
Receptor Tyrosine Kinase Signaling and Targeting in Glioblastoma Multiforme


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
1.1. Gliomas
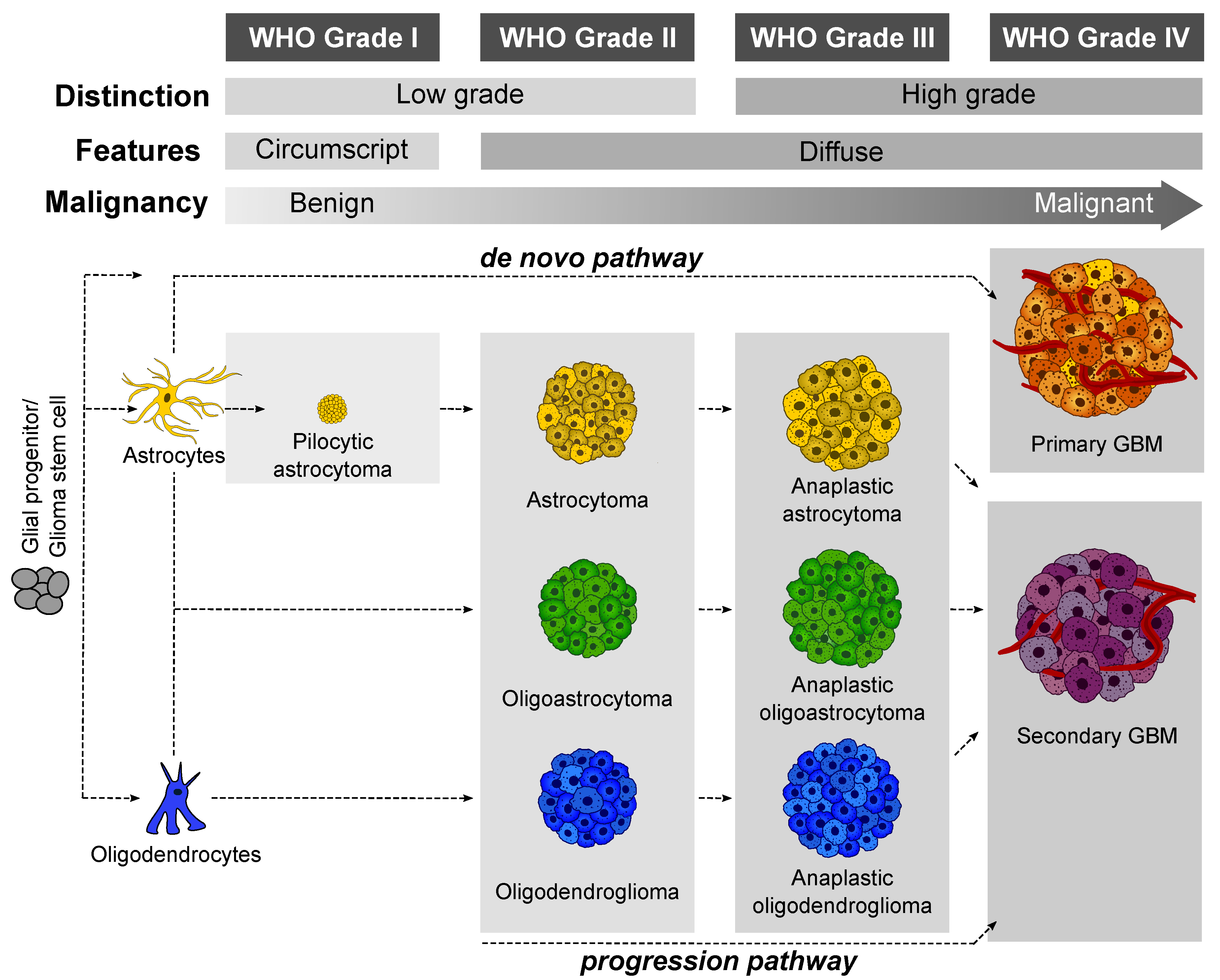
- Grade I: Low-grade benign lesions such as pilocytic astrocytomas that exhibit limited proliferation and are frequently seen in children;
- Grade II: Low-grade but infiltrative lesions with a tendency to show higher recurrence after surgical resection;
- Grade III: Intermediate–high-grade lesions with relatively higher mitotic activity and evidence of malignancy;
1.2. Glioblastoma Multiforme (GBM)
2. Molecular Classification of GBM
2.1. Molecular Subtypes
2.2. Biomarkers
3. Key Cellular Hallmarks of GBM
3.1. Uncontrolled Proliferation
3.2. Invasion and Metastasis
3.3. Cellular Heterogeneity
3.4. Angiogenesis
3.5. Deregulated Cell Energetics
3.6. Mutation and Genome Instability
4. Receptor Tyrosine Kinase (RTK) Pathways Altered in GBM
4.1. RTK Regulation and Oncogenic Activation
4.1.1. EGF Receptors
4.1.2. Insulin Receptors
4.1.3. PDGF Receptors
4.1.4. VEGF Receptors
4.1.5. FGF Receptors
4.1.6. Neurotrophin Receptors
4.1.7. MET (HGF) Receptor
4.1.8. Eph Receptors
4.1.9. Tie Receptors
4.1.10. DDR Receptors
4.1.11. RET Receptor
4.1.12. ROS Receptors
4.2. RTK Downstream Signaling Pathways
4.2.1. Ras/MAPK Pathway
4.2.2. PI3K/Akt/PTEN Pathway
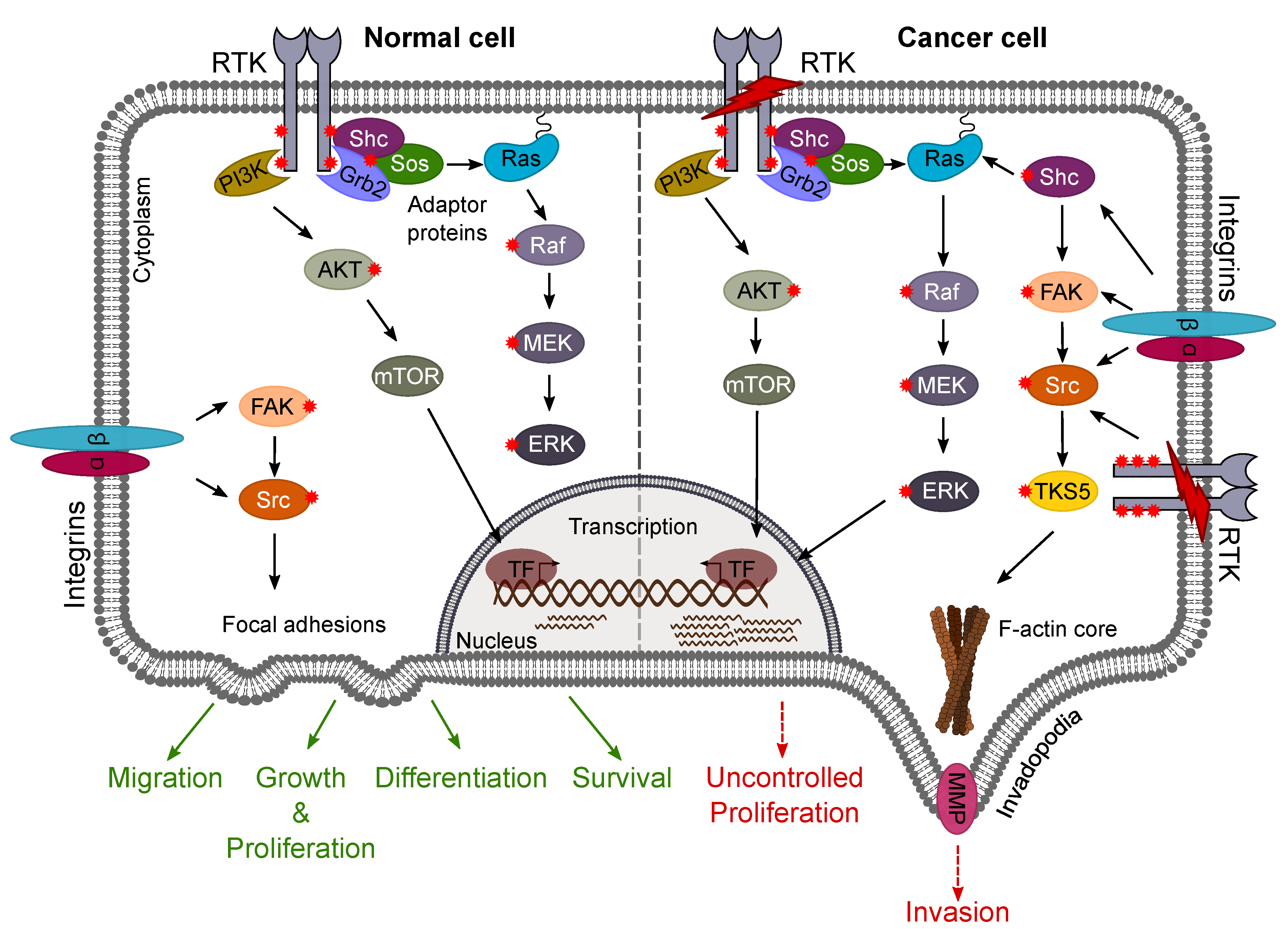
4.2.3. FAK/Src Pathway
4.2.4. Shc Adaptor Proteins
5. Non-Canonical Modes of RTK Activation in GBM
5.1. G-Protein-Coupled Receptor-RTK Signaling
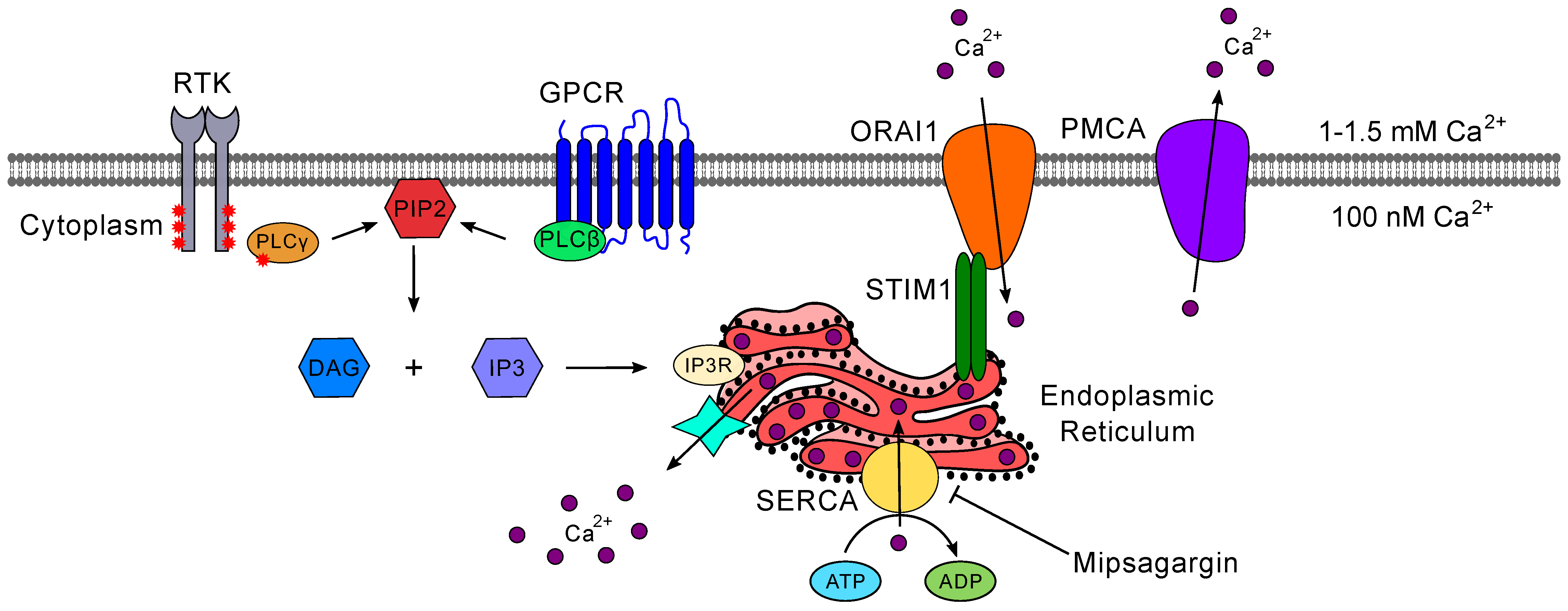
5.2. Calcium Signaling
5.2.1. Inositol 1,4,5-trisphosphate (IP3) Receptor-Mediated Calcium Signaling
5.2.2. Store-Operated Calcium Entry
6. RTK Inhibitors and Antagonists with Anti-GBM Properties
6.1. EGFR: Erlotinib, Gefitinib, and Afatinib; EGFRvIII: Rindopepimut
6.2. IGF1R: BMS-754807, KW-2450, and Picropodophyllin
6.3. Abl/PDGFR/c-KIT: Imatinib (Gleevec/Glivec)
6.4. VEGFs: Bevacizumab; VEGFRs: Axitinib, Cedarinib, Suritinib, and Sorafenib
6.5. FGFRs: AZD4547, Dovitinib, JNJ-42756493, Ponatinib, and Infigratinib
6.6. TrkB: ANA-12; Pan-Trk: Entrectinib and Larotrectinib
6.7. Tie2: Rebastinib, Bay-826, and Altiratinib
6.8. Other Inhibitors
7. Emerging Areas of GBM Investigation
8. Conclusions and Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| BDNF CDKN2A/B | Brain-derived neurotrophic factor Cyclin-dependent kinase inhibitors 2A/B |
| CNA | Copy number alterations |
| CNS | Central nervous system |
| DDR ECM | Discoidin domain receptor Extracellular matrix |
| EGFR EMT | Epidermal growth factor receptor Epithelial-mesenchymal transition |
| EPH | Erythropoeitin-producing human hepatocellular receptor |
| FAK | Focal adhesion kinase |
| FLT3 | FMS-like tyrosine kinase 3 |
| GBM | Glioblastoma multiforme |
| G-CIMP | Glioma-CpG-island methylator phenotype |
| GPCR | G-protein-coupled receptor |
| GSC | Glioma stem cell |
| IDH1/2 | Isocitrate dehydrogenase 1/2 |
| IGF1R | Insulin-like growth factor 1 receptor |
| INSR | Insulin receptor |
| INSRR | Insulin-receptor-related receptor |
| KIT/SCFR MAPK | c-KIT/stem cell growth factor receptor Mitogen-activated protein kinase |
| MET MGMT | MNNG-HOS transforming gene O6-methylguanine methyltransferase |
| mTOR | Mammalian target of rapamycin |
| PDGFR | Platelet-derived growth factor receptor |
| PI3K PLC | Phosphatidylinositol-3-kinase Phospholipase C |
| PTEN | Phosphatase and tensin homolog |
| RET ROS1 | Rearranged during transfection c-ROS oncogene 1 |
| RPPA | Reverse Phase Protein Array |
| RTK | Receptor tyrosine kinase |
| Shc SOCE SOX2 | Src homology and collagen Store operated Ca2+ entry Sex-determining region y-box 2 |
| TCGA | The Cancer Genome Atlas |
| TEM | Tie2-expressing monocytes/macrophages |
| TFP TKI TMZ TRK | Trifluoperazine Tyrosine kinase inhibitor Temozolomide Tropomyosin receptor kinase |
| VEGFR | Vascular endothelial growth factor receptor |
| WHO | World Health Organization |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Goodenberger, M.L.; Jenkins, R.B. Genetics of adult glioma. Cancer Genet. 2012, 205, 613–621. [Google Scholar] [CrossRef]
- Furnari, F.B.; Fenton, T.; Bachoo, R.M.; Mukasa, A.; Stommel, J.M.; Stegh, A.; Hahn, W.C.; Ligon, K.L.; Louis, D.N.; Brennan, C.; et al. Malignant astrocytic glioma: Genetics, biology, and paths to treatment. Genes Dev. 2007, 21, 2683–2710. [Google Scholar] [CrossRef] [Green Version]
- Ohgaki, H.; Kleihues, P. Epidemiology and etiology of gliomas. Acta Neuropathol. 2005, 109, 93–108. [Google Scholar] [CrossRef]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [Green Version]
- Ostrom, Q.T.; Cioffi, G.; Gittleman, H.; Patil, N.; Waite, K.; Kruchko, C.; Barnholtz-Sloan, J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012–2016. Neuro. Oncol. 2019, 21, V1–V100. [Google Scholar] [CrossRef] [PubMed]
- Gittleman, H.; Ostrom, Q.T.; Stetson, L.C.; Waite, K.; Hodges, T.R.; Wright, C.H.; Wright, J.; Rubin, J.B.; Berens, M.E.; Lathia, J.; et al. Sex is an important prognostic factor for glioblastoma but not for nonglioblastoma. Neuro-Oncol. Pract. 2019, 6, 451–462. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.H.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.; Uhrbom, L. On the origin of glioma. Upsala J. Med. Sci. 2012, 117, 113–121. [Google Scholar] [CrossRef] [Green Version]
- Ohgaki, H.; Kleihues, P. The definition of primary and secondary glioblastoma. Clin. Cancer Res. 2013, 19, 764–772. [Google Scholar] [CrossRef] [Green Version]
- Louis, D.N.; Perry, A.; Reifenberger, G.; von Deimling, A.; Figarella-Branger, D.; Cavenee, W.K.; Ohgaki, H.; Wiestler, O.D.; Kleihues, P.; Ellison, D.W. The 2016 World Health Organization Classification of Tumors of the Central Nervous System: A summary. Acta Neuropathol. 2016, 131, 803–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newlands, E.S.; Stevens, M.F.G.; Wedge, S.R.; Wheelhouse, R.T.; Brock, C. Temozolomide: A review of its discovery, chemical properties, pre-clinical development and clinical trials. Cancer Treat. Rev. 1997, 23, 35–61. [Google Scholar] [CrossRef]
- Stupp, R.; Mason, W.P.; Van Den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef] [PubMed]
- Stupp, R.; Hegi, M.E.; Mason, W.P.; van den Bent, M.J.; Taphoorn, M.J.; Janzer, R.C.; Ludwin, S.K.; Allgeier, A.; Fisher, B.; Belanger, K.; et al. Effects of radiotherapy with concomitant and adjuvant temozolomide versus radiotherapy alone on survival in glioblastoma in a randomised phase III study: 5-year analysis of the EORTC-NCIC trial. Lancet Oncol. 2009, 10, 459–466. [Google Scholar] [CrossRef]
- Hart, M.G.; Garside, R.; Rogers, G.; Stein, K.; Grant, R. Temozolomide for high grade glioma. Cochrane Database Syst. Rev. 2013, 2013, CD007415. [Google Scholar] [CrossRef] [Green Version]
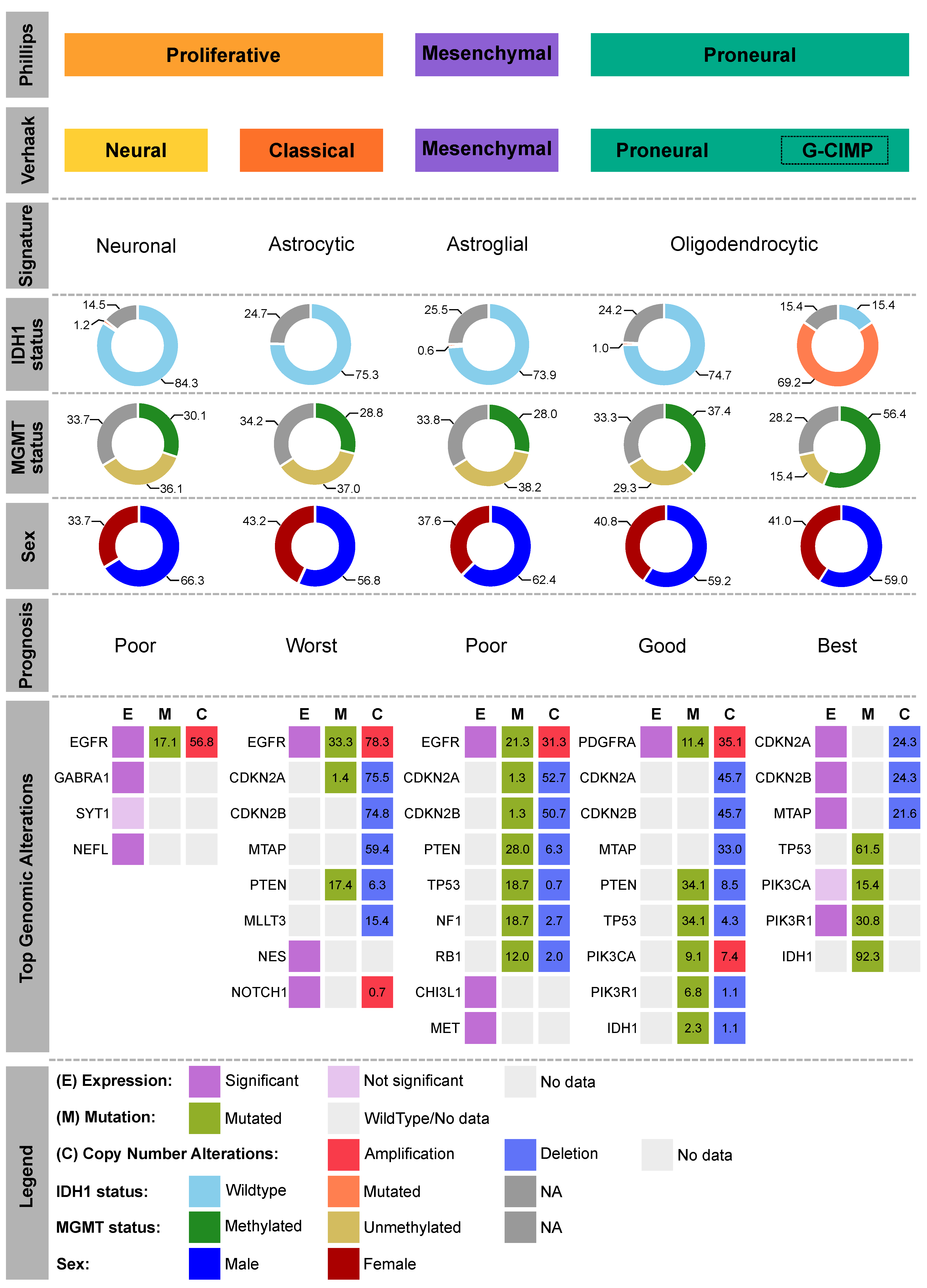
- Freije, W.A.; Castro-Vargas, F.E.; Fang, Z.; Horvath, S.; Cloughesy, T.; Liau, L.M.; Mischel, P.S.; Nelson, S.F. Gene expression profiling of gliomas strongly predicts survival. Cancer Res. 2004, 64, 6503–6510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Phillips, H.S.; Kharbanda, S.; Chen, R.; Forrest, W.F.; Soriano, R.H.; Wu, T.D.; Misra, A.; Nigro, J.M.; Colman, H.; Soroceanu, L.; et al. Molecular subclasses of high-grade glioma predict prognosis, delineate a pattern of disease progression, and resemble stages in neurogenesis. Cancer Cell 2006, 9, 157–173. [Google Scholar] [CrossRef] [Green Version]
- Verhaak, R.G.W.; Hoadley, K.A.; Purdom, E.; Wang, V.; Qi, Y.; Wilkerson, M.D.; Miller, C.R.; Ding, L.; Golub, T.; Mesirov, J.P.; et al. Integrated Genomic Analysis Identifies Clinically Relevant Subtypes of Glioblastoma Characterized by Abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell 2010, 17, 98–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Hu, B.; Hu, X.; Kim, H.; Squatrito, M.; Scarpace, L.; de Carvalho, A.C.; Lyu, S.; Li, P.; Li, Y.; et al. Tumor Evolution of Glioma-Intrinsic Gene Expression Subtypes Associates with Immunological Changes in the Microenvironment. Cancer Cell 2017, 32, 42–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brennan, C.W.; Verhaak, R.G.W.; McKenna, A.; Campos, B.; Noushmehr, H.; Salama, S.R.; Zheng, S.; Chakravarty, D.; Sanborn, J.Z.; Berman, S.H.; et al. The somatic genomic landscape of glioblastoma. Cell 2013, 155, 462. [Google Scholar] [CrossRef]
- Gao, J.; Aksoy, B.A.; Dogrusoz, U.; Dresdner, G.; Gross, B.; Sumer, S.O.; Sun, Y.; Jacobsen, A.; Sinha, R.; Larsson, E.; et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013, 6, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gómez-Rubio, V. ggplot2—Elegant Graphics for Data Analysis (2nd Edition). J. Stat. Softw. 2017, 77, 3–5. [Google Scholar] [CrossRef] [Green Version]
- McLendon, R.; Friedman, A.; Bigner, D.; Van Meir, E.G.; Brat, D.J.; Mastrogianakis, G.M.; Olson, J.J.; Mikkelsen, T.; Lehman, N.; Aldape, K.; et al. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 2008, 455, 1061–1068. [Google Scholar] [CrossRef]
- Noushmehr, H.; Weisenberger, D.J.; Diefes, K.; Phillips, H.S.; Pujara, K.; Berman, B.P.; Pan, F.; Pelloski, C.E.; Sulman, E.P.; Bhat, K.P.; et al. Identification of a CpG Island Methylator Phenotype that Defines a Distinct Subgroup of Glioma. Cancer Cell 2010, 17, 510–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malta, T.M.; De Souza, C.F.; Sabedot, T.S.; Silva, T.C.; Mosella, M.S.; Kalkanis, S.N.; Snyder, J.; Castro, A.V.B.; Noushmehr, H. Glioma CpG island methylator phenotype (G-CIMP): Biological and clinical implications. Neuro. Oncol. 2018, 20, 608–620. [Google Scholar] [CrossRef] [PubMed]
- Martinez, R.; Schackert, G.; Yaya-Tur, R.; Rojas-Marcos, I.; Herman, J.G.; Esteller, M. Frequent hypermethylation of the DNA repair gene MGMT in long-term survivors of glioblastoma multiforme. J. Neurooncol. 2007, 83, 91–93. [Google Scholar] [CrossRef] [PubMed]
- Erasimus, H.; Gobin, M.; Niclou, S.; Van Dyck, E. DNA repair mechanisms and their clinical impact in glioblastoma. Mutat. Res. Mutat. Res. 2016, 769, 19–35. [Google Scholar] [CrossRef] [PubMed]
- Kitange, G.J.; Carlson, B.L.; Schroeder, M.A.; Grogan, P.T.; Lamont, J.D.; Decker, P.A.; Wu, W.; James, C.D.; Sarkaria, J.N. Induction of MGMT expression is associated with temozolomide resistance in glioblastoma xenografts. Neuro. Oncol. 2009, 11, 281–291. [Google Scholar] [CrossRef] [Green Version]
