Swafford, A.; Powers, A.F.; Walker, J.; et al. Inhibition of DYRK1A and GSK3B induces human beta-cell proliferation. Nat. Commun. 2015, 6, 8372. [Google Scholar] [CrossRef]
- Wang, P.; Alvarez-Perez, J.C.; Felsenfeld, D.P.; Liu, H.; Sivendran, S.; Bender, A.; Kumar, A.; Sanchez, R.; Scott, D.K.; Garcia-Ocana, A.; et al. A high-throughput chemical screen reveals that harmine-mediated inhibition of DYRK1A increases human pancreatic beta cell replication. Nat. Med. 2015, 21, 383–388. [Google Scholar] [CrossRef] [Green Version]
- Aamodt, K.I.; Aramandla, R.; Brown, J.J.; Fiaschi-Taesch, N.; Wang, P.; Stewart, A.F.; Brissova, M.; Powers, A.C. Development of a reliable automated screening system to identify small molecules and biologics that promote human beta-cell regeneration. Am. J. Physiol. Endocrinol. Metab. 2016, 311, E859–E868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abdolazimi, Y.; Zhao, Z.; Lee, S.; Xu, H.; Allegretti, P.; Horton, T.M.; Yeh, B.; Moeller, H.P.; Nichols, R.J.; McCutcheon, D.; et al. CC-401 Promotes beta-Cell Replication via Pleiotropic Consequences of DYRK1A/B Inhibition. Endocrinology 2018, 159, 3143–3157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.J.; Golson, M.L.; Schug, J.; Traum, D.; Liu, C.; Vivek, K.; Dorrell, C.; Naji, A.; Powers, A.C.; Chang, K.M.; et al. Single-Cell Mass Cytometry Analysis of the Human Endocrine Pancreas. Cell Metab. 2016, 24, 616–626. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Karakose, E.; Liu, H.; Swartz, E.; Ackeifi, C.; Zlatanic, V.; Wilson, J.; Gonzalez, B.J.; Bender, A.; Takane, K.K.; et al. Combined Inhibition of DYRK1A, SMAD, and Trithorax Pathways Synergizes to Induce Robust Replication in Adult Human Beta Cells. Cell Metab. 2019, 29, 638–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simao, A.Y.; Goncalves, J.; Duarte, A.P.; Barroso, M.; Cristovao, A.C.; Gallardo, E. Toxicological Aspects and Determination of the Main Components of Ayahuasca: A Critical Review. Medicines 2019, 6, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hohmeier, H.E.; Zhang, L.; Taylor, B.; Stephens, S.; Lu, D.; McNamara, P.; Laffitte, B.; Newgard, C.B. Identification of a small molecule that stimulates human beta-cell proliferation and insulin secretion, and protects against cytotoxic stress in rat insulinoma cells. PLoS ONE 2020, 15, e0224344. [Google Scholar] [CrossRef] [Green Version]
- Boggi, U.; Vistoli, F.; Amorese, G.; Giannarelli, R.; Coppelli, A.; Mariotti, R.; Rondinini, L.; Barsotti, M.; Signori, S.; De Lio, N.; et al. Long-term (5 years) efficacy and safety of pancreas transplantation alone in type 1 diabetic patients. Transplantation 2012, 93, 842–846. [Google Scholar] [CrossRef]
- Gruessner, R.W.G.; Gruessner, A.C. Solid-organ Transplants From Living Donors: Cumulative United States Experience on 140,156 Living Donor Transplants Over 28 Years. Transplant. Proc. 2018, 50, 3025–3035. [Google Scholar] [CrossRef]
- Vardanyan, M.; Parkin, E.; Gruessner, C.; Rodriguez Rilo, H.L. Pancreas vs. islet transplantation: A call on the future. Curr. Opin. Organ. Transplant. 2010, 15, 124–130. [Google Scholar] [CrossRef]
- Shapiro, A.M.; Lakey, J.R.; Ryan, E.A.; Korbutt, G.S.; Toth, E.; Warnock, G.L.; Kneteman, N.M.; Rajotte, R.V. Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen. N. Engl. J. Med. 2000, 343, 230–238. [Google Scholar] [CrossRef] [PubMed]
