
Crosstalk between Macrophages and Pancreatic β-Cells in Islet Development, Homeostasis and Disease


Abstract
:1. Main
2. Macrophage Heterogeneity, Plasticity and Signalling
3. Involvement of Macrophages in β-Cell Differentiation, Proliferation and Homeostasis
4. Role of Macrophages in Type 1 Diabetes
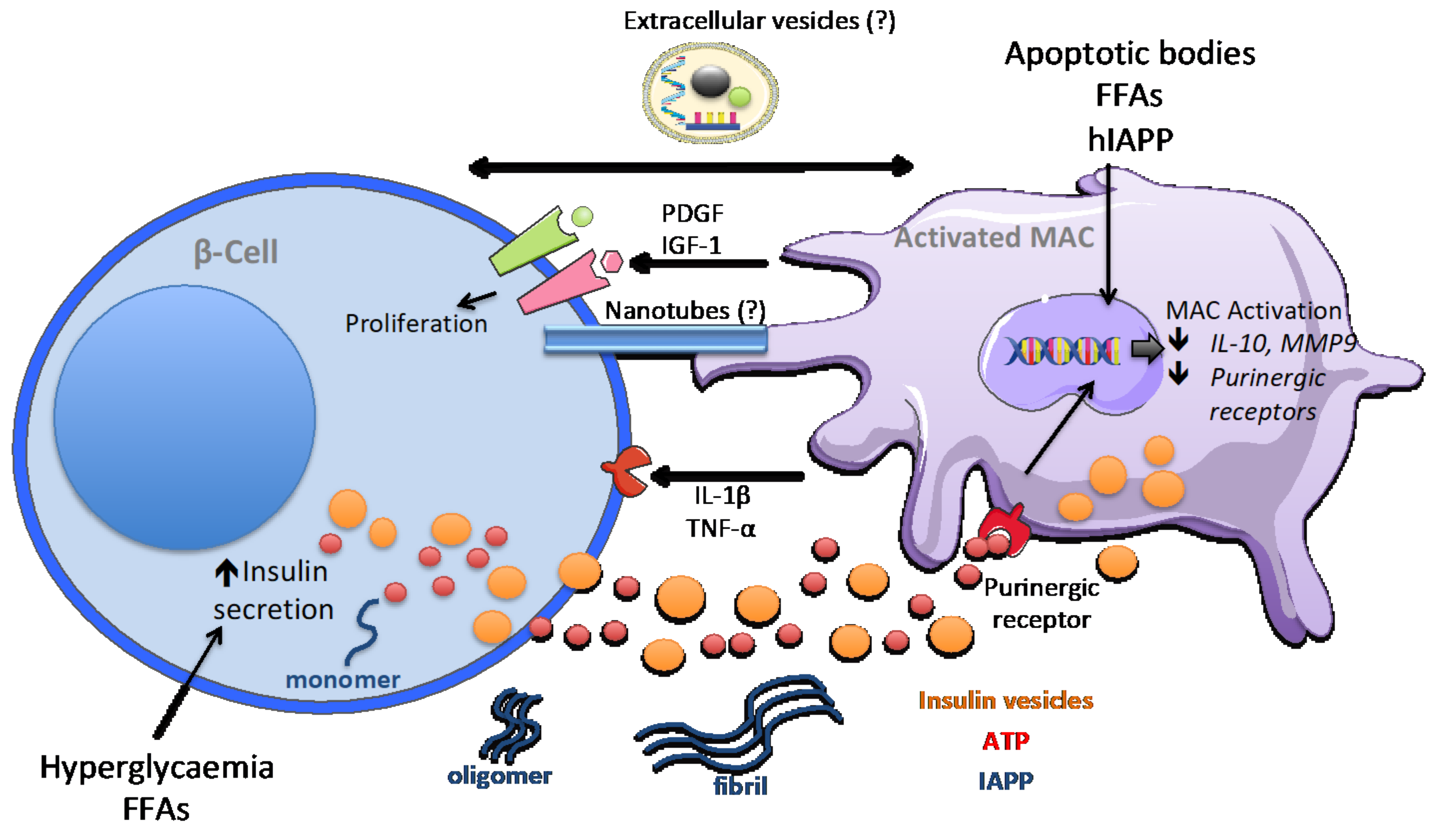
5. Macrophage Signalling in Obesity-Dependent Inflammation and Type 2 Diabetes
6. Discussion
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Eizirik, D.L.; Colli, M.L.; Ortis, F. The Role of Inflammation in Insulitis and β-Cell Loss in Type 1 Diabetes. Nat. Rev. Endocrinol. 2009, 5, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Donath, M.Y.; Dalmas, É.; Sauter, N.S.; Böni-Schnetzler, M. Inflammation in Obesity and Diabetes: Islet Dysfunction and Therapeutic Opportunity. Cell Metab. 2013, 17, 860–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, S.; Taylor, P.R. Monocyte and Macrophage Heterogeneity. Nat. Rev. Immunol. 2005, 5, 953–964. [Google Scholar] [CrossRef]
- Davies, L.C.; Jenkins, S.J.; Allen, J.E.; Taylor, P.R. Tissue-Resident Macrophages. Nat. Immunol. 2013, 14, 986–995. [Google Scholar] [CrossRef]
- Hoeffel, G.; Wang, Y.; Greter, M.; See, P.; Teo, P.; Malleret, B.; Leboeuf, M.; Low, D.; Oller, G.; Almeida, F.; et al. Adult Langerhans Cells Derive Predominantly from Embryonic Fetal Liver Monocytes with a Minor Contribution of Yolk Sac–Derived Macrophages. J. Exp. Med. 2012, 209, 1167–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ginhoux, F.; Greter, M.; Leboeuf, M.; Nandi, S.; See, P.; Gokhan, S.; Mehler, M.F.; Conway, S.J.; Ng, L.G.; Stanley, E.R.; et al. Fate Mapping Analysis Reveals That Adult Microglia Derive from Primitive Macrophages. Science 2010, 330, 841–845. [Google Scholar] [CrossRef] [Green Version]
- Schulz, C.; Perdiguero, E.G.; Chorro, L.; Szabo-Rogers, H.; Cagnard, N.; Kierdorf, K.; Prinz, M.; Wu, B.; Jacobsen, S.E.W.; Pollard, J.W.; et al. A Lineage of Myeloid Cells Independent of Myb and Hematopoietic Stem Cells. Science 2012, 336, 86–90. [Google Scholar] [CrossRef] [Green Version]
- Mosser, D.M.; Edwards, J.P. Exploring the Full Spectrum of Macrophage Activation. Nat. Rev. Immunol. 2008, 8, 958–969. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.D.; Qiu, Y.; Cui, X.; Goh, Y.P.S.; Mwangi, J.; David, T.; Mukundan, L.; Brombacher, F.; Locksley, R.M.; Chawla, A. Alternatively Activated Macrophages Produce Catecholamines to Sustain Adaptive Thermogenesis. Nat. Cell Biol. 2011, 480, 104–108. [Google Scholar] [CrossRef] [Green Version]
- Odegaard, J.I.; Gonzalez, R.R.R.; Goforth, M.H.; Morel, C.R.; Subramanian, V.; Makundan, L.; Eagle, A.R.; Vats, D.; Brombacher, F.; Ferrante, A.W.; et al. Macrophage-Specific PPARgamma Controls Alternative Activation and Improves Insulin Resistance. Nature 2007, 447, 1116–1120. [Google Scholar] [CrossRef] [Green Version]