- Ichimura, K.; Pearson, D.M.; Kocialkowski, S.; Bäcklund, L.M.; Chan, R.; Jones, D.T.W.; Collins, V.P. IDH1 mutations are present in the majority of common adult gliomas but rare in primary glioblastomas. Neuro. Oncol. 2009, 11, 341–347. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Zhao, J.; Xu, Z.; Peng, B.; Huang, Q.; Arnold, E.; Ding, J. Structures of human cytosolic NADP-dependent isocitrate dehydrogenase reveal a novel self-regulatory mechanism of activity. J. Biol. Chem. 2004, 279, 33946–33957. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, T.; Nobusawa, S.; Kleihues, P.; Ohgaki, H. IDH1 mutations are early events in the development of astrocytomas and oligodendrogliomas. Am. J. Pathol. 2009, 174, 1149–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, H.; Parsons, D.W.; Jin, G.; McLendon, R.; Rasheed, B.A.; Yuan, W.; Kos, I.; Batinic-Haberle, I.; Jones, S.; Riggins, G.J.; et al. IDH1 and IDH2 mutations in gliomas. N. Engl. J. Med. 2009, 360, 765–773. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Lin, Y.; Xu, W.; Jiang, W.; Zhai, Z.; Wang, P.; Yu, W.; Li, Z.; Gong, L.; Peng, Y.; et al. Glioma-derived mutations in IDH1 dominantly inhibit IDH1 catalytic activity and induce HIF-1α. Science 2009, 324, 261–265. [Google Scholar] [CrossRef] [Green Version]
- Butler, M.; Pongor, L.; Su, Y.T.; Xi, L.; Raffeld, M.; Quezado, M.; Trepel, J.; Aldape, K.; Pommier, Y.; Wu, J. MGMT Status as a Clinical Biomarker in Glioblastoma. Trends Cancer 2020, 6, 380–391. [Google Scholar] [CrossRef]
- Gullotta, F.; Schindler, F.; Schmutzler, R.; Weeks-Seifert, A. GFAP in Brain Tumor Diagnosis: Possibilities and Limitations. Pathol. Res. Pract. 1985, 180, 54–60. [Google Scholar] [CrossRef]
- Kajiwara, K.; Orita, T.; Nishizaki, T.; Kamiryo, T.; Nakayama, H.; Ito, H. Glial fibrillary acidic protein (GFAP) expression and nucleolar organizer regions (NORs) in human gliomas. Brain Res. 1992, 572, 314–318. [Google Scholar] [CrossRef]
- Ligon, K.L.; Alberta, J.A.; Kho, A.T.; Weiss, J.; Kwaan, M.R.; Nutt, C.L.; Louis, D.N.; Stiles, C.D.; Rowitch, D.H. The oligodendroglial lineage marker OLIG2 is universally expressed in diffuse gliomas. J. Neuropathol. Exp. Neurol. 2004, 63, 499–509. [Google Scholar] [CrossRef] [Green Version]
- Mokhtari, K.; Paris, S.; Aguirre-Cruz, L.; Privat, N.; Crinière, E.; Marie, Y.; Hauw, J.J.; Kujas, M.; Rowitch, D.; Hoang-Xuan, K.; et al. Olig2 expression, GFAP, p53 and 1p loss analysis contribute to glioma subclassification. Neuropathol. Appl. Neurobiol. 2005, 31, 62–69. [Google Scholar] [CrossRef]
- Ma, Y.H.; Mentlein, R.; Knerlich, F.; Kruse, M.L.; Mehdorn, H.M.; Held-Feindt, J. Expression of stem cell markers in human astrocytomas of different WHO grades. J. Neurooncol. 2008, 86, 31–45. [Google Scholar] [CrossRef]
- Takenobu, H.; Shimozato, O.; Nakamura, T.; Ochiai, H.; Yamaguchi, Y.; Ohira, M.; Nakagawara, A.; Kamijo, T. CD133 suppresses neuroblastoma cell differentiation via signal pathway modification. Oncogene 2011, 30, 97–105. [Google Scholar] [CrossRef] [Green Version]
- Berezovsky, A.D.; Poisson, L.M.; Cherba, D.; Webb, C.P.; Transou, A.D.; Lemke, N.W.; Hong, X.; Hasselbach, L.A.; Irtenkauf, S.M.; Mikkelsen, T.; et al. Sox2 promotes malignancy in glioblastoma by regulating plasticity and astrocytic differentiation. Neoplasia 2014, 16, 193–206. [Google Scholar] [CrossRef] [Green Version]
- Hattermann, K.; Flüh, C.; Engel, D.; Mehdorn, H.M.; Synowitz, M.; Mentlein, R.; Held-Feindt, J. Stem cell markers in glioma progression and recurrence. Int. J. Oncol. 2016, 49, 1899–1910. [Google Scholar] [CrossRef] [Green Version]
- Rashidian, J.; Copaciu, R.; Su, Q.; Merritt, B.; Johnson, C.; Yahyabeik, A.; French, E.; Cummings, K. Generation and Performance of R132H Mutant IDH1 Rabbit Monoclonal Antibody. Antibodies 2017, 6, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Agarwal, S.; Sharma, M.C.; Jha, P.; Pathak, P.; Suri, V.; Sarkar, C.; Chosdol, K.; Suri, A.; Kale, S.S.; Mahapatra, A.K.; et al. Comparative study of IDH1 mutations in gliomas by immunohistochemistry and DNA sequencing. Neuro. Oncol. 2013, 15, 718–726. [Google Scholar] [CrossRef] [Green Version]
- Reuss, D.E.; Sahm, F.; Schrimpf, D.; Wiestler, B.; Capper, D.; Koelsche, C.; Schweizer, L.; Korshunov, A.; Jones, D.T.W.; Hovestadt, V.; et al. ATRX and IDH1-R132H immunohistochemistry with subsequent copy number analysis and IDH sequencing as a basis for an “integrated” diagnostic approach for adult astrocytoma, oligodendroglioma and glioblastoma. Acta Neuropathol. 2015, 129, 133–146. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Giese, A.; Loo, M.A.; Tran, N.; Haskett, D.; Coons, S.W.; Berens, M.E. Dichotomy of astrocytoma migration and proliferation. Int. J. Cancer 1996, 67, 275–282. [Google Scholar] [CrossRef]
- Rajapakse, V.N.; Herrada, S.; Lavi, O. Phenotype stability under dynamic brain-tumor environment stimuli maps glioblastoma progression in patients. Sci. Adv. 2020, 6, eaaz4125. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, A.; Hill, R.; Pilkington, G.; Madureira, P. The Role of Hypoxia in Glioblastoma Invasion. Cells 2017, 6, 45. [Google Scholar] [CrossRef] [Green Version]
- Velásquez, C.; Mansouri, S.; Mora, C.; Nassiri, F.; Suppiah, S.; Martino, J.; Zadeh, G.; Fernández-Luna, J.L. Molecular and Clinical Insights into the Invasive Capacity of Glioblastoma Cells. J. Oncol. 2019, 2019, 1740763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bindal, A.K.; Hammoud, M.; Shi, W.M.; Wu, S.Z.; Sawaya, R.; Rao, J.S. Prognostic significance of proteolytic enzymes in human brain tumors. J. Neurooncol. 1994, 22, 101–110. [Google Scholar] [CrossRef]
- Iwadate, Y. Epithelial-mesenchymal transition in glioblastoma progression. Oncol. Lett. 2016, 11, 1615–1620. [Google Scholar] [CrossRef] [Green Version]
- Brabletz, T.; Jung, A.; Spaderna, S.; Hlubek, F.; Kirchner, T. Migrating cancer stem cells—An integrated concept of malignant tumour progression. Nat. Rev. Cancer 2005, 5, 744–749. [Google Scholar] [CrossRef]
- Ho, I.A.W.; Shim, W.S.N. Contribution of the microenvironmental niche to glioblastoma heterogeneity. BioMed Res. Int. 2017, 2017, 9634172. [Google Scholar] [CrossRef]
- Prager, B.C.; Xie, Q.; Bao, S.; Rich, J.N. Cancer Stem Cells: The Architects of the Tumor Ecosystem. Cell Stem Cell 2019, 24, 41–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilbertson, R.J.; Rich, J.N. Making a tumour’s bed: Glioblastoma stem cells and the vascular niche. Nat. Rev. Cancer 2007, 7, 733–736. [Google Scholar] [CrossRef]
- Aderetti, D.A.; Hira, V.V.V.; Molenaar, R.J.; van Noorden, C.J.F. The hypoxic peri-arteriolar glioma stem cell niche, an integrated concept of five types of niches in human glioblastoma. Biochim. Biophys. Acta Rev. Cancer 2018, 1869, 346–354. [Google Scholar] [CrossRef]
- Bouwens van der Vlis, T.A.M.; Kros, J.M.; Mustafa, D.A.M.; van Wijck, R.T.A.; Ackermans, L.; van Hagen, P.M.; van der Spek, P.J. The complement system in glioblastoma multiforme. Acta Neuropathol. Commun. 2018, 6, 91. [Google Scholar] [CrossRef] [PubMed]
- Dirkse, A.; Golebiewska, A.; Buder, T.; Nazarov, P.V.; Muller, A.; Poovathingal, S.; Brons, N.H.C.; Leite, S.; Sauvageot, N.; Sarkisjan, D.; et al. Stem cell-associated heterogeneity in Glioblastoma results from intrinsic tumor plasticity shaped by the microenvironment. Nat. Commun. 2019, 10, 1787. [Google Scholar] [CrossRef]
- Schiffer, D.; Mellai, M.; Bovio, E.; Bisogno, I.; Casalone, C.; Annovazzi, L. Glioblastoma niches: From the concept to the phenotypical reality. Neurol. Sci. 2018, 39, 1161–1168. [Google Scholar] [CrossRef] [PubMed]
- Vescovi, A.L.; Galli, R.; Reynolds, B.A. Brain tumour stem cells. Nat. Rev. Cancer 2006, 6, 425–436. [Google Scholar] [CrossRef]
- Lathia, J.D.; Mack, S.C.; Mulkearns-Hubert, E.E.; Valentim, C.L.L.; Rich, J.N. Cancer stem cells in glioblastoma. Genes Dev. 2015, 29, 1203–1217. [Google Scholar] [CrossRef] [Green Version]
- Cheng, L.; Wu, Q.; Guryanova, O.A.; Huang, Z.; Huang, Q.; Rich, J.N.; Bao, S. Elevated invasive potential of glioblastoma stem cells. Biochem. Biophys. Res. Commun. 2011, 406, 643–648. [Google Scholar] [CrossRef] [Green Version]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef] [PubMed]
- Suvà, M.L.; Tirosh, I. The Glioma Stem Cell Model in the Era of Single-Cell Genomics. Cancer Cell 2020, 37, 630–636. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.P.; Tirosh, I.; Trombetta, J.J.; Shalek, A.K.; Gillespie, S.M.; Wakimoto, H.; Cahill, D.P.; Nahed, B.V.; Curry, W.T.; Martuza, R.L.; et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 2014, 344, 1396–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neftel, C.; Laffy, J.; Filbin, M.G.; Hara, T.; Shore, M.E.; Rahme, G.J.; Richman, A.R.; Silverbush, D.; Shaw, M.L.; Hebert, C.M.; et al. An Integrative Model of Cellular States, Plasticity, and Genetics for Glioblastoma. Cell 2019, 178, 835–849. [Google Scholar] [CrossRef]
- Hardee, M.E.; Zagzag, D. Mechanisms of glioma-associated neovascularization. Am. J. Pathol. 2012, 181, 1126–1141. [Google Scholar] [CrossRef] [Green Version]
- Zadeh, G.; Koushan, K.; Pillo, L.; Shannon, P.; Guha, A. Role of Ang1 and its interaction with VEGF-A in astrocytomas. J. Neuropathol. Exp. Neurol. 2004, 63, 978–989. [Google Scholar] [CrossRef] [Green Version]
- Zagzag, D.; Hooper, A.; Friedlander, D.R.; Chan, W.; Holash, J.; Wiegand, S.J.; Yancopoulos, G.D.; Grumet, M. In situ expression of angiopoietins in astrocytomas identifies angiopoietin-2 as an early marker of tumor angiogenesis. Exp. Neurol. 1999, 159, 391–400. [Google Scholar] [CrossRef]
- Ricci-Vitiani, L.; Pallini, R.; Biffoni, M.; Todaro, M.; Invernici, G.; Cenci, T.; Maira, G.; Parati, E.A.; Stassi, G.; Larocca, L.M.; et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 2010, 468, 824–830. [Google Scholar] [CrossRef] [PubMed]
- Treps, L.; Perret, R.; Edmond, S.; Ricard, D.; Gavard, J. Glioblastoma stem-like cells secrete the pro-angiogenic VEGF-A factor in extracellular vesicles. J. Extracell. Vesicles 2017, 6, 1359479. [Google Scholar] [CrossRef] [PubMed]
- Salajegheh, A. Angiogenesis in Health, Disease and Malignancy; Springer: Berlin/Heidelberg, Germany, 2016. [Google Scholar]
- Onishi, M.; Ichikawa, T.; Kurozumi, K.; Date, I. Angiogenesis and invasion in glioma. Brain Tumor Pathol. 2011, 28, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Brahimi-Horn, C.; Berra, E.; Pouysségur, J. Hypoxia: The tumor’s gateway to progression along the angiogenic pathway. Trends Cell Biol. 2001, 11, S32–S36. [Google Scholar] [CrossRef]
- Ferrara, N. Vascular Endothelial Growth Factor: Basic Science and Clinical Progress. Endocr. Rev. 2004, 25, 581–611. [Google Scholar] [CrossRef]