- Bretzel, R.G.; Hering, B.J.; Schultz, A.O.; Geier, C.; Federlin, K. International islet transplant registry report. In Yearbook of Cell and Tissue Transplantation 1996–1997; Lanza, R.P., Chick, B.B., Eds.; Springer: Amsterdam, The Netherlands, 1996; pp. 153–160. [Google Scholar]
- Bennet, W.; Sundberg, B.; Groth, C.G.; Brendel, M.D.; Brandhorst, D.; Brandhorst, H.; Bretzel, R.G.; Elgue, G.; Larsson, R.; Nilsson, B.; et al. Incompatibility between human blood and isolated islets of Langerhans: A finding with implications for clinical intraportal islet transplantation? Diabetes 1999, 48, 1907–1914. [Google Scholar] [CrossRef]
- Kaufman, D.B.; Gores, P.F.; Field, M.J.; Farney, A.C.; Gruber, S.A.; Stephanian, E.; Sutherland, D.E. Effect of 15-deoxyspergualin on immediate function and long-term survival of transplanted islets in murine recipients of a marginal islet mass. Diabetes 1994, 43, 778–783. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, H.; Rawson, J.; Medrano, L.; Cook, C.A.; Barriga, A.; Gonzalez, N.; Salgado, M.; Omori, K.; Kandeel, F.; Tai, Y.C.; et al. Optimizing Temperature and Oxygen Supports Long-term Culture of Human Islets. Transplantation 2019, 103, 299–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pittenger, M.F.; Mackay, A.M.; Beck, S.C.; Jaiswal, R.K.; Douglas, R.; Mosca, J.D.; Moorman, M.A.; Simonetti, D.W.; Craig, S.; Marshak, D.R. Multilineage potential of adult human mesenchymal stem cells. Science 1999, 284, 143–147. [Google Scholar] [CrossRef] [Green Version]
- Porada, C.D.; Zanjani, E.D.; Almeida-Porad, G. Adult mesenchymal stem cells: A pluripotent population with multiple applications. Curr. Stem Cell Res. Ther. 2006, 1, 365–369. [Google Scholar] [CrossRef]
- Bhonde, R.R.; Sheshadri, P.; Sharma, S.; Kumar, A. Making surrogate beta-cells from mesenchymal stromal cells: Perspectives and future endeavors. Int. J. Biochem. Cell. Biol. 2014, 46, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Iskovich, S.; Goldenberg-Cohen, N.; Stein, J.; Yaniv, I.; Fabian, I.; Askenasy, N. Elutriated stem cells derived from the adult bone marrow differentiate into insulin-producing cells in vivo and reverse chemical diabetes. Stem Cells Dev. 2012, 21, 86–96. [Google Scholar] [CrossRef]
- Ianus, A.; Holz, G.G.; Theise, N.D.; Hussain, M.A. In vivo derivation of glucose-competent pancreatic endocrine cells from bone marrow without evidence of cell fusion. J. Clin. Invest. 2003, 111, 843–850. [Google Scholar] [CrossRef] [Green Version]
- Bell, G.I.; Broughton, H.C.; Levac, K.D.; Allan, D.A.; Xenocostas, A.; Hess, D.A. Transplanted human bone marrow progenitor subtypes stimulate endogenous islet regeneration and revascularization. Stem Cells Dev. 2012, 21, 97–109. [Google Scholar] [CrossRef]
- Choi, J.B.; Uchino, H.; Azuma, K.; Iwashita, N.; Tanaka, Y.; Mochizuki, H.; Migita, M.; Shimada, T.; Kawamori, R.; Watada, H. Little evidence of transdifferentiation of bone marrow-derived cells into pancreatic beta cells. Diabetologia 2003, 46, 1366–1374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ezquer, F.; Ezquer, M.; Contador, D.; Ricca, M.; Simon, V.; Conget, P. The antidiabetic effect of mesenchymal stem cells is unrelated to their transdifferentiation potential but to their capability to restore Th1/Th2 balance and to modify the pancreatic microenvironment. Stem Cells 2012, 30, 1664–1674. [Google Scholar] [CrossRef] [PubMed]
- Hess, D.; Li, L.; Martin, M.; Sakano, S.; Hill, D.; Strutt, B.; Thyssen, S.; Gray, D.A.; Bhatia, M. Bone marrow-derived stem cells initiate pancreatic regeneration. Nat. Biotechnol. 2003, 21, 763–770. [Google Scholar] [CrossRef]
