
Inhibition of NUPR1–Karyopherin β1 Binding Increases Anticancer Drug Sensitivity

 , ,
, ,
Abstract
:1. Introduction
2. Results
2.1. Dose- and Time-Dependent Cytotoxic Effects of Doxorubicin on MDA-MB 231 Breast Cancer Cells
2.2. Increased NUPR1 Nuclear Transportation after Doxorubicin Treatment on MDA-MB-231 Breast Cancer Cells
2.3. ATZ-502 Inhibits NUPR1 Activity in MDA-MB-231 Breast Cancer Cells by Inhibiting Nuclear Transportation
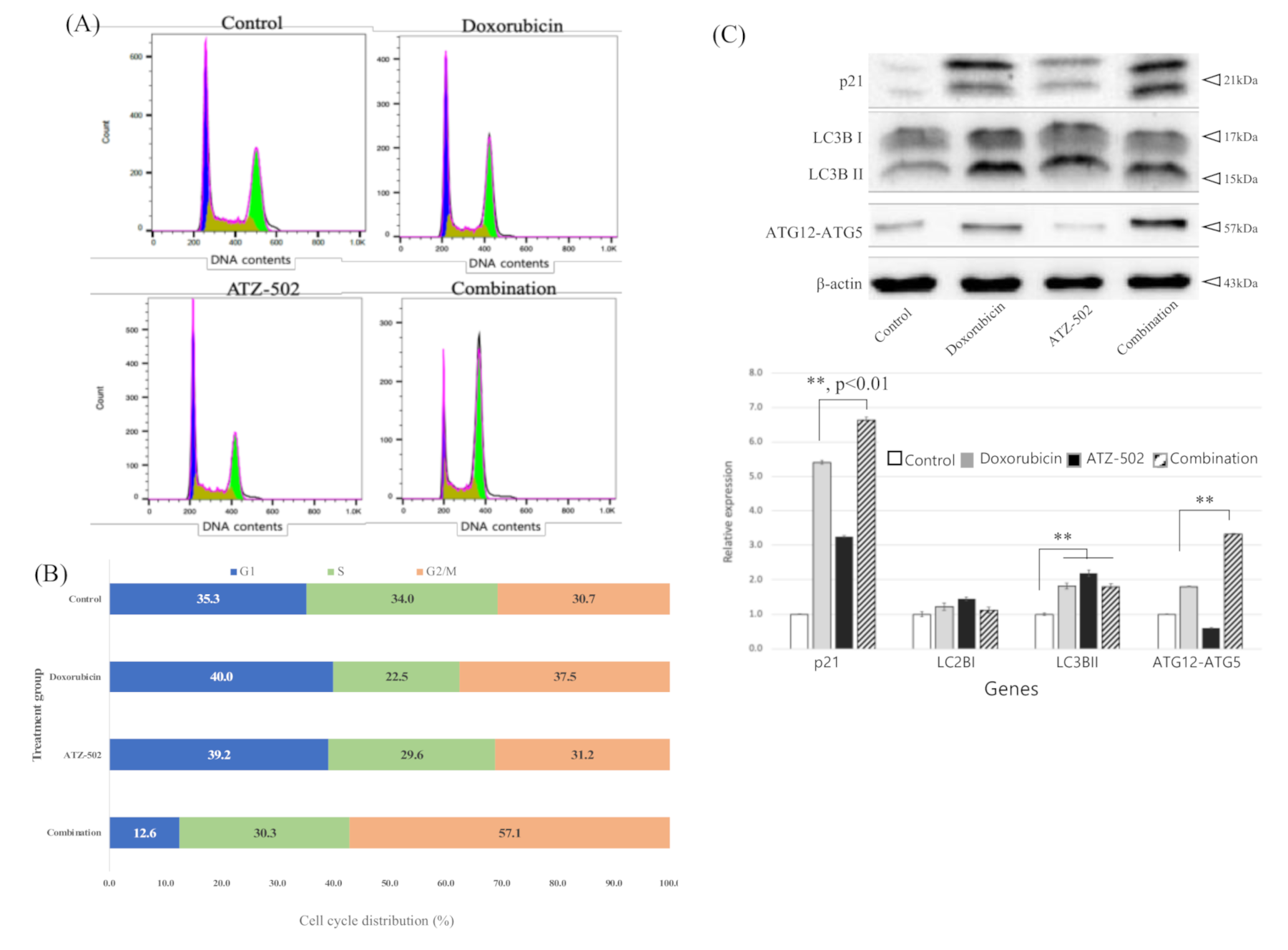
2.4. Inhibition of NUPR1 Nucleus Transportation Induces G2/M Phase Arrest and Increases Synergistic Cytotoxicity Effects
2.5. RNA Quant-Seq Analysis Revealed That Single and Combination Treatments Yielded Different mRNA Expression Patterns
2.6. Gene Ontology Analysis for DEGs
2.7. NUPR1-Induced Gene Expression Is Differentially Regulated between Single-Treatment and Combination-Treatment Groups
2.8. Karyopherin β1 Binds to NUPR1 with High Affinity
2.9. ATZ-502 Inhibits the NUPR1–Karyopherin β1 Interaction
3. Discussion
4. Materials and Methods
4.1. Cell Culture
4.2. Cell Viability Assay
4.3. Cell Cycle and Apoptosis with Fluorescence-Activated Cell Sorting (FACS) Analysis
4.4. Single-Molecule Binding Assay
4.5. Drug Inhibition Assay at the Single-Molecule Level
4.6. Immunofluorescence Assay
4.7. Gene Expression Profiling Analysis Using QuantSeq 3′ mRNA-seq
4.8. Data Analysis and Gene Selection
4.9. Quantitative Real-Time RT-PCR
4.10. Protein Expression, Immunoprecipitaton, and Western Blotting
4.11. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cano, C.E.; Hamidi, T.; Sandi, M.J.; Iovanna, J.L. Nupr1: The Swiss-knife of cancer. J. Cell. Physiol. 2011, 226, 1439–1443. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Liu, X.; Sengupta, J.; Bu, Y.; Yi, F.; Wang, C.; Shi, Y.; Zhu, Y.; Jiao, Q.; Song, F. Silencing of Bmi-1 gene by RNA interference enhances sensitivity to doxorubicin in breast cancer cells. Indian J. Exp. Biol. 2011, 49, 105–112. [Google Scholar] [PubMed]
- Yang, W.T.; Zhu, X.Z. The introduction of 2012 WHO classification of tumours of the breast. Chin. J. Pathol. 2013, 42, 78–80. [Google Scholar] [CrossRef]
- Su, S.B.; Motoo, Y.; Iovanna, J.L.; Berthezene, P.; Xie, M.J.; Mouri, H.; Ohtsubo, K.; Matsubara, F.; Sawabu, N. Overexpression of p8 is inversely correlated with apoptosis in pancreatic cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2001, 7, 1320–1324. [Google Scholar]