- Chambless, L.B.; Parker, S.L.; Hassam-Malani, L.; McGirt, M.J.; Thompson, R.C. Type 2 diabetes mellitus and obesity are independent risk factors for poor outcome in patients with high-grade glioma. J. Neurooncol. 2012, 106, 383–389. [Google Scholar] [CrossRef]
- Gong, Y.; Ma, Y.; Sinyuk, M.; Loganathan, S.; Thompson, R.C.; Sarkaria, J.N.; Chen, W.; Lathia, J.D.; Mobley, B.C.; Clark, S.W.; et al. Insulin-mediated signaling promotes proliferation and survival of glioblastoma through Akt activation. Neuro. Oncol. 2016, 18, 48–57. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Moore, L.M.; Li, X.; Yung, W.K.A.; Zhang, W. IDH1/2 mutations target a key hallmark of cancer by deregulating cellular metabolism in glioma. Neuro. Oncol. 2013, 15, 1114–1126. [Google Scholar] [CrossRef]
- Agnihotri, S.; Zadeh, G. Metabolic reprogramming in glioblastoma: The influence of cancer metabolism on epigenetics and unanswered questions. Neuro. Oncol. 2016, 18, 160–172. [Google Scholar] [CrossRef] [Green Version]
- Snuderl, M.; Fazlollahi, L.; Le, L.P.; Nitta, M.; Zhelyazkova, B.H.; Davidson, C.J.; Akhavanfard, S.; Cahill, D.P.; Aldape, K.D.; Betensky, R.A.; et al. Mosaic amplification of multiple receptor tyrosine kinase genes in glioblastoma. Cancer Cell 2011, 20, 810–817. [Google Scholar] [CrossRef] [Green Version]
- Van der Geer, P.; Hunter, T.; Lindberg, R.A. Receptor Protein-Tyrosine Kinases and Their Signal Transduction Pathways. Annu. Rev. Cell Biol. 1994, 10, 251–337. [Google Scholar] [CrossRef]
- Schlessinger, J. Cell signaling by receptor tyrosine kinases. Cell 2000, 103, 211–225. [Google Scholar] [CrossRef] [Green Version]
- Joshi, G.; Singh, P.K.; Negi, A.; Rana, A.; Singh, S.; Kumar, R. Growth factors mediated cell signalling in prostate cancer progression: Implications in discovery of anti-prostate cancer agents. Chem. Biol. Interact. 2015, 240, 120–133. [Google Scholar] [CrossRef]
- Normanno, N.; De Luca, A.; Bianco, C.; Strizzi, L.; Mancino, M.; Maiello, M.R.; Carotenuto, A.; De Feo, G.; Caponigro, F.; Salomon, D.S. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 2006, 366, 2–16. [Google Scholar] [CrossRef]
- Roberts, J.G.; Williams, M.; Henk, J.M.; Bligh, A.S.; Baum, M. The hypronosticon test in breast cancer. Clin.Oncol. 1975, 1, 33–43. [Google Scholar] [PubMed]
- Black, P.C.; Brown, G.A.; Dinney, C.P.; Kassouf, W.; Inamoto, T.; Arora, A.; Gallagher, D.; Munsell, M.F.; Bar-Eli, M.; McConkey, D.J.; et al. Receptor heterodimerization: A new mechanism for platelet-derived growth factor induced resistance to anti-epidermal growth factor receptor therapy for bladder cancer. J. Urol. 2011, 185, 693–700. [Google Scholar] [CrossRef] [PubMed]
- Erikson, J.; Griffin, C.A.; Ar-Rushdi, A.; Valtieri, M.; Hoxie, J.; Finan, J.; Emanuel, B.S.; Rovera, G.; Nowell, P.C.; Croce, C.M. Heterogeneity of chromosome 22 breakpoint in Philadelphia-positive (Ph+) acute lymphocytic leukemia. Proc. Natl. Acad. Sci. USA 1986, 83, 1807–1811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fantl, W.J.; Johnson, D.E.; Williams, L.T. Signalling by Receptor Tyrosine Kinases. Annu. Rev. Biochem. 1993, 62, 453–481. [Google Scholar] [CrossRef]
- Carvalho-Silva, D.; Pierleoni, A.; Pignatelli, M.; Ong, C.K.; Fumis, L.; Karamanis, N.; Carmona, M.; Faulconbridge, A.; Hercules, A.; McAuley, E.; et al. Open Targets Platform: New developments and updates two years on. Nucleic Acids Res. 2019, 47, D1056–D1065. [Google Scholar] [CrossRef]
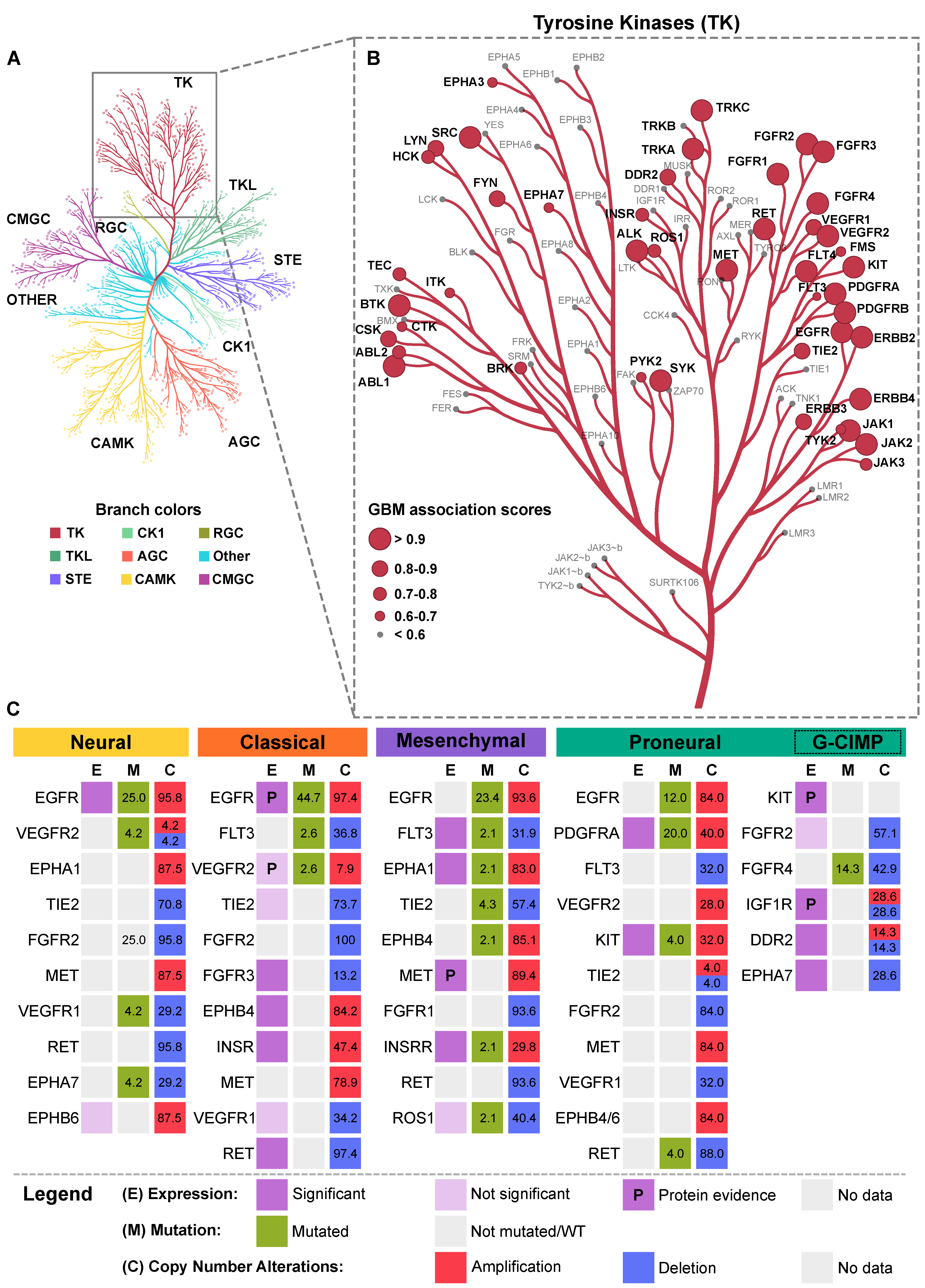
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [Green Version]
- Metz, K.S.; Deoudes, E.M.; Berginski, M.E.; Jimenez-Ruiz, I.; Aksoy, B.A.; Hammerbacher, J.; Gomez, S.M.; Phanstiel, D.H. Coral: Clear and Customizable Visualization of Human Kinome Data. Cell Syst. 2018, 7, 347–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burgess, A.W. EGFR family: Structure physiology signalling and therapeutic target. Growth Factors 2008, 26, 263–274. [Google Scholar] [CrossRef]
- Plowman, G.D.; Culouscou, J.M.; Whitney, G.S.; Green, J.M.; Carlton, G.W.; Foy, L.; Neubauer, M.G.; Shoyab, M. Ligand-specific activation of HER4/p180erbB4, a fourth member of the epidermal growth factor receptor family. Proc. Natl. Acad. Sci. USA 1993, 90, 1746–1750. [Google Scholar] [CrossRef] [Green Version]
- Plowman, G.D.; Whitney, G.S.; Neubauer, M.G.; Green, J.M.; McDonald, V.L.; Todaro, G.J.; Shoyab, M. Molecular cloning and expression of an additional epidermal growth factor receptor-related gene. Proc. Natl. Acad. Sci. USA 1990, 87, 4905–4909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stern, D.F.; Heffernan, P.A.; Weinberg, R.A. P185, a Product of the Neu Proto-Oncogene, Is a Receptorlike Protein Associated with Tyrosine Kinase Activity. Mol. Cell. Biol. 1986, 6, 1729–1740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bargmann, C.I.; Weinberg, R.A. Increased tyrosine kinase activity associated with the protein encoded by the activated neu oncogene. Proc. Natl. Acad. Sci. USA 1988, 85, 5394–5398. [Google Scholar] [CrossRef] [Green Version]
- Schlessinger, J. Ligand-induced, receptor-mediated dimerization and activation of EGF receptor. Cell 2002, 110, 669–672. [Google Scholar] [CrossRef] [Green Version]
- Blume-Jensen, P.; Hunter, T. Oncogenic kinase signalling. Nature 2001, 411, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Hatanpaa, K.J.; Burma, S.; Zhao, D.; Habib, A.A. Epidermal growth factor receptor in glioma: Signal transduction, neuropathology, imaging, and radioresistance1. Neoplasia 2010, 12, 675–684. [Google Scholar] [CrossRef] [Green Version]
- Rowinsky, E.K. The erbB Family: Targets for Therapeutic Development Against Cancer and Therapeutic Strategies Using Monoclonal Antibodies and Tyrosine Kinase Inhibitors. Annu. Rev. Med. 2004, 55, 433–457. [Google Scholar] [CrossRef] [Green Version]
- Zadeh, G.; Bhat, K.L.; Aldape, K. EGFR and EGFRvIII in Glioblastoma: Partners in Crime. Cancer Cell 2013, 24, 403–404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, Q.W.; Cheng, C.K.; Gustafson, W.C.; Charron, E.; Zipper, P.; Wong, R.A.; Chen, J.; Lau, J.; Knobbe-Thomsen, C.; Weller, M.; et al. EGFR Phosphorylates Tumor-Derived EGFRvIII Driving STAT3/5 and Progression in Glioblastoma. Cancer Cell 2013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koboldt, D.C.; Fulton, R.S.; McLellan, M.D.; Schmidt, H.; Kalicki-Veizer, J.; McMichael, J.F.; Fulton, L.L.; Dooling, D.J.; Ding, L.; Mardis, E.R.; et al. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar]
- Schlegel, J.; Stumm, G.; Brändle, K.; Merdes, A.; Mechtersheimer, G.; Hynes, N.E.; Kiessling, M. Amplification and differential expression of members of the erbB-gene family in human glioblastoma. J. Neurooncol. 1994, 22, 201–207. [Google Scholar] [CrossRef]
- Zhang, C.; Burger, M.C.; Jennewein, L.; Genßler, S.; Schönfeld, K.; Zeiner, P.; Hattingen, E.; Harter, P.N.; Mittelbronn, M.; Tonn, T.; et al. ErbB2/HER2-Specific NK Cells for Targeted Therapy of Glioblastoma. J. Natl. Cancer Inst. 2016, 108. [Google Scholar] [CrossRef] [PubMed]
- Andersson, U.; Guo, D.; Malmer, B.; Bergenheim, A.T.; Brännström, T.; Hedman, H.; Henriksson, R. Epidermal growth factor receptor family (EGFR, ErbB2-4) in gliomas and meningiomas. Acta Neuropathol. 2004. [Google Scholar] [CrossRef] [PubMed]
- Birchmeier, C. ErbB receptors and the development of the nervous system. Exp. Cell Res. 2009, 315, 611–618. [Google Scholar] [CrossRef]
- Donoghue, J.F.; Kerr, L.T.; Alexander, N.W.; Greenall, S.A.; Longano, A.B.; Gottardo, N.G.; Wang, R.; Tabar, V.; Adams, T.E.; Mischel, P.S.; et al. Activation of ERBB4 in glioblastoma can contribute to increased tumorigenicity and influence therapeutic response. Cancers 2018, 10, 243. [Google Scholar] [CrossRef] [Green Version]
- Belfiore, A.; Malaguarnera, R.; Vella, V.; Lawrence, M.C.; Sciacca, L.; Frasca, F.; Morrione, A.; Vigneri, R. Insulin receptor isoforms in physiology and disease: An updated view. Endocr. Rev. 2017, 38, 1–84. [Google Scholar] [CrossRef]
- Taniguchi, C.M.; Emanuelli, B.; Kahn, C.R. Critical nodes in signalling pathways: Insights into insulin action. Nat. Rev. Mol. Cell Biol. 2006, 7, 85–96. [Google Scholar] [CrossRef] [PubMed]
- Payankaulam, S.; Raicu, A.M.; Arnosti, D.N. Transcriptional regulation of INSR, the insulin receptor gene. Genes 2019, 10, 984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trojan, J.; Cloix, J.F.; Ardourel, M.Y.; Chatel, M.; Anthony, D.D. Insulin-like growth factor type I biology and targeting in malignant gliomas. Neuroscience 2007, 145, 795–811. [Google Scholar] [CrossRef] [PubMed]
- Deyev, I.E.; Rzhevsky, D.I.; Berchatova, A.A.; Serova, O.V.; Popova, N.V.; Murashev, A.N.; Petrenko, A.G. Deficient Response to Experimentally Induced Alkalosis in Mice with the Inactivated insrr Gene. Acta Nat. 2011, 3, 114–117. [Google Scholar] [CrossRef]
- Maris, C.; D’Haene, N.; Trépant, A.L.; Le Mercier, M.; Sauvage, S.; Allard, J.; Rorive, S.; Demetter, P.; Decaestecker, C.; Salmon, I. IGF-IR: A new prognostic biomarker for human glioblastoma. Br. J. Cancer 2015, 113, 729–737. [Google Scholar] [CrossRef]
- Girnita, L.; Worrall, C.; Takahashi, S.I.; Seregard, S.; Girnita, A. Something old, something new and something borrowed: Emerging paradigm of insulin-like growth factor type 1 receptor (IGF-1R) signaling regulation. Cell. Mol. Life Sci. 2014, 71, 2403–2427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fruttiger, M.; Karlsson, L.; Hall, A.C.; Abramsson, A.; Calver, A.R.; Boström, H.; Willetts, K.; Bertold, C.H.; Heath, J.K.; Betsholtz, C.; et al. Defective oligodendrocyte development and severe hypomyelination in PDGF-A knockout mice. Development 1999, 126, 457–467. [Google Scholar]
- Hellström, M.; Kalén, M.; Lindahl, P.; Abramsson, A.; Betsholtz, C. Role of PDGF-B and PDGFR-β in recruitment of vascular smooth muscle cells and pericytes during embryonic blood vessel formation in the mouse. Development 1999, 126, 3047–3055. [Google Scholar]
- Ozawa, T.; Brennan, C.W.; Wang, L.; Squatrito, M.; Sasayama, T.; Nakada, M.; Huse, J.T.; Pedraza, A.; Utsuki, S.; Yasui, Y.; et al. PDGFRA gene rearrangements are frequent genetic events in PDGFRA-amplified glioblastomas. Genes Dev. 2010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.; Kim, E.; Wu, Q.; Guryanova, O.; Hitomi, M.; Lathia, J.D.; Serwanski, D.; Sloan, A.E.; Weil, R.J.; Lee, J.; et al. Platelet-derived growth factor receptors differentially inform intertumoral and intratumoral heterogeneity. Genes Dev. 2012, 26, 1247–1262. [Google Scholar] [CrossRef] [Green Version]