- Gordon, S.; Martinez, F.O. Alternative Activation of Macrophages: Mechanism and Functions. Immunology 2010, 32, 593–604. [Google Scholar] [CrossRef] [Green Version]
- O’Shea, J.J.; Murray, P.J. Cytokine Signaling Modules in Inflammatory Responses. Immunology 2008, 28, 477–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenkins, S.J.; Ruckerl, D.; Cook, P.C.; Jones, L.H.; Finkelman, F.D.; Rooijen, v.N.; MacDonald, A.S.; Allen, J.E. Local Macrophage Proliferation, Rather than Recruitment from the Blood, Is a Signature of Inflammation. Science 2011, 332, 1284–1288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amano, S.U.; Cohen, J.L.; Vangala, P.; Tencerova, M.; Nicoloro, S.M.; Yawe, J.C.; Shen, Y.; Czech, M.P.; Aouadi, M. Local Proliferation of Macrophages Contributes to Obesity-Associated Adipose Tissue Inflammation. Cell Metab. 2014, 19, 162–171. [Google Scholar] [CrossRef] [Green Version]
- Robbins, C.S.; Hilgendorf, I.; Weber, G.F.; Theurl, I.; Iwamoto, Y.; Figueiredo, J.-L.; Gorbatov, R.; Sukhova, G.K.; Gerhardt, L.M.; Smyth, D.; et al. Local Proliferation Dominates Lesional Macrophage Accumulation in Atherosclerosis. Nat. Med. 2013, 19, 1166–1172. [Google Scholar] [CrossRef] [PubMed]
- Arango Duque, G.; Descoteaux, A. Macrophage Cytokines: Involvement in Immunity and Infectious Diseases. Front. Immunol. 2014, 5, 491. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Zhao, M.; Liu, S.; Guo, J.; Lu, Y.; Cheng, J.; Liu, J. Macrophage-Derived Extracellular Vesicles: Diverse Mediators of Pathology and Therapeutics in Multiple Diseases. Cell Death Dis. 2020, 11, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Wiktor-Jedrzejczak, W.; Bartocci, A.; Ferrante, A.W.; Ahmed-Ansari, A.; Sell, K.W.; Pollard, J.W.; Stanley, E.R. Total absence of colony-stimulating factor 1 in the macrophage-deficient osteopetrotic (op/op) mouse. Proc. Natl. Acad. Sci. USA 1990, 87, 4828–4832. [Google Scholar] [CrossRef] [Green Version]
- Bouchareb, B.L.; Evans, V.G.; Boustani, D.S.; Castelloti, M.C.; Czerinichow, P.; Pollard, J.W.; Polak, M. Insulin Cell Mass is Altered in Csf1op/Csf1op Macrophage-Deficient Mice. J. Leukoc. Biol. 2004, 76, 359–367. [Google Scholar] [CrossRef]
- Mussar, K.; Tucker, A.; McLennan, L.; Gearhart, A.; Jimenez-Caliani, A.J.; Cirulli, V.; Crisa, L. Macrophage/Epithelium Cross-Talk Regulates Cell Cycle Progression and Migration in Pancreatic Progenitors. PLoS ONE 2014, 9, e89492. [Google Scholar] [CrossRef] [Green Version]
- Geutskens, S.B.; Otonkoski, T.; Pulkkinen, M.-A.; Drexhage, H.A.; Leenen, P.J.M. Macrophages in the Murine Pancreas and Their Involvement in Fetal Endocrine Development in Vitro. J. Leukoc. Biol. 2005, 78, 845–852. [Google Scholar] [CrossRef] [PubMed]