- Taneera, J.; Rosengren, A.; Renstrom, E.; Nygren, J.M.; Serup, P.; Rorsman, P.; Jacobsen, S.E. Failure of transplanted bone marrow cells to adopt a pancreatic beta-cell fate. Diabetes 2006, 55, 290–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, T.; Itakura, S.; Todorov, I.; Rawson, J.; Asari, S.; Shintaku, J.; Nair, I.; Ferreri, K.; Kandeel, F.; Mullen, Y. Mesenchymal stem cell and islet co-transplantation promotes graft revascularization and function. Transplantation 2010, 89, 1438–1445. [Google Scholar] [CrossRef]
- Thomson, J.A.; Itskovitz-Eldor, J.; Shapiro, S.S.; Waknitz, M.A.; Swiergiel, J.J.; Marshall, V.S.; Jones, J.M. Embryonic stem cell lines derived from human blastocysts. Science 1998, 282, 1145–1147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Pagliuca, F.W.; Millman, J.R.; Gurtler, M.; Segel, M.; Van Dervort, A.; Ryu, J.H.; Peterson, Q.P.; Greiner, D.; Melton, D.A. Generation of functional human pancreatic beta cells in vitro. Cell 2014, 159, 428–439. [Google Scholar] [CrossRef] [Green Version]
- Rezania, A.; Bruin, J.E.; Arora, P.; Rubin, A.; Batushansky, I.; Asadi, A.; O’Dwyer, S.; Quiskamp, N.; Mojibian, M.; Albrecht, T.; et al. Reversal of diabetes with insulin-producing cells derived in vitro from human pluripotent stem cells. Nat. Biotechnol. 2014, 32, 1121–1133. [Google Scholar] [CrossRef]
- Russ, H.A.; Parent, A.V.; Ringler, J.J.; Hennings, T.G.; Nair, G.G.; Shveygert, M.; Guo, T.; Puri, S.; Haataja, L.; Cirulli, V.; et al. Controlled induction of human pancreatic progenitors produces functional beta-like cells in vitro. EMBO J. 2015, 34, 1759–1772. [Google Scholar] [CrossRef] [Green Version]
- Tremmel, D.M.; Mitchell, S.A.; Sackett, S.D.; Odorico, J.S. Mimicking nature-made beta cells: Recent advances towards stem cell-derived islets. Curr. Opin. Organ. Transplant. 2019, 24, 574–581. [Google Scholar] [CrossRef]
- Nair, G.G.; Liu, J.S.; Russ, H.A.; Tran, S.; Saxton, M.S.; Chen, R.; Juang, C.; Li, M.L.; Nguyen, V.Q.; Giacometti, S.; et al. Recapitulating endocrine cell clustering in culture promotes maturation of human stem-cell-derived beta cells. Nat. Cell. Biol. 2019, 21, 263–274. [Google Scholar] [CrossRef]
- Velazco-Cruz, L.; Song, J.; Maxwell, K.G.; Goedegebuure, M.M.; Augsornworawat, P.; Hogrebe, N.J.; Millman, J.R. Acquisition of Dynamic Function in Human Stem Cell-Derived beta Cells. Stem Cell Rep. 2019, 12, 351–365. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Chen, P.; Vaughan, J.; Blount, A.; Chen, A.; Jamieson, P.M.; Rivier, J.; Smith, M.S.; Vale, W. Urocortin III is expressed in pancreatic beta-cells and stimulates insulin and glucagon secretion. Endocrinology 2003, 144, 3216–3224. [Google Scholar] [CrossRef] [Green Version]
- Li, C.; Chen, P.; Vaughan, J.; Lee, K.F.; Vale, W. Urocortin 3 regulates glucose-stimulated insulin secretion and energy homeostasis. Proc. Natl. Acad. Sci. USA 2007, 104, 4206–4211. [Google Scholar] [CrossRef] [Green Version]
- van der Meulen, T.; Xie, R.; Kelly, O.G.; Vale, W.W.; Sander, M.; Huising, M.O. Urocortin 3 marks mature human primary and embryonic stem cell-derived pancreatic alpha and beta cells. PLoS ONE 2012, 7, e52181. [Google Scholar] [CrossRef]
- Ghazizadeh, Z.; Kao, D.I.; Amin, S.; Cook, B.; Rao, S.; Zhou, T.; Zhang, T.; Xiang, Z.; Kenyon, R.; Kaymakcalan, O.; et al. ROCKII inhibition promotes the maturation of human pancreatic beta-like cells. Nat. Commun. 2017, 8, 298. [Google Scholar] [CrossRef] [Green Version]