- Matsui, J.; Wakabayashi, T.; Asada, M.; Yoshimatsu, K.; Okada, M. Stem Cell Factor/c-kit Signaling Promotes the Survival, Migration, and Capillary Tube Formation of Human Umbilical Vein Endothelial Cells. J. Biol. Chem. 2004, 279, 18600–18607. [Google Scholar] [CrossRef] [Green Version]
- Gomes, A.L.; Reis-Filho, J.S.; Lopes, J.M.; Martinho, O.; Lambros, M.B.K.; Martins, A.; Schmitt, F.; Pardal, F.; Reis, R.M. Molecular alterations of KIT oncogene in gliomas. Cell. Oncol. 2007, 29, 399–408. [Google Scholar] [CrossRef] [PubMed]
- Grafone, T.; Palmisano, M.; Nicci, C.; Storti, S. An overview on the role of FLT3-tyrosine kinase receptor in acute myeloid leukemia: Biology and treatment. Oncol. Rev. 2012, 6, 64–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eßbach, C.; Andrae, N.; Pachow, D.; Warnke, J.P.; Wilisch-Neumann, A.; Kirches, E.; Mawrin, C. Abundance of Flt3 and its ligand in astrocytic tumors. Onco. Targets Ther. 2013, 6, 555–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shibuya, M.; Yamaguchi, S.; Yamane, A.; Ikeda, T.; Tojo, A.; Matsushime, H.; Sato, M. Nucleotide sequence and expression of a novel human receptor-type tyrosine kinase gene (flt) closely relatd to the fms family. Oncogene 1990, 5, 519–524. [Google Scholar]
- Matthews, W.; Jordan, C.T.; Gavin, M.; Jenkins, N.A.; Copeland, N.G.; Lemischka, I.R. A receptor tyrosine kinase cDNA isolated from a population of enriched primitive hematopoietic cells and exhibiting close genetic linkage to c-kit. Proc. Natl. Acad. Sci. USA 1991, 88, 9026–9030. [Google Scholar] [CrossRef] [Green Version]
- Galland, F.; Karamysheva, A.; Pebusque, M.J.; Borg, J.P.; Rottapel, R.; Dubreuil, P.; Rosnet, O.; Birnbaum, D. The FLT4 gene encodes a transmembrane tyrosine kinase related to the vascular endothelial growth factor receptor. Oncogene 1993, 8, 1233–1240. [Google Scholar]
- Shibuya, M. Vascular Endothelial Growth Factor (VEGF) and Its Receptor (VEGFR) Signaling in Angiogenesis: A Crucial Target for Anti- and Pro-Angiogenic Therapies. Genes Cancer 2011, 2, 1097–1105. [Google Scholar] [CrossRef]
- Takahashi, H.; Shibuya, M. The vascular endothelial growth factor (VEGF)/VEGF receptor system and its role under physiological and pathological conditions. Clin. Sci. 2005, 109, 227–241. [Google Scholar] [CrossRef] [Green Version]
- Loureiro, L.V.M.; Neder, L.; Callegaro-Filho, D.; de Oliveira Koch, L.; Stavale, J.N.; Malheiros, S.M.F. The immunohistochemical landscape of the VEGF family and its receptors in glioblastomas. Surg. Exp. Pathol. 2020, 3, 9. [Google Scholar] [CrossRef]
- Guarnaccia, L.; Navone, S.E.; Trombetta, E.; Cordiglieri, C.; Cherubini, A.; Crisà, F.M.; Rampini, P.; Miozzo, M.; Fontana, L.; Caroli, M.; et al. Angiogenesis in human brain tumors: Screening of drug response through a patient-specific cell platform for personalized therapy. Sci. Rep. 2018, 8, 8748. [Google Scholar] [CrossRef]
- Siveen, K.S.; Prabhu, K.; Krishnankutty, R.; Kuttikrishnan, S.; Tsakou, M.; Alali, F.Q.; Dermime, S.; Mohammad, R.M.; Uddin, S. Vascular Endothelial Growth Factor (VEGF) Signaling in Tumour Vascularization: Potential and Challenges. Curr. Vasc. Pharmacol. 2017, 15. [Google Scholar] [CrossRef] [PubMed]
- Zimna, A.; Kurpisz, M. Hypoxia-Inducible factor-1 in physiological and pathophysiological angiogenesis: Applications and therapies. BioMed Res. Int. 2015, 2015. [Google Scholar] [CrossRef] [Green Version]
- Turner, N.; Grose, R. Fibroblast growth factor signalling: From development to cancer. Nat. Rev. Cancer 2010, 10, 116–129. [Google Scholar] [CrossRef]
- Collette, J.C.; Choubey, L.; Smith, K.M. Glial and stem cell expression of murine fibroblast growth factor receptor 1 in the embryonic and perinatal nervous system. PeerJ 2017, 2017, e3519. [Google Scholar] [CrossRef] [Green Version]
- Kang, W.; Hébert, J.M. FGF signaling is necessary for neurogenesis in young mice and sufficient to reverse its decline in old mice. J. Neurosci. 2015, 35, 10217–10223. [Google Scholar] [CrossRef]
- Touat, M.; Ileana, E.; Postel-Vinay, S.; André, F.; Soria, J.C. Targeting FGFR signaling in cancer. Clin. Cancer Res. 2015, 21, 2684–2694. [Google Scholar] [CrossRef] [Green Version]
- Darmanis, S.; Sloan, S.A.; Croote, D.; Mignardi, M.; Chernikova, S.; Samghababi, P.; Zhang, Y.; Neff, N.; Kowarsky, M.; Caneda, C.; et al. Single-Cell RNA-Seq Analysis of Infiltrating Neoplastic Cells at the Migrating Front of Human Glioblastoma. Cell Rep. 2017, 21, 1399–1410. [Google Scholar] [CrossRef] [Green Version]
- Huang, E.J.; Reichardt, L.F. Trk Receptors: Roles in Neuronal Signal Transduction. Annu. Rev. Biochem. 2003, 72, 609–642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen-Cory, S.; Kidane, A.H.; Shirkey, N.J.; Marshak, S. Brain-derived neurotrophic factor and the development of structural neuronal connectivity. Dev. Neurobiol. 2010, 70, 271–288. [Google Scholar] [CrossRef] [Green Version]
- Segal, R.A. Selectivity in neurotrophin signaling: Theme and variations. Annu. Rev. Neurosci. 2003, 26, 299–330. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.; Jing, S.; Nanduri, V.; O’Rourke, E.; Barbacid, M. The trk proto-oncogene encodes a receptor for nerve growth factor. Cell 1991, 65, 189–197. [Google Scholar] [CrossRef]
- Singer, H.S.; Hansen, B.; Martinie, D.; Karp, C.L. Mitogenesis in glioblastoma multiforme cell lines: A role for NGF and its TrkA receptors. J. Neurooncol. 1999, 45, 1–8. [Google Scholar] [CrossRef]
- Lu, B.; Figurov, A. Role of neurotrophins in synapse development and plasticity. Rev. Neurosci. 1997, 8, 1–12. [Google Scholar] [CrossRef]
- Guo, W.; Nagappan, G.; Lu, B. Differential effects of transient and sustained activation of BDNF-TrkB signaling. Dev. Neurobiol. 2018, 78, 647–659. [Google Scholar] [CrossRef]
- Lawn, S.; Krishna, N.; Pisklakova, A.; Qu, X.; Fenstermacher, D.A.; Fournier, M.; Vrionis, F.D.; Tran, N.; Chan, J.A.; Kenchappa, R.S.; et al. Neurotrophin Signaling via TrkB and TrkC Receptors Promotes the Growth of Brain Tumor-initiating Cells. J. Biol. Chem. 2015, 290, 3814–3824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinheiro, K.V.; Alves, C.; Buendia, M.; Gil, M.S.; Thomaz, A.; Schwartsmann, G.; De Farias, C.B.; Roesler, R. Targeting tyrosine receptor kinase B in gliomas. Neuro. Oncol. 2017, 19, 138–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, G.; Broniscer, A.; McEachron, T.A.; Lu, C.; Paugh, B.S.; Becksfort, J.; Qu, C.; Ding, L.; Huether, R.; Parker, M.; et al. Somatic histone H3 alterations in pediatric diffuse intrinsic pontine gliomas and non-brainstem glioblastomas. Nat. Genet. 2012, 44, 251–253. [Google Scholar]
- Petrini, I. Biology of MET: A double life between normal tissue repair and tumor progression. Ann. Transl. Med. 2015, 3, 82. [Google Scholar] [PubMed]
- Bladt, F.; Riethmacher, D.; Isenmann, S.; Aguzzi, A.; Birchmeier, C. Essential role for the c-met receptor in the migration of myogenic precursor cells into the limb bud. Nature 1995, 376, 768–771. [Google Scholar] [CrossRef]
- Cheng, F.; Guo, D. MET in glioma: Signaling pathways and targeted therapies. J. Exp. Clin. Cancer Res. 2019, 38, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Li, A.; Glas, M.; Lal, B.; Ying, M.; Sang, Y.; Xia, S.; Trageser, D.; Guerrero-Cázares, H.; Eberhart, C.G.; et al. c-Met signaling induces a reprogramming network and supports the glioblastoma stem-like phenotype. Proc. Natl. Acad. Sci. USA 2011, 108, 9951–9956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajadurai, C.V.; Havrylov, S.; Zaoui, K.; Vaillancourt, R.; Stuible, M.; Naujokas, M.; Zuo, D.; Tremblay, M.L.; Park, M. Met receptor tyrosine kinase signals through a cortactin-Gab1 scaffold complex, to mediate invadopodia. J. Cell Sci. 2012, 125, 2940–2953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petterson, S.A.; Dahlrot, R.H.; Hermansen, S.K.; Munthe, S.K.A.; Gundesen, M.T.; Wohlleben, H.; Rasmussen, T.; Beier, C.P.; Hansen, S.; Kristensen, B.W. High levels of c-Met is associated with poor prognosis in glioblastoma. J. Neurooncol. 2015, 122, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.S.; Song, S.Y.; Kim, D.H.; Kyeung, M.J.; Yoo, J.S.; Jong, S.K.; Seung, M.D.; Suh, Y.L.; Lee, J.I.; Park, K.; et al. Prognostic significance of c-Met expression in glioblastomas. Cancer 2009, 115, 140–148. [Google Scholar] [CrossRef] [PubMed]
- Gale, N.W.; Holland, S.J.; Valenzuela, D.M.; Flenniken, A.; Pan, L.; Ryan, T.E.; Henkemeyer, M.; Strebhardt, K.; Hirai, H.; Wilkinson, D.G.; et al. Eph receptors and ligands comprise two major specificity subclasses and are reciprocally compartmentalized during embryogenesis. Neuron 1996, 17, 9–19. [Google Scholar] [CrossRef] [Green Version]
- Pasquale, E.B. Eph-Ephrin Bidirectional Signaling in Physiology and Disease. Cell 2008, 133, 38–52. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, Y.; Pasquale, E.B. Eph receptors in the adult brain. Curr. Opin. Neurobiol. 2004, 14, 288–296. [Google Scholar] [CrossRef] [PubMed]
- Wilkinson, D.G. Eph receptors and ephrins: Regulators of guidance and assembly. Int. Rev. Cytol. 2000, 196, 177–244. [Google Scholar]
- Wilkinson, D.G. Multiple roles of eph receptors and ephrins in neural development. Nat. Rev. Neurosci. 2001, 2, 155–164. [Google Scholar] [CrossRef]
- Ferluga, S.; Tomé, C.M.L.; Herpai, D.M.; D’Agostino, R.; Debinski, W. Simultaneous targeting of Eph receptors in glioblastoma. Oncotarget 2016, 7, 59860–59876. [Google Scholar] [CrossRef] [Green Version]
- Binda, E.; Visioli, A.; Giani, F.; Lamorte, G.; Copetti, M.; Pitter, K.L.; Huse, J.T.; Cajola, L.; Zanetti, N.; DiMeco, F.; et al. The EphA2 Receptor Drives Self-Renewal and Tumorigenicity in Stem-like Tumor-Propagating Cells from Human Glioblastomas. Cancer Cell 2012, 22, 765–780. [Google Scholar] [CrossRef] [Green Version]
- Day, B.W.; Stringer, B.W.; Al-Ejeh, F.; Ting, M.J.; Wilson, J.; Ensbey, K.S.; Jamieson, P.R.; Bruce, Z.C.; Lim, Y.C.; Offenhäuser, C.; et al. EphA3 Maintains Tumorigenicity and Is a Therapeutic Target in Glioblastoma Multiforme. Cancer Cell 2013, 23, 238–248. [Google Scholar] [CrossRef] [Green Version]
- Qazi, M.A.; Vora, P.; Venugopal, C.; Adams, J.; Singh, M.; Hu, A.; Gorelik, M.; Subapanditha, M.K.; Savage, N.; Yang, J.; et al. Cotargeting ephrin receptor tyrosine kinases A2 and A3 in cancer stem cells reduces growth of recurrent glioblastoma. Cancer Res. 2018, 78, 5023–5037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dumont, D.J.; Yamaguchi, T.P.; Conlon, R.A.; Rossant, J.; Breitman, M.L. Tek, a novel tyrosine kinase gene located on mouse chromosome 4, is expressed in endothelial cells and their presumptive precursors. Oncogene 1992, 7, 1471–1480. [Google Scholar] [PubMed]