- Calderon, B.; Carrero, J.A.; Ferris, S.T.; Sojka, D.K.; Moore, L.; Epelman, S.; Murphy, K.M.; Yokoyama, W.M.; Randolph, G.J.; Unanue, E.R. The Pancreas Anatomy Conditions the Origin and Properties of Resident Macrophages. J. Exp. Med. 2015, 212, 1497–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riley, K.G.; Pasek, R.C.; Maulis, M.F.; Peek, J.; Thorel, F.; Brigstock, D.R.; Herrera, P.L.; Gannon, M. Connective Tissue Growth Factor Modulates Adult β-Cell Maturity and Proliferation to Promote β-Cell Regeneration in Mice. Diabetes 2014, 64, 1284–1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riley, K.G.; Pasek, R.C.; Maulis, M.F.; Dunn, J.C.; Bolus, W.R.; Kendall, P.L.; Hasty, A.H.; Gannon, M. Macrophages Are Essential for CTGF-Mediated Adult β-Cell Proliferation after Injury. Mol. Metab. 2015, 4, 584–591. [Google Scholar] [CrossRef] [PubMed]
- Brissova, M.; Aamodt, K.; Brahmachary, P.; Prasad, N.; Hong, J.-Y.; Dai, C.; Mellati, M.; Shostak, A.; Poffenberger, G.; Aramandla, R.; et al. Islet Microenvironment, Modulated by Vascular Endothelial Growth Factor-A Signaling, Promotes β Cell Regeneration. Cell Metab. 2014, 19, 498–511. [Google Scholar] [CrossRef] [Green Version]
- Criscimanna, A.; Coudriet, G.M.; Gittes, G.K.; Coudriet, G.M.; Esni, F. Activated Macrophages Create Lineage-Specific Microenvironments for Pancreatic Acinar-and β-Cell Regeneration in Mice. Gastroenterology 2014, 147, 1106–1118.e11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nackiewicz, D.; Dan, M.; Speck, M.; Chow, S.Z.; Chen, Y.-C.; Pospisilik, J.A.; Verchere, C.B.; Ehses, J.A. Islet Macrophages Shift to a Reparative State following Pancreatic Beta-Cell Death and Are a Major Source of Islet Insulin-like Growth Factor-1. iScience 2020, 23, 100775. [Google Scholar] [CrossRef] [Green Version]
- Xiao, X.; Gaffar, I.; Guo, P.; Wiersch, J.; Fischbach, S.; Peirish, L.; Song, Z.; El-Gohary, Y.; Prasadan, K.; Shiota, C.; et al. M2 Macrophages Promote Beta-Cell Proliferation by up-Regulation of SMAD7. Proc. Natl. Acad. Sci. USA 2014, 111, E1211–E1220. [Google Scholar] [CrossRef] [Green Version]
- Weitz, J.R.; Makhmutova, M.; Almaça, J.; Stertmann, J.; Aamodt, K.; Brissova, M.; Speier, S.; Rodriguez-Diaz, R.; Caicedo, A. Mouse Pancreatic Islet Macrophages Use Locally Released ATP to Monitor Beta Cell Activity. Diabetology 2018, 61, 182–192. [Google Scholar] [CrossRef]
- Obermüller, S.; Lindqvist, A.; Karanauskaite, J.; Galvanovskis, J.; Rorsman, P.; Barg, S. Selective NucleotiDe-release from Dense-Core Granules in Insulin-Secreting Cells. J. Cell Sci. 2005, 118, 4271–4282. [Google Scholar] [CrossRef] [Green Version]
- Hazama, S.H.; Okada, Y. Cell Surface Measurements of ATP Release from Single Pancreatic Beta Cells Using a Novel Biosensor Technique. Pflugers Arch. 1998, 437, 31–35. [Google Scholar] [CrossRef]
- Zinselmeyer, B.H.; Vomund, A.N.; Saunders, B.T.; Johnson, M.W.; Carrero, J.A.; Unanue, E.R. The Resident Macrophages in Murine Pancreatic Islets Are Constantly Probing Their Local Environment, Capturing Beta Cell Granules and Blood Particles. Diabetology 2018, 61, 1374–1383. [Google Scholar] [CrossRef] [Green Version]