- Hamidi, T.; Cano, C.E.; Grasso, D.; Garcia, M.N.; Sandi, M.J.; Calvo, E.L.; Dagorn, J.C.; Lomberk, G.; Goruppi, S.; Urrutia, R.; et al. NUPR1 works against the metabolic stress-induced autophagy-associated cell death in pancreatic cancer cells. Autophagy 2013, 9, 95–97. [Google Scholar] [CrossRef] [Green Version]
- Mallo, G.V.; Fiedler, F.; Calvo, E.L.; Ortiz, E.M.; Vasseur, S.; Keim, V.; Morisset, J.; Iovanna, J.L. Cloning and expression of the rat p8 cDNA, a new gene activated in pancreas during the acute phase of pancreatitis, pancreatic development, and regeneration, and which promotes cellular growth. J. Biol. Chem. 1997, 272, 32360–32369. [Google Scholar] [CrossRef] [Green Version]
- Vasseur, S.; Folch-Puy, E.; Hlouschek, V.; Garcia, S.; Fiedler, F.; Lerch, M.M.; Dagorn, J.C.; Closa, D.; Iovanna, J.L. p8 improves pancreatic response to acute pancreatitis by enhancing the expression of the anti-inflammatory protein pancreatitis-associated protein I. J. Biol. Chem. 2004, 279, 7199–7207. [Google Scholar] [CrossRef] [Green Version]
- Vincent, A.J.; Ren, S.; Harris, L.G.; Devine, D.J.; Samant, R.S.; Fodstad, O.; Shevde, L.A. Cytoplasmic translocation of p21 mediates NUPR1-induced chemoresistance: NUPR1 and p21 in chemoresistance. FEBS Lett. 2012, 586, 3429–3434. [Google Scholar] [CrossRef] [Green Version]
- Clark, D.W.; Mitra, A.; Fillmore, R.A.; Jiang, W.G.; Samant, R.S.; Fodstad, O.; Shevde, L.A. NUPR1 interacts with p53, transcriptionally regulates p21 and rescues breast epithelial cells from doxorubicin-induced genotoxic stress. Curr. Cancer Drug Targets 2008, 8, 421–430. [Google Scholar] [CrossRef]
- Emma, M.R.; Iovanna, J.L.; Bachvarov, D.; Puleio, R.; Loria, G.R.; Augello, G.; Candido, S.; Libra, M.; Gulino, A.; Cancila, V.; et al. NUPR1, a new target in liver cancer: Implication in controlling cell growth, migration, invasion and sorafenib resistance. Cell Death Dis. 2016, 7, e2269. [Google Scholar] [CrossRef]
- Cervello, M.; Bachvarov, D.; Lampiasi, N.; Cusimano, A.; Azzolina, A.; McCubrey, J.A.; Montalto, G. Molecular mechanisms of sorafenib action in liver cancer cells. Cell Cycle 2012, 11, 2843–2855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malicet, C.; Hoffmeister, A.; Moreno, S.; Closa, D.; Dagorn, J.C.; Vasseur, S.; Iovanna, J.L. Interaction of the stress protein p8 with Jab1 is required for Jab1-dependent p27 nuclear-to-cytoplasm translocation. Biochem. Biophys. Res. Commun. 2006, 339, 284–289. [Google Scholar] [CrossRef]