- Jones, N.; Iljin, K.; Dumont, D.J.; Alitalo, K. Tie receptors: New modulators of angiogenic and lymphangiogenic responses. Nat. Rev. Mol. Cell Biol. 2001, 2, 257–267. [Google Scholar] [CrossRef] [PubMed]
- Lee, O.H.; Xu, J.; Fueyo, J.; Fuller, G.N.; Aldape, K.D.; Alonso, M.M.; Piao, Y.; Liu, T.J.; Lang, F.F.; Bekele, B.N.; et al. Expression of the receptor tyrosine kinase Tie2 in neoplastic glial cells is associated with integrin β1-dependent adhesion to the extracellular matrix. Mol. Cancer Res. 2006, 4, 915–926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serrano Cardona, L.; Muñoz Mata, E. Paraninfo Digital. Early Hum. Dev. 2013, 83, 1–11. [Google Scholar] [CrossRef]
- Teichert-Kuliszewska, K.; Maisonpierre, P.C.; Jones, N.; Campbell, A.I.M.; Master, Z.; Bendeck, M.P.; Alitalo, K.; Dumont, D.J.; Yancopoulos, G.D.; Stewart, D.J. Biological action of angiopoietin-2 in a fibrin matrix model of angiogenesis is associated with activation of Tie2. Cardiovasc. Res. 2001, 49, 659–670. [Google Scholar] [CrossRef] [Green Version]
- Daly, C.; Eichten, A.; Castanaro, C.; Pasnikowski, E.; Adler, A.; Lalani, A.S.; Papadopoulos, N.; Kyle, A.H.; Minchinton, A.I.; Yancopoulos, G.D.; et al. Angiopoietin-2 functions as a Tie2 agonist in tumor models, where it limits the effects of VEGF inhibition. Cancer Res. 2013, 73, 108–118. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Guo, P.; Fang, Q.; Tao, H.Q.; Wang, D.; Nagane, M.; Huang, H.J.S.; Gunji, Y.; Nishikawa, R.; Alitalo, K.; et al. Angiopoietin-2 induces human glioma invasion through the activation of matrix metalloprotease-2. Proc. Natl. Acad. Sci. USA 2003, 100, 8904–8909. [Google Scholar] [CrossRef] [Green Version]
- Hu, B.; Jarzynka, M.J.; Guo, P.; Imanishi, Y.; Schlaepfer, D.D.; Cheng, S.Y. Angiopoietin 2 induces glioma cell invasion by stimulating matrix metalloprotease 2 expression through the αvβ1 integrin and focal adhesion kinase signaling pathway. Cancer Res. 2006, 66, 775–783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coffelt, S.B.; Tal, A.O.; Scholz, A.; De Palma, M.; Patel, S.; Urbich, C.; Biswas, S.K.; Murdoch, C.; Plate, K.H.; Reiss, Y.; et al. Angiopoietin-2 regulates gene expression in TIE2-expressing monocytes and augments their inherent proangiogenic functions. Cancer Res. 2010, 70, 5270–5280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gabrusiewicz, K.; Liu, D.; Cortes-Santiago, N.; Hossain, M.B.; Conrad, C.A.; Aldape, K.D.; Fuller, G.N.; Marini, F.C.; Alonso, M.M.; Idoate, M.A.; et al. Anti-vascular endothelial growth factor therapy-induced glioma invasion is associated with accumulation of Tie2-expressing monocytes. Oncotarget 2014, 5, 2208–2220. [Google Scholar] [CrossRef] [Green Version]
- Cortes-Santiago, N.; Hossain, M.B.; Gabrusiewicz, K.; Fan, X.; Gumin, J.; Marini, F.C.; Alonso, M.M.; Lang, F.; Yung, W.K.; Fueyo, J.; et al. Soluble Tie2 overrides the heightened invasion induced by anti-angiogenesis therapies in gliomas. Oncotarget 2016, 7, 16146–16157. [Google Scholar] [CrossRef] [Green Version]
- Carafoli, F.; Hohenester, E. Collagen recognition and transmembrane signalling by discoidin domain receptors. Biochim. Biophys. Acta Proteins Proteom. 2013, 1834, 2187–2194. [Google Scholar] [CrossRef] [Green Version]
- Vogel, W.; Gish, G.D.; Alves, F.; Pawson, T. The Discoidin Domain Receptor Tyrosine Kinases Are Activated by Collagen. Mol. Cell 1997, 1, 13–23. [Google Scholar] [CrossRef]
- Valiathan, R.R.; Marco, M.; Leitinger, B.; Kleer, C.G.; Fridman, R. Discoidin domain receptor tyrosine kinases: New players in cancer progression. Cancer Metastasis Rev. 2012, 31, 295–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Payne, L.S.; Huang, P.H. The pathobiology of collagens in glioma. Mol. Cancer Res. 2013, 11, 1129–1140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiner, H.L.; Rothman, M.; Miller, D.C.; Ziff, E.B. Pediatric brain tumors express multiple receptor tyrosine kinases including novel cell adhesion kinases. Pediatr. Neurosurg. 1996, 25, 64–72. [Google Scholar] [CrossRef]
- Weiner, H.L.; Huang, H.; Zagzag, D.; Boyce, H.; Lichtenbaum, R.; Ziff, E.B. Consistent and Selective Expression of the Discoidin Domain Receptor-1 Tyrosine Kinase in Human Brain Tumors. Neurosurgery 2000, 47, 1400–1409. [Google Scholar] [CrossRef] [PubMed]
- Ram, R.; Lorente, G.; Nikolich, K.; Urfer, R.; Foehr, E.; Nagavarapu, U. Discoidin Domain Receptor-1a (DDR1a) Promotes Glioma Cell Invasion and Adhesion in Association with Matrix Metalloproteinase-2. J. Neurooncol. 2006, 76, 239–248. [Google Scholar] [CrossRef] [PubMed]
- Yamanaka, R.; Arao, T.; Yajima, N.; Tsuchiya, N.; Homma, J.; Tanaka, R.; Sano, M.; Oide, A.; Sekijima, M.; Nishio, K. Identification of expressed genes characterizing long-term survival in malignant glioma patients. Oncogene 2006, 25, 5994–6002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- El Husseini, K.; Marguet, F.; Lamy, A.; Magne, N.; Fontanilles, M. Major response to temozolomide as first-line treatment for newly-diagnosed DDR2-mutated glioblastoma: A case report. Rev. Neurol. 2020, 176, 402–404. [Google Scholar] [CrossRef] [PubMed]
- Arighi, E.; Borrello, M.G.; Sariola, H. RET tyrosine kinase signaling in development and cancer. Cytokine Growth Factor Rev. 2005, 16, 441–467. [Google Scholar] [CrossRef]
- Pachnis, V.; Mankoo, B.; Costantini, F. Expression of the c-ret proto-oncogene during mouse embryogenesis. Development 1993, 119, 1005–1017. [Google Scholar]
- Bonanomi, D.; Chivatakarn, O.; Bai, G.; Abdesselem, H.; Lettieri, K.; Marquardt, T.; Pierchala, B.A.; Pfaff, S.L. Ret Is a Multifunctional Coreceptor that Integrates Diffusible- and Contact-Axon Guidance Signals. Cell 2012, 148, 568–582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pierchala, B.A. Glial Cell Line-Derived Neurotrophic Factor-Dependent Recruitment of Ret into Lipid Rafts Enhances Signaling by Partitioning Ret from Proteasome-Dependent Degradation. J. Neurosci. 2006, 26, 2777–2787. [Google Scholar] [CrossRef] [Green Version]
- Wiesenhofer, B.; Stockhammer, G.; Kostron, H.; Maier, H.; Hinterhuber, H.; Humpel, C. Glial cell line-derived neurotrophic factor (GDNF) and its receptor (GFR-α1) are strongly expressed in human gliomas. Acta Neuropathol. 2000, 99, 131–137. [Google Scholar] [CrossRef]
- Kato, S.; Subbiah, V.; Marchlik, E.; Elkin, S.K.; Carter, J.L.; Kurzrock, R. RET Aberrations in Diverse Cancers: Next-Generation Sequencing of 4,871 Patients. Clin. Cancer Res. 2017, 23, 1988–1997. [Google Scholar] [CrossRef] [Green Version]
- Birchmeier, C.; O’Neill, K.; Riggs, M.; Wigler, M. Characterization of ROS1 cDNA from a human glioblastoma cell line. Proc. Natl. Acad. Sci. USA 1990, 87, 4799–4803. [Google Scholar] [CrossRef] [Green Version]
- El-Deeb, I.M.; Yoo, K.H.; Lee, S.H. ROS receptor tyrosine kinase: A new potential target for anticancer drugs. Med. Res. Rev. 2010, 31, 794–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birchmeier, C.; Sharma, S.; Wigler, M. Expression and rearrangement of the ROS1 gene in human glioblastoma cells. Proc. Natl. Acad. Sci. USA 1987, 84, 9270–9274. [Google Scholar] [CrossRef] [Green Version]
- Jun, H.J.; Woolfenden, S.; Coven, S.; Lane, K.; Bronson, R.; Housman, D.; Charest, A. Epigenetic Regulation of c-ROS Receptor Tyrosine Kinase Expression in Malignant Gliomas. Cancer Res. 2009, 69, 2180–2184. [Google Scholar] [CrossRef] [Green Version]
- Pawson, T. Dynamic control of signaling by modular adaptor proteins. Curr. Opin. Cell Biol. 2007, 19, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Batistatou, A.; Zioga, A.; Panelos, J.; Massi, D.; Agnantis, N.J.; Charalabopoulos, K. A new concept of melanocytic neoplasia pathogenesis based on the phenotype of common acquired nevi. Med. Hypotheses 2007, 69, 1334–1339. [Google Scholar] [CrossRef]
- Hall, B.E.; Bar-Sagi, D.; Nassar, N. The structural basis for the transition from Ras-GTP to Ras-GDP. Proc. Natl. Acad. Sci. USA 2002, 99, 12138–12142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajasekhar, V.K.; Viale, A.; Socci, N.D.; Wiedmann, M.; Hu, X.; Holland, E.C. Oncogenic Ras and Akt signaling contribute to glioblastoma formation by differential recruitment of existing mRNAs to polysomes. Mol. Cell 2003, 12, 889–901. [Google Scholar] [CrossRef]
- Cheng, C.K.; Fan, Q.W.; Weiss, W.A. PI3K signaling in glioma—Animal models and therapeutic challenges. Brain Pathol. 2009, 19, 112–120. [Google Scholar] [CrossRef] [Green Version]
- Kim, I.; Kim, H.G.; Moon, S.O.; Chae, S.W.; So, J.N.; Koh, K.N.; Ahn, B.C.; Koh, G.Y. Angiopoietin-1 induces endothelial cell sprouting through the activation of focal adhesion kinase and plasmin secretion. Circ. Res. 2000, 86, 952–959. [Google Scholar] [CrossRef] [Green Version]
- Kim, I.; Kim, J.H.; Moon, S.O.; Kwak, H.J.; Kim, N.G.; Koh, G.Y. Angiopoietin-2 at high concentration can enhance enthelial cell survival through the phosphatidylinositol 3′-kinase/Akt signal transduction pathway. Oncogene 2000, 19, 4549–4552. [Google Scholar] [CrossRef] [Green Version]
- Vivanco, I.; Sawyers, C.L. The phosphatidylinositol 3-kinase-AKT pathway in humancancer. Nat. Rev. Cancer 2002, 2, 489–501. [Google Scholar] [CrossRef]
- Sunayama, J.; Matsuda, K.I.; Sato, A.; Tachibana, K.; Suzuki, K.; Narita, Y.; Shibui, S.; Sakurada, K.; Kayama, T.; Tomiyama, A.; et al. Crosstalk between the PI3K/mTOR and MEK/ERK pathways involved in the maintenance of self-renewal and tumorigenicity of glioblastoma stem-like cells. Stem Cells 2010, 28, 1930–1939. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, M.C.; Er, E.E.; Blenis, J. The Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends Biochem. Sci. 2011, 36, 320–328. [Google Scholar] [CrossRef] [Green Version]
- Jeuken, J.; van de Broecke, C.; Gijsen, S.; Boots-Sprenger, S.; Wesseling, P. RAS/RAF pathway activation in gliomas: The result of copy number gains rather than activating mutations. Acta Neuropathol. 2007, 114, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Laplante, M.; Sabatini, D.M. mTOR signaling at a glance. J. Cell Sci. 2009, 122, 3589–3594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sen, B.; Xie, Z.; Case, N.; Thompson, W.R.; Uzer, G.; Styner, M.; Rubin, J. MTORC2 regulates mechanically induced cytoskeletal reorganization and lineage selection in marrow-derived mesenchymal stem cells. J. Bone Miner. Res. 2014, 29, 78–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mecca, C.; Giambanco, I.; Donato, R.; Arcuri, C. Targeting mTOR in glioblastoma: Rationale and preclinical/clinical evidence. Dis. Markers 2018, 2018. [Google Scholar] [CrossRef] [Green Version]