- Weitz, J.R.; Silva, C.J.; Qadir, M.M.F.; Umland, O.; Pereira, E.; Qureshi, F.; Tamayo, A.; Bendala, J.D.; Diaz, R.R.; Almaca, J.; et al. Secretory Functions of Macrophages in the Human Pancreatic Islet Are Regulated by Endogenous Purinergic Signaling. Diabetes 2020, 69, 1206–1218. [Google Scholar] [CrossRef]
- Diana, J.; Gahzarian, L.; Simoni, Y.; Lehuen, A. Innate Immunity in Type 1 Diabetes. Discov. Med. 2011, 11, 513–520. [Google Scholar] [PubMed]
- Oschilewski, U.; Kiesel, U.; Kolb, H. Administration of Silica Prevents Diabetes in BB-Rats. Diabetes 1985, 34, 197–199. [Google Scholar] [CrossRef] [Green Version]
- Charlton, B.; Bacelj, A.; Mandel, T.E. Administration of Silica Particles or Anti-Lyt2 Antibody Prevents Beta-Cell Destruction in NOD Mice Given Cyclophosphamide. Diabetes 1988, 37, 930–935. [Google Scholar] [CrossRef]
- Hutchings, P.; Rosen, H.; O’Reilly, L.; Simpson, E.; Gordon, S.; Cooke, A. Transfer of Diabetes in Mice Prevented by Blockade of Adhesion-Promoting Receptor on Macrophages. Nat. Cell Biol. 1990, 348, 639–642. [Google Scholar] [CrossRef]
- Chen, M.C.; Proost, P.; Gysemans, C.; Mathieu, C.; Eizirik, D.L. Monocyte Chemoattractant Protein-1 Is Expressed in Pancreatic Islets from Prediabetic NOD Mice and in Interleukin-1 Be-Ta-Exposed Human and Rat Islet Cells. Diabetologia 2001, 44, 325–332. [Google Scholar] [CrossRef] [Green Version]
- Martin, A.P.; Grisotto, M.G.; Canasto-Chibuque, C.; Kunkel, S.L.; Bromberg, J.S.; Furtado, G.C.; Lira, S.A. Islet Expression of M3 Uncovers a Key Role for Chemokines in the Development and Recruitment of Diabetogenic Cells in NOD Mice. Diabetes 2007, 57, 387–394. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Jia, S.; Geoffrey, R.; Alemzadeh, R.; Ghosh, S.; Hessner, M.J. Identification of a Molecular Signature in Human Type 1 Diabetes Mellitus Using Serum and Functional Genomics. J. Immunol. 2008, 180, 1929–1937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Willcox, A.; Richardson, S.J.; Bone, A.; Foulis, A.K.; Morgan, N.G. Analysis of Islet Inflammation in Human Type 1 Diabetes. Clin. Exp. Immunol. 2009, 155, 173–181. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.J.; Traum, D.; Schug, J.; Gao, L.; Liu, C.; Atkinson, M.A.; Powers, A.C.; Feldman, M.D.; Naji, A.; Chang, K.-M.; et al. Multiplexed in Situ Imaging Mass Cytometry Analysis of the Human Endocrine Pancreas and Immune System in Type 1 Diabetes. Cell Metab. 2019, 29, 769–783.e4. [Google Scholar] [CrossRef] [Green Version]
- Ortis, F.; Miani, M.; Colli, M.L.; Cunha, D.A.; Gurzov, E.N.; Allagnat, F.; Chariot, A.; Eizirik, D.L. Differential Usage of NF-κB Activating Signals by IL-1β and TNF-α in Pancreatic Beta Cells. FEBS Lett. 2012, 586, 984–989. [Google Scholar] [CrossRef] [Green Version]