- Augsornworawat, P.; Maxwell, K.G.; Velazco-Cruz, L.; Millman, J.R. Single-Cell Transcriptome Profiling Reveals beta Cell Maturation in Stem Cell-Derived Islets after Transplantation. Cell. Rep. 2020, 32, 108067. [Google Scholar] [CrossRef]
- Veres, A.; Faust, A.L.; Bushnell, H.L.; Engquist, E.N.; Kenty, J.H.; Harb, G.; Poh, Y.C.; Sintov, E.; Gurtler, M.; Pagliuca, F.W.; et al. Charting cellular identity during human in vitro beta-cell differentiation. Nature 2019, 569, 368–373. [Google Scholar] [CrossRef]
- Faleo, G.; Russ, H.A.; Wisel, S.; Parent, A.V.; Nguyen, V.; Nair, G.G.; Freise, J.E.; Villanueva, K.E.; Szot, G.L.; Hebrok, M.; et al. Mitigating Ischemic Injury of Stem Cell-Derived Insulin-Producing Cells after Transplant. Stem Cell Rep. 2017, 9, 807–819. [Google Scholar] [CrossRef] [Green Version]
- Cunha, J.P.; Leuckx, G.; Sterkendries, P.; Korf, H.; Bomfim-Ferreira, G.; Overbergh, L.; Vaes, B.; Heimberg, H.; Gysemans, C.; Mathieu, C. Human multipotent adult progenitor cells enhance islet function and revascularisation when co-transplanted as a composite pellet in a mouse model of diabetes. Diabetologia 2017, 60, 134–142. [Google Scholar] [CrossRef] [Green Version]
- Gamble, A.; Pawlick, R.; Pepper, A.R.; Bruni, A.; Adesida, A.; Senior, P.A.; Korbutt, G.S.; Shapiro, A.M.J. Improved islet recovery and efficacy through co-culture and co-transplantation of islets with human adipose-derived mesenchymal stem cells. PLoS ONE 2018, 13, e0206449. [Google Scholar] [CrossRef]
- Tremmel, D.M.; Odorico, J.S. Rebuilding a better home for transplanted islets. Organogenesis 2018, 14, 163–168. [Google Scholar] [CrossRef]
- Shapiro, A.M.; Pokrywczynska, M.; Ricordi, C. Clinical pancreatic islet transplantation. Nat. Rev. Endocrinol. 2017, 13, 268–277. [Google Scholar] [CrossRef]
- Espona-Noguera, A.; Ciriza, J.; Canibano-Hernandez, A.; Orive, G.; Hernandez, R.M.M.; Saenz Del Burgo, L.; Pedraz, J.L. Review of Advanced Hydrogel-Based Cell Encapsulation Systems for Insulin Delivery in Type 1 Diabetes Mellitus. Pharmaceutics 2019, 11, 597. [Google Scholar] [CrossRef] [Green Version]
- O’Sullivan, E.S.; Vegas, A.; Anderson, D.G.; Weir, G.C. Islets transplanted in immunoisolation devices: A review of the progress and the challenges that remain. Endocr. Rev. 2011, 32, 827–844. [Google Scholar] [CrossRef] [Green Version]
- Yoshihara, E.; O’Connor, C.; Gasser, E.; Wei, Z.; Oh, T.G.; Tseng, T.W.; Wang, D.; Cayabyab, F.; Dai, Y.; Yu, R.T.; et al. Immune-evasive human islet-like organoids ameliorate diabetes. Nature 2020, 586, 606–611. [Google Scholar] [CrossRef]
- Cai, E.P.; Ishikawa, Y.; Zhang, W.; Leite, N.C.; Li, J.; Hou, S.; Kiaf, B.; Hollister-Lock, J.; Yilmaz, N.K.; Schiffer, C.A.; et al. Genome-scale in vivo CRISPR screen identifies RNLS as a target for beta cell protection in type 1 diabetes. Nat. Metab. 2020, 2, 934–945. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Docherty, F.M.; Sussel, L. Islet Regeneration: Endogenous and Exogenous Approaches. Int. J. Mol. Sci. 2021, 22, 3306. https://doi.org/10.3390/ijms22073306
Docherty FM, Sussel L. Islet Regeneration: Endogenous and Exogenous Approaches. International Journal of Molecular Sciences. 2021; 22(7):3306. https://doi.org/10.3390/ijms22073306
Chicago/Turabian StyleDocherty, Fiona M., and Lori Sussel. 2021. "Islet Regeneration: Endogenous and Exogenous Approaches" International Journal of Molecular Sciences 22, no. 7: 3306. https://doi.org/10.3390/ijms22073306