- Hauck, C.R.; Hsia, D.A.; Schlaepfer, D.D. The focal adhesion kinase—A regulator of cell migration and invasion. IUBMB Life 2002, 53, 115–119. [Google Scholar] [CrossRef] [Green Version]
- Guo, W.; Giancotti, F.G. Integrin signalling during tumour progression. Nat. Rev. Mol. Cell Biol. 2004, 5, 816–826. [Google Scholar] [CrossRef]
- Hu, J.; Mukhopadhyay, A.; Truesdell, P.; Chander, H.; Mukhopadhyay, U.K.; Mak, A.S.; Craig, A.W.B. Cdc42-interacting protein 4 is a Src substrate that regulates invadopodia and invasiveness of breast tumors by promoting MT1-MMP endocytosis. J. Cell Sci. 2011, 124, 1739–1751. [Google Scholar] [CrossRef] [Green Version]
- Hecker, T.P.; Grammer, J.R.; Gillespie, G.Y.; Stewart, J.; Gladson, C.L. Focal adhesion kinase enhances signaling through the Shc/extracellular signal-regulated kinase pathway in anaplastic astrocytoma tumor biopsy samples. Cancer Res. 2002, 62, 2699–2707. [Google Scholar] [PubMed]
- Golubovskaya, V.M.; Huang, G.; Ho, B.; Yemma, M.; Morrison, C.D.; Lee, J.; Eliceiri, B.P.; Cance, W.G. Pharmacologic blockade of FAK autophosphorylation decreases human glioblastoma tumor growth and synergizes with temozolomide. Mol. Cancer Ther. 2013, 12, 162–172. [Google Scholar] [CrossRef] [Green Version]
- Wills, M.K.B.; Jones, N. Teaching an old dogma new tricks: Twenty years of Shc adaptor signalling. Biochem. J. 2012, 447, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Wills, M.K.B.; Chahi, A.K.; Lau, H.R.; Tilak, M.; Guild, B.D.; New, L.A.; Lu, P.; Jacquet, K.; Meakin, S.O.; Bisson, N.; et al. Signaling adaptor ShcD suppresses extracellular signal-regulated kinase (Erk) phosphorylation distal to the Ret and Trk neurotrophic receptors. J. Biol. Chem. 2017, 292, 5748–5759. [Google Scholar] [CrossRef] [Green Version]
- Audero, E.; Cascone, I.; Maniero, F.; Napione, L.; Arese, M.; Lanfrancone, L.; Bussolino, F. Adaptor ShcA Protein Binds Tyrosine Kinase Tie2 Receptor and Regulates Migration and Sprouting but Not Survival of Endothelial Cells. J. Biol. Chem. 2004, 279, 13224–13233. [Google Scholar] [CrossRef] [Green Version]
- Jones, N.; Master, Z.; Jones, J.; Bouchard, D.; Gunji, Y.; Sasaki, H.; Daly, R.; Alitalo, K.; Dumont, D.J. Identification of Tek/Tie2 binding partners. Binding to a multifunctional docking site mediates cell survival and migration. J. Biol. Chem. 1999, 274, 30896–30905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayer, B.J. Perspective: Dynamics of receptor tyrosine kinase signaling complexes. FEBS Lett. 2012, 586, 2575–2579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jadwin, J.A.; Curran, T.G.; Lafontaine, A.T.; White, F.M.; Mayer, B.J. Src homology 2 domains enhance tyrosine phosphorylation in vivo by protecting binding sites in their target proteins from dephosphorylation. J. Biol. Chem. 2018, 293, 623–627. [Google Scholar] [CrossRef] [Green Version]
- Wills, M.K.B.; Tong, J.; Tremblay, S.L.; Moran, M.F.; Jones, N. The ShcD signaling adaptor facilitates ligand-independent phosphorylation of the EGF receptor. Mol. Biol. Cell 2014, 25, 739–752. [Google Scholar] [CrossRef]
- Tilak, M.; Alural, B.; Wismer, S.E.; Brasher, M.I.; New, L.A.; Sheridan, S.D.; Perlis, R.H.; Coppolino, M.G.; Lalonde, J.; Jones, N. Adaptor protein ShcD interacts with Tie2 receptor to synergistically promote glioma cell invasion. Mol. Cancer Res. 2021. [Google Scholar] [CrossRef]
- Weis, W.I.; Kobilka, B.K. The Molecular Basis of G Protein-Coupled Receptor Activation. Annu. Rev. Biochem. 2018, 87, 897–919. [Google Scholar] [CrossRef]
- Daub, H.; Weiss, F.U.; Wallasch, C.; Ullrich, A. Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature 1996, 379, 557–560. [Google Scholar] [CrossRef]
- Cattaneo, F.; Guerra, G.; Parisi, M.; De Marinis, M.; Tafuri, D.; Cinelli, M.; Ammendola, R. Cell-surface receptors transactivation mediated by G protein-coupled receptors. Int. J. Mol. Sci. 2014, 15, 19700. [Google Scholar] [CrossRef] [Green Version]
- Prenzel, N.; Zwick, E.; Daub, H.; Leserer, M.; Abraham, R.; Wallasch, C.; Ullrich, A. EGF receptor transactivation by G-protein-coupled receptors requires metalloproteinase cleavage of proHB-EGF. Nature 1999, 402, 884–888. [Google Scholar] [CrossRef] [PubMed]
- Sternlicht, M.D.; Werb, Z. How matrix metalloproteinases regulate cell behavior. Annu. Rev. Cell Dev. Biol. 2001, 17, 463–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borroto-Escuela, D.O.; Romero-Fernandez, W.; Mudó, G.; Pérez-Alea, M.; Ciruela, F.; Tarakanov, A.O.; Narvaez, M.; Di Liberto, V.; Agnati, L.F.; Belluardo, N.; et al. Fibroblast growth factor receptor 1 5-hydroxytryptamine 1A heteroreceptor complexes and their enhancement of hippocampal plasticity. Biol. Psychiatry 2012, 71, 84–91. [Google Scholar] [CrossRef] [PubMed]
- El-Shewy, H.M.; Abdel-Samie, S.A.; al Qalam, A.M.; Lee, M.H.; Kitatani, K.; Anelli, V.; Jaffa, A.A.; Obeid, L.M.; Luttrell, L.M. Phospholipase C and protein kinase C-β 2 mediate insulin-like growth factor ii-dependent sphingosine kinase 1 activation. Mol. Endocrinol. 2011, 25, 2144–2156. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.-K.; Chen, J.-K.; Harris, R.C. Angiotensin II Induces Epithelial-to-Mesenchymal Transition in Renal Epithelial Cells through Reactive Oxygen Species/Src/Caveolin-Mediated Activation of an Epidermal Growth Factor Receptor-Extracellular Signal-Regulated Kinase Signaling Pathway. Mol. Cell. Biol. 2012, 32, 981–991. [Google Scholar] [CrossRef] [Green Version]
- Belcheva, M.M.; Haas, P.D.; Tan, Y.; Heaton, V.M.; Coscia, C.J. The fibroblast growth factor receptor is at the site of convergence between μ-opioid receptor and growth factor signaling pathways in rat C6 glioma cells. J. Pharmacol. Exp. Ther. 2002, 303, 909–918. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Hu, J.; Bian, X.; Chen, K.; Gong, W.; Dunlop, N.M.; Howard, O.M.Z.; Ji, M.W. Transactivation of the epidermal growth factor receptor by formylpeptide receptor exacerbates the malignant behavior of human glioblastoma cells. Cancer Res. 2007, 67, 5906–5913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sciaccaluga, M.; D’Alessandro, G.; Pagani, F.; Ferrara, G.; Lopez, N.; Warr, T.; Gorello, P.; Porzia, A.; Mainiero, F.; Santoro, A.; et al. Functional cross talk between CXCR4 and PDGFR on glioblastoma cells is essential for migration. PLoS ONE 2013, 8, e73426. [Google Scholar] [CrossRef]
- Delcourt, N.; Bockaert, J.; Marin, P. GPCR-jacking: From a new route in RTK signalling to a new concept in GPCR activation. Trends Pharmacol. Sci. 2007, 28, 602–607. [Google Scholar] [CrossRef] [PubMed]
- Clapham, D.E. Calcium Signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [Green Version]
- Leclerc, C.; Haeich, J.; Aulestia, F.J.; Kilhoffer, M.C.; Miller, A.L.; Néant, I.; Webb, S.E.; Schaeffer, E.; Junier, M.P.; Chneiweiss, H.; et al. Calcium signaling orchestrates glioblastoma development: Facts and conjunctures. Biochim. Biophys. Acta Mol. Cell Res. 2016, 1863, 1447–1459. [Google Scholar] [CrossRef] [PubMed]
- Maklad, A.; Sharma, A.; Azimi, I. Calcium signaling in brain cancers: Roles and therapeutic targeting. Cancers 2019, 11, 145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, S.S.; Han, K.S.; Ku, B.M.; Lee, Y.K.; Hong, J.; Shin, H.Y.; Almonte, A.G.; Woo, D.H.; Brat, D.J.; Hwang, E.M.; et al. Caffeine-mediated inhibition of calcium release channel inositol 1,4,5-trisphosphate receptor subtype 3 blocks glioblastoma invasion and extends survival. Cancer Res. 2010, 70, 1173–1183. [Google Scholar] [CrossRef] [Green Version]
- Kang, S.; Hong, J.; Lee, J.M.; Moon, H.E.; Jeon, B.; Choi, J.; Yoon, N.A.; Paek, S.H.; Roh, E.J.; Lee, C.J.; et al. Trifluoperazine, a well-known antipsychotic, inhibits glioblastoma invasion by binding to calmodulin and disinhibiting calcium release channel IP3R. Mol. Cancer Ther. 2017, 16, 217–227. [Google Scholar] [CrossRef] [Green Version]
- Mahalingam, D.; Wilding, G.; Denmeade, S.; Sarantopoulas, J.; Cosgrove, D.; Cetnar, J.; Azad, N.; Bruce, J.; Kurman, M.; Allgood, V.E.; et al. Mipsagargin, a novel thapsigargin-based PSMA-Activated prodrug: Results of a first-in-man phase i clinical trial in patients with refractory, advanced or metastatic solid tumours. Br. J. Cancer 2016, 114, 986–994. [Google Scholar] [CrossRef]
- Mahalingam, D.; Peguero, J.; Cen, P.; Arora, S.P.; Sarantopoulos, J.; Rowe, J.; Allgood, V.; Tubb, B.; Campos, L. A phase II, multicenter, single-arm study of mipsagargin (G-202) as a second-line therapy following sorafenib for adult patients with progressive advanced hepatocellular carcinoma. Cancers 2019, 11, 833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prakriya, M.; Lewis, R.S. Store-operated calcium channels. Physiol. Rev. 2015, 95, 1383–1436. [Google Scholar] [CrossRef] [Green Version]
- Xie, J.; Pan, H.; Yao, J.; Zhou, Y.; Han, W. SOCE and cancer: Recent progress and new perspectives. Int. J. Cancer 2016, 138, 2067–2077. [Google Scholar] [CrossRef]
- Chen, Y.F.; Lin, P.C.; Yeh, Y.M.; Chen, L.H.; Shen, M.R. Store-Operated Ca2+ entry in tumor progression: From molecular mechanisms to clinical implications. Cancers 2019, 11, 899. [Google Scholar] [CrossRef] [Green Version]
- Motiani, R.K.; Hyzinski-García, M.C.; Zhang, X.; Henkel, M.M.; Abdullaev, I.F.; Kuo, Y.H.; Matrougui, K.; Mongin, A.A.; Trebak, M. STIM1 and Orai1 mediate CRAC channel activity and are essential for human glioblastoma invasion. Pflug. Arch. Eur. J. Physiol. 2013, 465, 1249–1260. [Google Scholar] [CrossRef] [Green Version]
- Zhu, M.; Chen, L.; Zhao, P.; Zhou, H.; Zhang, C.; Yu, S.; Lin, Y.; Yang, X. Store-operated Ca2+ entry regulates glioma cell migration and invasion via modulation of Pyk2 phosphorylation. J. Exp. Clin. Cancer Res. 2014, 33, 98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, F.; Yi, L.; Hai, L.; Wang, Y.; Yang, Y.; Li, T.; Tong, L.; Ma, H.; Liu, P.; Ming, H.; et al. Identification of Key Pathways and Genes in the Orai2 Mediated Classical and Mesenchymal Subtype of Glioblastoma by Bioinformatic Analyses. Dis. Markers 2019, 2019, 7049294. [Google Scholar] [CrossRef] [PubMed]
- Shen, T.; Guo, Q. Role of Pyk2 in human cancers. Med. Sci. Monit. 2018, 24, 8172–8182. [Google Scholar] [CrossRef]