- Ortis, F.; Pirot, P.; Naamane, N.; Kreins, A.Y.; Rasschaert, J.; Moore, F.; Theatre, E.; Verhaeghe, C.; Magnusson, N.E.; Chariot, A.; et al. Induction of Nuclear Factor-κB and Its Downstream Genes by TNF-α and IL-1β Has a Pro-Apoptotic Role in Pancreatic Beta Cells. Diabetologia 2008, 51, 1213. [Google Scholar] [CrossRef] [Green Version]
- Unanue, E.R.; Ferris, S.T.; Carrero, J.A. The Role of Islet Antigen Presenting Cells and the Presentation of Insulin in the Initiation of Autoimmune Diabetes in the NOD Mouse. Immunol. Rev. 2016, 272, 183–201. [Google Scholar] [CrossRef]
- Vomund, A.N.; Zinselmeyer, B.H.; Hughes, J.; Calderon, B.; Valderrama, C.; Ferris, S.T.; Wan, X.; Kanekura, K.; Carrero, J.A.; Urano, F.; et al. Beta Cells Transfer Vesicles Containing Insulin to Phagocytes for Presentation to T Cells. Proc. Natl. Acad. Sci. USA 2015, 112, E5496–E5502. [Google Scholar] [CrossRef] [Green Version]
- Calderon, B.; Suri, A.; Unanue, E.R. In CD4+ T-Cell-Induced Diabetes, Macrophages Are the Final Effector Cells that Mediate Islet β-Cell Killing: Studies from an Acute Model, Am. J. Pathol. 2006, 169, 2137–2147. [Google Scholar]
- Carrero, J.A.; McCarthy, D.; Ferris, S.; Wan, X.; Hu, H.; Zinzelmeyer, B.; Vomund, A.; Unanue, E. Depletion of Islet Resident Macrophages Protects Mice from Type 1 Diabetes. J. Immunol. 2018, 200, 41.13. [Google Scholar]
- Zakharov, P.N.; Hu, H.; Wan, X.; Unanue, E.R. Single-Cell RNA Sequencing of Murine Islets Shows High Cellular Complexity at All Stages of Autoimmune Diabetes. J. Exp. Med. 2020, 217, 217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marroqui, L.; Santos, R.S.D. TYK2, a Candidate Gene for Type 1 Diabetes, Modulates Apoptosis and the Innate Immune Response in Human Pancreatic β-Cells. Diabetes 2015, 64, 3808 LP–3817. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarkar, S.A.; Lee, C.E.; Victorino, F.; Nguyen, T.T.; Walters, J.A.; Burrack, A.; Eberlein, J.; Hildemann, S.K.; Homann, D. Expression and Regulation of Chemokines in Murine and Human Type 1 Diabetes. Diabetes 2011, 61, 436–446. [Google Scholar] [CrossRef] [Green Version]
- Cardozo, A.K.; Proost, P.; Gysemans, C.; Chen, M.-C.; Mathieu, C.; Eizirik, D.L. IL-1beta and IFN-Gamma Induce the Expression of Diverse Chemokines and IL-15 in Human and Rat Pancreatic Islet Cells, and in Islets from Pre-Diabetic NOD Mice. Diabetologia 2003, 46, 255–266. [Google Scholar] [CrossRef]
- Colli, M.L.; Paula, F.M.; Marselli, L.; Marchetti, P.; Roivainen, M.; Eizirik, D.L.; De Beeck, A.O. Coxsackievirus B Tailors the Unfolded Protein Response to Favour Viral Amplification in Pancreatic β Cells. J. Innate Immun. 2019, 11, 375–390. [Google Scholar] [CrossRef]