- Harel, A.; Forbes, D.J. Importin beta: Conducting a much larger cellular symphony. Mol. Cell 2004, 16, 319–330. [Google Scholar] [CrossRef] [PubMed]
- Nachury, M.V.; Maresca, T.J.; Salmon, W.C.; Waterman-Storer, C.M.; Heald, R.; Weis, K. Importin beta is a mitotic target of the small GTPase Ran in spindle assembly. Cell 2001, 104, 95–106. [Google Scholar] [CrossRef] [Green Version]
- Ciciarello, M.; Mangiacasale, R.; Thibier, C.; Guarguaglini, G.; Marchetti, E.; Di Fiore, B.; Lavia, P. Importin beta is transported to spindle poles during mitosis and regulates Ran-dependent spindle assembly factors in mammalian cells. J. Cell Sci. 2004, 117, 6511–6522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, C.; Hutchins, J.R.; Muhlhausser, P.; Kutay, U.; Clarke, P.R. Role of importin-beta in the control of nuclear envelope assembly by Ran. Curr. Biol. CB 2002, 12, 498–502. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Shi, Y.; Lu, Q.; Ma, Y.; Luo, J.; Wang, Q.; Ji, J.; Jiang, Q.; Zhang, C. Requirement for lamin B receptor and its regulation by importin {beta} and phosphorylation in nuclear envelope assembly during mitotic exit. J. Biol. Chem. 2010, 285, 33281–33293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ha, S.; Jeong, J.; Oh, J.; Rhee, S.; Ham, S.W. A Small Organic Molecule Blocks EGFR Transport into the Nucleus by the Nonclassical Pathway Resulting in Repression of Cancer Invasion. Chembiochem 2018, 19, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Ha, S.; Choi, J.; Min, N.Y.; Lee, K.H.; Ham, S.W. Inhibition of Importin beta1 With a 2-Aminothiazole Derivative Resulted in G2/M Cell-cycle Arrest and Apoptosis. Anticancer. Res. 2017, 37, 2373–2379. [Google Scholar] [CrossRef] [Green Version]
- Ha, S.; Oh, J.; Kim, Y.H.; Ham, S.W. Determination of the binding site of 2-aminothiazole derivative with importin beta1 by UV-crosslinking experiment. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2017, 1060, 71–75. [Google Scholar] [CrossRef]
- Kim, Y.H.; Ha, S.; Kim, J.; Ham, S.W. Identification of KPNB1 as a Cellular Target of Aminothiazole Derivatives with Anticancer Activity. ChemMedChem 2016, 11, 1406–1409. [Google Scholar] [CrossRef]
- Gradishar, W.J.; Anderson, B.O.; Balassanian, R.; Blair, S.L.; Burstein, H.J.; Cyr, A.; Elias, A.D.; Farrar, W.B.; Forero, A.; Giordano, S.H.; et al. Invasive Breast Cancer Version 1.2016, NCCN Clinical Practice Guidelines in Oncology. JNCCN 2016, 14, 324–354. [Google Scholar] [CrossRef] [Green Version]