- Emeriau, N.; de Clippele, M.; Gailly, P.; Tajeddine, N. Store operated calcium entry is altered by the inhibition of receptors tyrosine kinase. Oncotarget 2018, 9, 16059–16073. [Google Scholar] [CrossRef] [Green Version]
- Van Tellingen, O.; Yetkin-Arik, B.; De Gooijer, M.C.; Wesseling, P.; Wurdinger, T.; De Vries, H.E. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist. Updates 2015, 19, 1–12. [Google Scholar] [CrossRef]
- Talasila, K.M.; Soentgerath, A.; Euskirchen, P.; Rosland, G.V.; Wang, J.; Huszthy, P.C.; Prestegarden, L.; Skaftnesmo, K.O.; Sakariassen, P.Ø.; Eskilsson, E.; et al. EGFR wild-type amplification and activation promote invasion and development of glioblastoma independent of angiogenesis. Acta Neuropathol. 2013, 125, 683–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orellana, L. Convergence of EGFR glioblastoma mutations: Evolution and allostery rationalizing targeted therapy. Mol. Cell. Oncol. 2019, 6, 1–3. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Liang, R.; Song, C.; Xiang, Y.; Liu, Y. Prognostic significance of epidermal growth factor receptor expression in glioma patients. Onco. Targets Ther. 2018, 11, 731–742. [Google Scholar] [CrossRef] [Green Version]
- Krawczyk, P.; Kowalski, D.M.; Ramlau, R.; Kalinka-Warzocha, E.; Winiarczyk, K.; Stencel, K.; Powrózek, T.; Reszka, K.; Wojas-Krawczyk, K.; Bryl, M.; et al. Comparison of the effectiveness of erlotinib, gefitinib, and afatinib for treatment of non-small cell lung cancer in patients with common and rare EGFR gene mutations. Oncol. Lett. 2017, 13, 4433–4444. [Google Scholar] [CrossRef] [PubMed]
- Prados, M.D.; Lamborn, K.R.; Chang, S.; Burton, E.; Butowski, N.; Malec, M.; Kapadia, A.; Rabbit, J.; Page, M.S.; Fedoroff, A.; et al. Phase 1 study of erlotinib HCl alone and combined with temozolomide in patients with stable or recurrent malignant glioma. Neuro. Oncol. 2006, 8, 67–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halatsch, M.E.; Gehrke, E.E.; Vougioukas, V.I.; Bötefür, I.C.; Borhani, F.A.; Efferth, T.; Gebhart, E.; Domhof, S.; Schmidt, U.; Buchfelder, M. Inverse correlation of epidermal growth factor receptor messenger RNA induction and suppression of anchorage-independent growth by OSI-774, an epidermal growth factor receptor tyrosine kinase inhibitor, in glioblastoma multiforme cell lines. J. Neurosurg. 2004, 100, 523–533. [Google Scholar] [CrossRef] [PubMed]
- Griffero, F.; Daga, A.; Marubbi, D.; Capra, M.C.; Melotti, A.; Pattarozzi, A.; Gatti, M.; Bajetto, A.; Porcile, C.; Barbieri, F.; et al. Different response of human glioma tumor-initiating cells to epidermal growth factor receptor kinase inhibitors. J. Biol. Chem. 2009, 284, 7138–7148. [Google Scholar] [CrossRef] [Green Version]
- Van Den Bent, M.J.; Brandes, A.A.; Rampling, R.; Kouwenhoven, M.C.M.; Kros, J.M.; Carpentier, A.F.; Clement, P.M.; Frenay, M.; Campone, M.; Baurain, J.F.; et al. Randomized phase II trial of erlotinib versus temozolomide or carmustine in recurrent glioblastoma: EORTC brain tumor group study 26034. J. Clin. Oncol. 2009, 27, 1268–1274. [Google Scholar] [CrossRef] [Green Version]
- Peereboom, D.M.; Shepard, D.R.; Ahluwalia, M.S.; Brewer, C.J.; Agarwal, N.; Stevens, G.H.J.; Suh, J.H.; Toms, S.A.; Vogelbaum, M.A.; Weil, R.J.; et al. Phase II trial of erlotinib with temozolomide and radiation in patients with newly diagnosed glioblastoma multiforme. J. Neurooncol. 2010, 98, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Arif, S.; Pandith, A.; Tabasum, R.; Ramzan, A.; Singh, S.; Siddiqi, M.; Bhat, A. Significant effect of anti-tyrosine kinase inhibitor (Gefitinib) on overall survival of the glioblastoma multiforme patients in the backdrop of mutational status of epidermal growth factor receptor and PTEN Genes. Asian J. Neurosurg. 2018, 13, 46. [Google Scholar] [CrossRef]
- Bethune, G.; Bethune, D.; Ridgway, N.; Xu, Z. Epidermal growth factor receptor (EGFR) in lung cancer: An overview and update. J. Thorac. Dis. 2010, 2, 48. [Google Scholar] [PubMed]
- Binder, Z.A.; Thorne, A.H.; Bakas, S.; Wileyto, E.P.; Bilello, M.; Akbari, H.; Rathore, S.; Ha, S.M.; Zhang, L.; Ferguson, C.J.; et al. Epidermal Growth Factor Receptor Extracellular Domain Mutations in Glioblastoma Present Opportunities for Clinical Imaging and Therapeutic Development. Cancer Cell 2018, 34, 163–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vivanco, I.; Ian Robins, H.; Rohle, D.; Campos, C.; Grommes, C.; Nghiemphu, P.L.; Kubek, S.; Oldrini, B.; Chheda, M.G.; Yannuzzi, N.; et al. Differential sensitivity of glioma- versus lung cancer-specific EGFR mutations to EGFR kinase inhibitors. Cancer Discov. 2012, 2, 458–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reardon, D.A.; Nabors, L.B.; Mason, W.P.; Perry, J.R.; Shapiro, W.; Kavan, P.; Mathieu, D.; Phuphanich, S.; Cseh, A.; Fu, Y.; et al. Phase I/randomized phase II study of afatinib, an irreversible ErbB family blocker, with or without protracted temozolomide in adults with recurrent glioblastoma. Neuro. Oncol. 2015, 17, 430–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vengoji, R.; Macha, M.A.; Nimmakayala, R.K.; Rachagani, S.; Siddiqui, J.A.; Mallya, K.; Gorantla, S.; Jain, M.; Ponnusamy, M.P.; Batra, S.K.; et al. Afatinib and Temozolomide combination inhibits tumorigenesis by targeting EGFRvIII-cMet signaling in glioblastoma cells. J. Exp. Clin. Cancer Res. 2019, 38, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chen, X.; Shi, L.; Shan, Q.; Cao, Q.; Yue, C.; Li, H.; Li, S.; Wang, J.; Gao, S.; et al. The third-generation EGFR inhibitor AZD9291 overcomes primary resistance by continuously blocking ERK signaling in glioblastoma. J. Exp. Clin. Cancer Res. 2019, 38, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Elsamadicy, A.A.; Chongsathidkiet, P.; Desai, R.; Woroniecka, K.; Farber, S.H.; Fecci, P.E.; Sampson, J.H. Prospect of rindopepimut in the treatment of glioblastoma. Expert Opin. Biol. Ther. 2017, 17, 507–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carboni, J.M.; Wittman, M.; Yang, Z.; Lee, F.; Greer, A.; Hurlburt, W.; Hillerman, S.; Cao, C.; Cantor, G.H.; Dell-John, J.; et al. BMS-754807, a small molecule inhibitor of insulin-like growth factor-1R/IR. Mol. Cancer Ther. 2009, 8, 3341–3349. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, G.K.; Dickson, M.A.; Lorusso, P.M.; Sausville, E.A.; Maekawa, Y.; Watanabe, Y.; Kashima, N.; Nakashima, D.; Akinaga, S. Preclinical and first-in-human phase I studies of KW-2450, An oral tyrosine kinase inhibitor with insulin-like growth factor receptor-1/insulin receptor selectivity. Cancer Sci. 2016, 107, 499–506. [Google Scholar] [CrossRef] [Green Version]
- Simpson, A.; Petnga, W.; Macaulay, V.M.; Weyer-Czernilofsky, U.; Bogenrieder, T. Insulin-Like Growth Factor (IGF) Pathway Targeting in Cancer: Role of the IGF Axis and Opportunities for Future Combination Studies. Target. Oncol. 2017, 12, 571–597. [Google Scholar] [CrossRef] [Green Version]
- Yin, S.; Girnita, A.; Strömberg, T.; Khan, Z.; Andersson, S.; Zheng, H.; Ericsson, C.; Axelson, M.; Nistér, M.; Larsson, O.; et al. Targeting the insulin-like growth factor-1 receptor by picropodophyllin as a treatment option for glioblastoma. Neuro. Oncol. 2010, 12, 19–27. [Google Scholar] [CrossRef]
- Iqbal, N.; Iqbal, N. Imatinib: A Breakthrough of Targeted Therapy in Cancer. Chemother. Res. Pract. 2014, 2014, 1–9. [Google Scholar] [CrossRef]
- Raymond, E.; Brandes, A.A.; Dittrich, C.; Fumoleau, P.; Coudert, B.; Clement, P.M.J.; Frenay, M.; Rampling, R.; Stupp, R.; Kros, J.M.; et al. Phase II study of imatinib in patients with recurrent gliomas of various histologies: A European organisation for research and treatment of cancer brain tumor group study. J. Clin. Oncol. 2008, 26, 4659–4665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, P.Y.; Yung, W.K.A.; Lamborn, K.R.; Dahia, P.L.; Wang, Y.; Peng, B.; Abrey, L.E.; Raizer, J.; Cloughesy, T.F.; Fink, K.; et al. Phase I/II study of imatinib mesylate for recurrent malignant gliomas: North American Brain Tumor Consortium Study 99-08. Clin. Cancer Res. 2006, 12, 4899–4907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frolov, A.; Evans, I.M.; Li, N.; Sidlauskas, K.; Paliashvili, K.; Lockwood, N.; Barrett, A.; Brandner, S.; Zachary, I.C.; Frankel, P. Imatinib and Nilotinib increase glioblastoma cell invasion via Abl-independent stimulation of p130Cas and FAK signalling. Sci. Rep. 2016, 6, 27378. [Google Scholar] [CrossRef] [PubMed]
- Oertel, S.; Krempien, R.; Lindel, K.; Zabel, A.; Milker-Zabel, S.; Bischof, M.; Lipson, K.E.; Peschke, P.; Debus, J.; Abdollahi, A.; et al. Human glioblastoma and carcinoma xenograft tumors treated by combined radiation and imatinib (Gleevec®). Strahlenther. Onkol. 2006, 182, 400–407. [Google Scholar] [CrossRef]
- Odia, Y.; Sul, J.; Shih, J.H.; Kreisl, T.N.; Butman, J.A.; Iwamoto, F.M.; Fine, H.A. A Phase II trial of tandutinib (MLN 518) in combination with bevacizumab for patients with recurrent glioblastoma. CNS Oncol. 2016, 5, 59–67. [Google Scholar] [CrossRef] [Green Version]
- Griswold, I.J.; Shen, L.J.; La Rosée, P.; Demehri, S.; Heinrich, M.C.; Braziel, R.M.; McGreevey, L.; Haley, A.D.; Giese, N.; Druker, B.J.; et al. Effects of MLN518, a dual FLT3 and KTT inhibitor, on normal and malignant hematopoiesis. Blood 2004, 104, 2912–2918. [Google Scholar] [CrossRef]
- Ferrara, N.; Hillan, K.J.; Gerber, H.P.; Novotny, W. Discovery and development of bevacizumab, an anti-VEGF antibody for treating cancer. Nat. Rev. Drug Discov. 2004, 3, 391–400. [Google Scholar] [CrossRef]
- Hu-Lowe, D.D.; Zou, H.Y.; Grazzini, M.L.; Hallin, M.E.; Wickman, G.R.; Amundson, K.; Chen, J.H.; Rewolinski, D.A.; Yamazaki, S.; Wu, E.Y.; et al. Nonclinical antiangiogenesis and antitumor activities of axitinib (AG-013736), an oral, potent, and selective inhibitor of vascular endothelial growth factor receptor tyrosine kinases 1, 2, 3. Clin. Cancer Res. 2008, 14, 7272–7283. [Google Scholar] [CrossRef] [Green Version]
- Duerinck, J.; Du Four, S.; Vandervorst, F.; D’Haene, N.; Le Mercier, M.; Michotte, A.; Van Binst, A.M.; Everaert, H.; Salmon, I.; Bouttens, F.; et al. Randomized phase II study of axitinib versus physicians best alternative choice of therapy in patients with recurrent glioblastoma. J. Neurooncol. 2016, 128, 147–155. [Google Scholar] [CrossRef]
- Siegelin, M.D.; Raskett, C.M.; Gilbert, C.A.; Ross, A.H.; Altieri, D.C. Sorafenib exerts anti-glioma activity in vitro and in vivo. Neurosci. Lett. 2010, 478, 165–170. [Google Scholar] [CrossRef] [Green Version]