- Marroqui, L.; Dos Santos, R.S.; De Beeck, A.O.; De Brachène, A.C.; Marselli, L.; Marchetti, P.; Eizirik, D.L. Interferon-α Mediates Human Beta Cell HLA Class I Overexpression, Endoplasmic Reticulum Stress and Apoptosis, Three Hallmarks of Early Human Type 1 Diabetes. Diabetology 2017, 60, 656–667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Demine, S.; Schiavo, A.A.; Marín-Cañas, S.; Marchetti, P.; Cnop, M.; Eizirik, D.L. Pro-inflammatory Cytokines Induce Cell Death, Inflammatory Responses, and Endoplasmic Reticulum Stress in Human iPSC-Derived Beta Cells. Stem Cell Res. Ther. 2020, 11, 7. [Google Scholar] [CrossRef] [Green Version]
- Wen, L.; Peng, J.; Li, Z.; Wong, F.S. The Effect of Innate Immunity on Autoimmune Diabetes and the Expression of Toll-like Receptors on Pancreatic Islets. J. Immunol. 2004, 172, 3173–3180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vives-Pi, M.; Somoza, N.; Fernández-Alvarez, J.; Vargas, F.; Caro, P.; Alba, A.; Gomis, R.; Labeta, M.O.; Pujol-Borrell, R. Evidence of Expression of Endotoxin Receptors CD14, Toll-Like Receptors TLR4 and TLR2 and Associated Molecule MD-2 and of Sensitivity to Endotoxin (LPS) in Islet Beta Cells. Clin. Exp. Immunol. 2003, 133, 208–218. [Google Scholar] [CrossRef] [PubMed]
- Schulthess, F.T.; Paroni, F.; Sauter, N.S.; Shu, L.; Ribaux, P.; Haataja, L.; Strieter, R.M.; Oberholzer, J.; King, C.C.; Maedler, K. CXCL10 Impairs β Cell Function and Viability in Diabetes through TLR4 Signaling. Cell Metab. 2009, 9, 125–139. [Google Scholar] [CrossRef] [Green Version]
- Bollyky, P.L.; Bice, J.B.; Sweet, I.R.; Falk, B.A.; Gebe, J.A.; Clark, A.E.; Gersuk, V.H.; Aderem, A.; Hawn, T.R.; Nepom, G.T. The Toll-Like Receptor Signaling Molecule Myd88 Contributes to Pancreatic Beta-Cell Homeostasis in Response to Injury. PLoS ONE 2009, 4, e5063. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasschaert, J.; Ladriere, L.; Urbain, M.; Dogusan, Z.; Katabua, B.; Sato, S.; Akira, S.; Gysemans, C.; Mathieu, C.; Eizirik, D.L. Toll-like Receptor 3 and STAT-1 Contribute to Double-Stranded RNA+ Interferon-Gamma-Induced Apoptosis in Primary Pancreatic Beta-Cells. J. Biol. Chem. 2005, 280, 33984–33991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nackiewicz, D.; Dan, M.; He, W.; Kim, R.; Salmi, A.; Rutti, S.; Roper, C.W.; Cunningham, A.; Speck, M.; Clein, C.S.; et al. TLR2/6 and TLR4-Activated Macrophages Contribute to Islet Inflammation and Impair Beta Cell Insulin Gene Expression via IL-1 and IL-6. Diabetologia 2014, 57, 1645–1654. [Google Scholar] [CrossRef] [PubMed]
- Hanley, A.J.; Festa, A.; D’Agostino, R.B.; Wagenknecht, L.E.; Savage, P.J.; Tracy, R.P.; Saad, M.F.; Haffner, S.M. Metabolic and Inflammation Variable Clusters and Prediction of Type 2 Diabetes: Factor Analysis Using Directly Measured Insulin Sensitivity. Diabetes 2004, 53, 1773–1781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodríguez-Hernández, H.; Simental-Mendía, L.E.; Rodríguez, R.G.; Romero, M.A.R. Obesity and Inflammation: Epidemiology, Risk Factors, and Markers of Inflammation. Int. J. Endocrinol. 2013, 2013, 678159. [Google Scholar] [CrossRef] [Green Version]