- Minotti, G.; Menna, P.; Salvatorelli, E.; Cairo, G.; Gianni, L. Anthracyclines: Molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacol. Rev. 2004, 56, 185–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Figueroa-Magalhaes, M.C.; Jelovac, D.; Connolly, R.; Wolff, A.C. Treatment of HER2-positive breast cancer. Breast 2014, 23, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Lovitt, C.J.; Shelper, T.B.; Avery, V.M. Doxorubicin resistance in breast cancer cells is mediated by extracellular matrix proteins. BMC Cancer 2018, 18, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlisi, D.; De Blasio, A.; Drago-Ferrante, R.; Di Fiore, R.; Buttitta, G.; Morreale, M.; Scerri, C.; Vento, R.; Tesoriere, G. Parthenolide prevents resistance of MDA-MB231 cells to doxorubicin and mitoxantrone: The role of Nrf2. Cell Death Discov. 2017, 3, 17078. [Google Scholar] [CrossRef] [Green Version]
- Galichon, P.; Bataille, A.; Vandermeersch, S.; Wetzstein, M.; Xu-Dubois, Y.C.; Legouis, D.; Hertig, A.; Buob, D.; Placier, S.; Bige, N.; et al. Stress Response Gene Nupr1 Alleviates Cyclosporin A Nephrotoxicity In Vivo. J. Am. Soc. Nephrol. 2017, 28, 545–556. [Google Scholar] [CrossRef] [Green Version]
- He, S.; Miao, X.; Wu, Y.; Zhu, X.; Miao, X.; Yin, H.; He, Y.; Li, C.; Liu, Y.; Lu, X.; et al. Upregulation of nuclear transporter, Kpnbeta1, contributes to accelerated cell proliferation- and cell adhesion-mediated drug resistance (CAM-DR) in diffuse large B-cell lymphoma. J. Cancer Res. Clin. Oncol. 2016, 142, 561–572. [Google Scholar] [CrossRef]
- Zhu, Z.C.; Liu, J.W.; Li, K.; Zheng, J.; Xiong, Z.Q. KPNB1 inhibition disrupts proteostasis and triggers unfolded protein response-mediated apoptosis in glioblastoma cells. Oncogene 2018, 37, 2936–2952. [Google Scholar] [CrossRef] [PubMed]
- Van der Watt, P.J.; Chi, A.; Stelma, T.; Stowell, C.; Strydom, E.; Carden, S.; Angus, L.; Hadley, K.; Lang, D.; Wei, W.; et al. Targeting the Nuclear Import Receptor Kpn beta 1 as an Anticancer Therapeutic. Mol. Cancer Ther. 2016, 15, 560–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.Q.; Erikson, R.L. Polo-like kinase (Plk)1 depletion induces apoptosis in cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 5789–5794. [Google Scholar] [CrossRef] [Green Version]