- Mendel, D.B.; Douglas Laird, A.; Xin, X.; Louie, S.G.; Christensen, J.G.; Li, G.; Schreck, R.E.; Abrams, T.J.; Ngai, T.J.; Lee, L.B.; et al. In vivo antitumor activity of SU11248, a novel tyrosine kinase inhibitor targeting vascular endothelial growth factor and platelet-derived growth factor receptors: Determination of a pharmacokinetic/pharmacodynamic relationship. Clin. Cancer Res. 2003, 9, 327–337. [Google Scholar]
- Hottinger, A.F.; Aissa, A.B.; Espeli, V.; Squiban, D.; Dunkel, N.; Vargas, M.I.; Hundsberger, T.; MacH, N.; Schaller, K.; Weber, D.C.; et al. Phase i study of sorafenib combined with radiation therapy and temozolomide as first-line treatment of high-grade glioma. Br. J. Cancer 2014, 110, 2655–2661. [Google Scholar] [CrossRef]
- Neyns, B.; Sadones, J.; Chaskis, C.; Dujardin, M.; Everaert, H.; Lv, S.; Duerinck, J.; Tynninen, O.; Nupponen, N.; Michotte, A.; et al. Phase II study of sunitinib malate in patients with recurrent high-grade glioma. J. Neurooncol. 2011, 103, 491–501. [Google Scholar] [CrossRef] [PubMed]
- Katoh, M. FGFR inhibitors: Effects on cancer cells, tumor microenvironment and whole-body homeostasis (Review). Int. J. Mol. Med. 2016, 38, 3–15. [Google Scholar] [CrossRef] [Green Version]
- Lassman, A.; Sepúlveda-Sánchez, J.; Cloughesy, T.; Gil-Gil, J.; Puduvalli, V.; Raizer, J.; De Vos, F.; Wen, P.; Butowski, N.; Clement, P.; et al. Actr-33. Infigratinib (Bgj398) in Patients With Recurrent Gliomas with Fibroblast Growth Factor Receptor (Fgfr) Alterations: A Multicenter Phase Ii Study. Neuro. Oncol. 2019, 21, vi20. [Google Scholar] [CrossRef]
- Wadhwa, S.; Nag, T.C.; Jindal, A.; Kushwaha, R.; Mahapatra, A.K.; Sarkar, C. Expression of the neurotrophin receptors Trk A and Trk B in adult human astrocytoma and glioblastoma. J. Biosci. 2003, 28, 181–188. [Google Scholar] [CrossRef] [PubMed]
- Assimakopoulou, M.; Kondyli, M.; Gatzounis, G.; Maraziotis, T.; Varakis, J. Neurotrophin receptors expression and JNK pathway activation in human astrocytomas. BMC Cancer 2007, 7, 202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinet, S.; Bessette, B.; Vedrenne, N.; Lacroix, A.; Richard, L.; Jauberteau, M.O.; Battu, S.; Lalloué, F. TrkB-containing exosomes promote the transfer of glioblastoma aggressiveness to YKL-40-inactivated glioblastoma cells. Oncotarget 2016, 7, 50349–50364. [Google Scholar] [CrossRef] [Green Version]
- Cazorla, M.; Prémont, J.; Mann, A.; Girard, N.; Kellendonk, C.; Rognan, D. Identification of a low-molecular weight TrkB antagonist with anxiolytic and antidepressant activity in mice. J. Clin. Investig. 2011, 121, 1846–1857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, J.; Xie, S.; Ramkissoon, S.H.; Luu, V.; Sun, Y.; Bandopadhayay, P.; Beroukhim, R.; Roberts, T.M.; Stiles, C.D.; Segal, R.A.; et al. Tyrosine receptor kinase B is a drug target in astrocytomas. Neuro. Oncol. 2017, 19, 22–30. [Google Scholar] [CrossRef] [Green Version]
- Cocco, E.; Scaltriti, M.; Drilon, A. NTRK fusion-positive cancers and TRK inhibitor therapy. Nat. Rev. Clin. Oncol. 2018, 15, 731–747. [Google Scholar] [CrossRef]
- Xu, T.; Wang, H.; Huang, X.; Li, W.; Huang, Q.; Yan, Y.; Chen, J. Gene Fusion in Malignant Glioma: An Emerging Target for Next-Generation Personalized Treatment. Transl. Oncol. 2018, 11, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Ardini, E.; Menichincheri, M.; Banfi, P.; Bosotti, R.; De Ponti, C.; Pulci, R.; Ballinari, D.; Ciomei, M.; Texido, G.; Degrassi, A.; et al. Entrectinib, a Pan-TRK, ROS1, and ALK inhibitor with activity in multiple molecularly defined cancer indications. Mol. Cancer Ther. 2016, 15, 628–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doebele, R.C.; Davis, L.E.; Vaishnavi, A.; Le, A.T.; Estrada-Bernal, A.; Keysar, S.; Jimeno, A.; Varella-Garcia, M.; Aisner, D.L.; Li, Y.; et al. An oncogenic NTRK fusion in a patient with soft-tissue sarcoma with response to the tropomyosin-related kinase inhibitor LOXO-101. Cancer Discov. 2015, 5, 1049–1057. [Google Scholar] [CrossRef] [Green Version]
- Drilon, A.; Siena, S.; Ou, S.H.I.; Patel, M.; Ahn, M.J.; Lee, J.; Bauer, T.M.; Farago, A.F.; Wheler, J.J.; Liu, S.V.; et al. Safety and antitumor activity of the multitargeted pan-TRK, ROS1, and ALK inhibitor entrectinib: Combined results from two phase I trials (ALKA-372-001 and STARTRK-1). Cancer Discov. 2017, 7, 400–409. [Google Scholar] [CrossRef] [Green Version]
- Drilon, A.; Laetsch, T.W.; Kummar, S.; Dubois, S.G.; Lassen, U.N.; Demetri, G.D.; Nathenson, M.; Doebele, R.C.; Farago, A.F.; Pappo, A.S.; et al. Efficacy of larotrectinib in TRK fusion-positive cancers in adults and children. N. Engl. J. Med. 2018, 378, 731–739. [Google Scholar] [CrossRef] [PubMed]
- Laetsch, T.W.; DuBois, S.G.; Mascarenhas, L.; Turpin, B.; Federman, N.; Albert, C.M.; Nagasubramanian, R.; Davis, J.L.; Rudzinski, E.; Feraco, A.M.; et al. Larotrectinib for paediatric solid tumours harbouring NTRK gene fusions: Phase 1 results from a multicentre, open-label, phase 1/2 study. Lancet Oncol. 2018, 19, 705–714. [Google Scholar] [CrossRef]
- Drilon, A.; Nagasubramanian, R.; Blake, J.F.; Ku, N.; Tuch, B.B.; Ebata, K.; Smith, S.; Lauriault, V.; Kolakowski, G.R.; Brandhuber, B.J.; et al. A next-generation TRK kinase inhibitor overcomes acquired resistance to prior trk kinase inhibition in patients with TRK fusion-positive solid tumors. Cancer Discov. 2017, 7, 963–972. [Google Scholar] [CrossRef] [Green Version]
- Drilon, A.; Ou, S.H.I.; Cho, B.C.; Kim, D.W.; Lee, J.; Lin, J.J.; Zhu, V.W.; Ahn, M.J.; Camidge, D.R.; Nguyen, J.; et al. Repotrectinib (Tpx-0005) is a next-generation ros1/trk/alk inhibitor that potently inhibits ros1/trk/alk solvent-front mutations. Cancer Discov. 2018, 8, 1227–1236. [Google Scholar] [CrossRef] [Green Version]
- Park, J.S.; Kim, I.K.; Han, S.; Park, I.; Kim, C.; Bae, J.; Oh, S.J.; Lee, S.; Kim, J.H.; Woo, D.C.; et al. Normalization of Tumor Vessels by Tie2 Activation and Ang2 Inhibition Enhances Drug Delivery and Produces a Favorable Tumor Microenvironment. Cancer Cell 2016, 30, 953–967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reusch, P.; Barleon, B.; Weindel, K.; Martiny-Baron, G.; Gödde, A.; Siemeister, G.; Marmé, D. Identification of a soluble form of the angiopoietin receptor TIE-2 released from endothelial cells and present in human blood. Angiogenesis 2001, 4, 123–131. [Google Scholar] [CrossRef] [PubMed]
- Harney, A.S.; Karagiannis, G.S.; Pignatelli, J.; Smith, B.D.; Kadioglu, E.; Wise, S.C.; Hood, M.M.; Kaufman, M.D.; Leary, C.B.; Lu, W.P.; et al. The selective Tie2 inhibitor rebastinib blocks recruitment and function of Tie2Hi macrophages in breast cancer and pancreatic neuroendocrine tumors. Mol. Cancer Ther. 2017, 16, 2486–2501. [Google Scholar] [CrossRef] [Green Version]
- Cam, M.; Charan, M.; Welker, A.M.; Dravid, P.; Studebaker, A.W.; Leonard, J.R.; Pierson, C.R.; Nakano, I.; Beattie, C.E.; Hwang, E.I.; et al. ΔNp73/ETS2 complex drives glioblastoma pathogenesis- targeting downstream mediators by rebastinib prolongs survival in preclinical models of glioblastoma. Neuro. Oncol. 2020, 22, 345–356. [Google Scholar] [CrossRef] [PubMed]
- Schneider, H.; Szabo, E.; Machado, R.A.C.; Broggini-Tenzer, A.; Walter, A.; Lobell, M.; Heldmann, D.; Süssmeier, F.; Grünewald, S.; Weller, M. Novel TIE-2 inhibitor BAY-826 displays in vivo efficacy in experimental syngeneic murine glioma models. J. Neurochem. 2017, 140, 170–182. [Google Scholar] [CrossRef]
- Piao, Y.; Park, S.Y.; Henry, V.; Smith, B.D.; Tiao, N.; Flynn, D.L.; De Groot, J.F. Novel MET/TIE2/VEGFR2 inhibitor altiratinib inhibits tumor growth and invasiveness in bevacizumab-resistant glioblastoma mouse models. Neuro. Oncol. 2016, 18, 1230–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearson, J.R.D.; Regad, T. Targeting cellular pathways in glioblastoma multiforme. Signal Transduct. Target. Ther. 2017, 2, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Monje, M.; Borniger, J.C.; D’Silva, N.J.; Deneen, B.; Dirks, P.B.; Fattahi, F.; Frenette, P.S.; Garzia, L.; Gutmann, D.H.; Hanahan, D.; et al. Roadmap for the Emerging Field of Cancer Neuroscience. Cell 2020, 181, 219–222. [Google Scholar] [CrossRef]
- Venkatesh, H.S.; Johung, T.B.; Caretti, V.; Noll, A.; Tang, Y.; Nagaraja, S.; Gibson, E.M.; Mount, C.W.; Polepalli, J.; Mitra, S.S.; et al. Neuronal activity promotes glioma growth through neuroligin-3 secretion. Cell Rep. 2019, 26, 803–816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Venkatesh, H.S.; Morishita, W.; Geraghty, A.C.; Silverbush, D.; Gillespie, S.M.; Arzt, M.; Tam, L.T.; Espenel, C.; Ponnuswami, A.; Ni, L.; et al. Electrical and synaptic integration of glioma into neural circuits. Nature 2019, 573, 539–545. [Google Scholar] [CrossRef]
- Venkataramani, V.; Tanev, D.I.; Strahle, C.; Studier-Fischer, A.; Fankhauser, L.; Kessler, T.; Körber, C.; Kardorff, M.; Ratliff, M.; Xie, R.; et al. Glutamatergic synaptic input to glioma cells drives brain tumour progression. Nature 2019, 573, 532–538. [Google Scholar] [CrossRef]
- Zeng, Q.; Michael, I.P.; Zhang, P.; Saghafinia, S.; Knott, G.; Jiao, W.; McCabe, B.D.; Galván, J.A.; Robinson, H.P.C.; Zlobec, I.; et al. Synaptic proximity enables NMDAR signalling to promote brain metastasis. Nature 2019, 573, 526–531. [Google Scholar] [CrossRef]
- Tuveson, D.; Clevers, H. Cancer modeling meets human organoid technology. Science 2019, 364, 952–955. [Google Scholar] [CrossRef] [PubMed]
- Linkous, A.; Balamatsias, D.; Snuderl, M.; Edwards, L.; Miyaguchi, K.; Milner, T.; Reich, B.; Cohen-Gould, L.; Storaska, A.; Nakayama, Y.; et al. Modeling Patient-Derived Glioblastoma with Cerebral Organoids. Cell Rep. 2019, 26, 3203–3211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogawa, J.; Pao, G.M.; Shokhirev, M.N.; Verma, I.M. Glioblastoma Model Using Human Cerebral Organoids. Cell Rep. 2018, 23, 1220–1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tilak, M.; Holborn, J.; New, L.A.; Lalonde, J.; Jones, N. Receptor Tyrosine Kinase Signaling and Targeting in Glioblastoma Multiforme. Int. J. Mol. Sci. 2021, 22, 1831. https://doi.org/10.3390/ijms22041831
Tilak M, Holborn J, New LA, Lalonde J, Jones N. Receptor Tyrosine Kinase Signaling and Targeting in Glioblastoma Multiforme. International Journal of Molecular Sciences. 2021; 22(4):1831. https://doi.org/10.3390/ijms22041831
Chicago/Turabian StyleTilak, Manali, Jennifer Holborn, Laura A. New, Jasmin Lalonde, and Nina Jones. 2021. "Receptor Tyrosine Kinase Signaling and Targeting in Glioblastoma Multiforme" International Journal of Molecular Sciences 22, no. 4: 1831. https://doi.org/10.3390/ijms22041831