- Lumeng, C.N.; Bodzin, J.L.; Saltiel, A.R. Obesity Induces a Phenotypic Switch in Adipose Tissue Macrophage Polarization. J. Clin. Investig. 2007, 117, 175–184. [Google Scholar] [CrossRef] [Green Version]
- Reilly, S.M.; Saltiel, A.R. Adapting to Obesity with Adipose Tissue Inflammation. Nat. Rev. Endocrinol. 2017, 13, 633–643. [Google Scholar] [CrossRef] [PubMed]
- Zatterale, F.; Longo, M.; Naderi, J.; Raciti, G.A.; Desiderio, A.; Miele, C.; Beguinot, F. Chronic Adipose Tissue Inflammation Linking Obesity to Insulin Resistance and Type 2 Diabetes. Front. Physiol. 2020, 10, 1607. [Google Scholar] [CrossRef]
- Ying, W.; Riopel, M.; Bandyopadhyay, G.; Dong, Y.; Birmingham, A.; Seo, J.B.; Ofrecio, J.M.; Wollam, J.; Hernandez-Carretero, A.; Fu, W.; et al. Adipose Tissue Macrophage-Derived Exosomal miRNAs Can Modulate In Vivo and In Vitro Insulin Sensitivity. Cell 2017, 171, 372–384. [Google Scholar] [CrossRef]
- Richardson, S.J.; Willcox, A.; Bone, A.; Foulis, A.K.; Morgan, N.G. Islet-Associated Macrophages in Type 2 Diabetes. Diabetology 2009, 52, 1686–1688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ehses, J.A.; Perren, A.; Eppler, E.; Ribaux, P.; Pospisilik, J.A.; Maor-Cahn, R.; Gueripel, X.; Ellingsgaard, H.; Schneider, M.K.; Biollaz, G.; et al. Increased Number of Islet-Associated Macrophages in Type 2 Diabetes. Diabetes 2007, 56, 2356–2370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homo-Delarche, F.; Calderari, S.; Irminger, J.-C.; Gangnerau, M.-N.; Coulaud, J.; Rickenbach, K.; Dolz, M.; Halban, P.; Portha, B.; Serradas, P. Islet Inflammation and Fibrosis in a Spontaneous Model of Type 2 Diabetes, the GK Rat. Diabetes 2006, 55, 1625–1633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, W.; Lee, Y.S.; Dong, Y.; Seidman, J.S.; Yang, M.; Isaac, R.; Seo, J.B.; Yang, B.-H.; Wollam, J.; Riopel, M.; et al. Expansion of Islet-Resident Macrophages Leads to Inflammation Affecting β Cell Proliferation and Function in Obesity. Cell Metab. 2019, 29, 457–474.e5. [Google Scholar] [CrossRef] [Green Version]
- Larsen, C.M.; Faulenbach, M.; Vaag, A.; Vølund, A.; Ehses, J.A.; Seifert, B.; Mandrup-Poulsen, T.; Donath, M.Y. Interleukin-1–Receptor Antagonist in Type 2 Diabetes Mellitus. N. Engl. J. Med. 2007, 356, 1517–1526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnetzler, B.M.; Boller, S.; Debray, S.; Bouzakri, K.; Meier, D.T.; Prazak, R.; Kerr-Conte, J.; Pattou, F.; Ehses, J.A.; Schuit, F.C.; et al. Free Fatty Acids Induce a Proinflammatory Response in Islets via the Abundantly Expressed Interleukin-1 Receptor, I. Endocrinology 2009, 150, 5218–5229. [Google Scholar] [CrossRef] [PubMed]