- Shi, R.; Zhang, C.R.; Wu, Y.Q.; Wang, X.; Sun, Q.; Sun, J.; Xia, W.J.; Dong, G.C.; Wang, A.P.; Jiang, F.; et al. CDCA2 promotes lung adenocarcinoma cell proliferation and predicts poor survival in lung adenocarcinoma patients. Oncotarget 2017, 8, 19768–19779. [Google Scholar] [CrossRef] [Green Version]
- Ashton, J.C. Drug combination studies and their synergy quantification using the Chou-Talalay method--letter. Cancer Res. 2015, 75, 2400. [Google Scholar] [CrossRef] [Green Version]
- Roy, R.; Hohng, S.; Ha, T. A practical guide to single-molecule FRET. Nat. Methods 2008, 5, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Gommeaux, J.; Cano, C.; Garcia, S.; Gironella, M.; Pietri, S.; Culcasi, M.; Pebusque, M.J.; Malissen, B.; Dusetti, N.; Iovanna, J.; et al. Colitis and colitis-associated cancer are exacerbated in mice deficient for tumor protein 53-induced nuclear protein 1. Mol. Cell. Biol. 2007, 27, 2215–2228. [Google Scholar] [CrossRef] [Green Version]
- Taieb, D.; Roignot, J.; Andre, F.; Garcia, S.; Masson, B.; Pierres, A.; Iovanna, J.L.; Soubeyran, P. ArgBP2-dependent signaling regulates pancreatic cell migration, adhesion, and tumorigenicity. Cancer Res. 2008, 68, 4588–4596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moll, P.; Ante, M.; Seitz, A.; Reda, T. QuantSeq 3′ mRNA sequencing for RNA quantification. Nat. Methods 2014, 11, 972. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}

| Cell Lines | ATZ-50 (μM) | IC50 (μM) | Fold Difference | |
|---|---|---|---|---|
| Single | Combination | |||
| MDA-MB-231 | 0.50 | 1.767 | 0.438 | 4.0 |
| SiHa | 0.50 | 0.624 | 0.239 | 2.6 |
| MCF-7/ADR | 0.05 | 39.62 | 2.00 | 19.8 |
| SK-OV-3 | 0.01 | 0.250 | 0.175 | 1.4 |
| PC-3 | 0.01 | 0.357 | 0.018 | 19.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, C.; Oh, J.; Lee, W.M.; Koh, H.R.; Sohn, U.D.; Ham, S.W.; Oh, K. Inhibition of NUPR1–Karyopherin β1 Binding Increases Anticancer Drug Sensitivity. Int. J. Mol. Sci. 2021, 22, 2794. https://doi.org/10.3390/ijms22062794
Park C, Oh J, Lee WM, Koh HR, Sohn UD, Ham SW, Oh K. Inhibition of NUPR1–Karyopherin β1 Binding Increases Anticancer Drug Sensitivity. International Journal of Molecular Sciences. 2021; 22(6):2794. https://doi.org/10.3390/ijms22062794
Chicago/Turabian StylePark, Chanhee, Jiwon Oh, Won Mo Lee, Hye Ran Koh, Uy Dong Sohn, Seung Wook Ham, and Kyungsoo Oh. 2021. "Inhibition of NUPR1–Karyopherin β1 Binding Increases Anticancer Drug Sensitivity" International Journal of Molecular Sciences 22, no. 6: 2794. https://doi.org/10.3390/ijms22062794