- Esteve, M.I.; Marselli, L.; Da Cunha, D.A.; Ladrière, L.; Ortis, F.; Grieco, F.A.; Dotta, F.; Weir, G.C.; Marchetti, P.; Eizirik, D.L.; et al. Palmitate Induces a Pro-inflammatory Response in Human Pancreatic Islets That Mimics CCL2 Expression by Beta Cells in Type 2 Diabetes. Diabetol. 2010, 53, 1395–1405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jurgens, C.A.; Toukatly, M.N.; Fligner, C.L.; Udayasankar, J.; Subramanian, S.L.; Zraika, S.; Aston-Mourney, K.; Carr, D.B.; Westermark, P.; Westermark, G.T.; et al. β-Cell Loss and β-Cell Apoptosis in Human Type 2 Diabetes Are Related to Islet Amyloid Deposition. Am. J. Pathol. 2011, 178, 2632–2640. [Google Scholar] [CrossRef] [Green Version]
- Matveyenko, A.V.; Butler, P.C. Islet Amyloid Polypeptide (IAPP) Transgenic Rodents as Models for Type 2 Diabetes. ILAR J. 2006, 47, 225–233. [Google Scholar] [CrossRef] [Green Version]
- Lorenzo, A.; Razzaboni, B.; Weir, G.C.; Yankner, B.A. Pancreatic Islet Cell Toxicity of Amylin Associated with Type-2 Diabetes Mellitus. Nat. Cell Biol. 1994, 368, 756–760. [Google Scholar] [CrossRef]
- Westwell-Roper, C.Y.; Ehses, J.A.; Verchere, C.B. Resident Macrophages Mediate Islet Amyloid Polypeptide–Induced Islet IL-1β Production and β-Cell Dysfunction. Diabetes 2014, 63, 1698 LP–1711. [Google Scholar] [CrossRef] [Green Version]
- Ying, W.; Fu, W.; Lee, Y.S.; Olefsky, J.M. The Role of Macrophages in Obesity-Associated Islet Inflammation and β-Cell Abnormalities. Nat. Rev. Endocrinol. 2020, 16, 81–90. [Google Scholar] [CrossRef] [Green Version]
- Yin, N.; Xu, J.; Ginhoux, F.; Randolph, G.J.; Merad, M.; Ding, Y.; Bromberg, J.S. Functional Specialization of Islet Dendritic Cell Subsets. J. Immunol. 2012, 188, 4921–4930. [Google Scholar] [CrossRef] [PubMed]
- Hume, D.A.; Halpin, D.; Charlton, H.; Gordon, S. The Mononuclear Phagocyte System of the Mouse Defined by Immunohistochemical Localization of Antigen F4/80: Macrophages of Endocrine Organs. Proc. Natl. Acad. Sci. USA 1984, 81, 4174–4177. [Google Scholar] [CrossRef] [Green Version]
- Butcher, M.J.; Hallinger, D.; Garcia, E.; Machida, Y.; Chakrabarti, S.; Nadler, J.; Galkina, E.V.; Imai, Y. Association of Proinflammatory Cytokines and Islet Resident Leucocytes with Islet Dysfunction in Type 2 Diabetes. Diabetology 2014, 57, 491–501. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eguchi, K.; Manabe, I.; Oishi-Tanaka, Y.; Ohsugi, M.; Kono, N.; Ogata, F.; Yagi, N.; Ohto, U.; Kimoto, M.; Miyake, K.; et al. Saturated Fatty Acid and TLR Signaling Link β Cell Dysfunction and Islet Inflammation. Cell Metab. 2012, 15, 518–533. [Google Scholar] [CrossRef] [Green Version]
- Parsa, R.; Andresen, P.; Gillett, A.; Mia, S.; Zhang, X.-M.; Mayans, S.; Holmberg, D.; Harris, R.A. Adoptive Transfer of Immunomodulatory M2 Macrophages Prevents Type 1 Diabetes in NOD Mice. Diabetes 2012, 61, 2881–2892. [Google Scholar] [CrossRef] [Green Version]
- Padgett, L.E.; Burg, A.R.; Lei, W.; Tse, H.M. Loss of NADPH Oxidase–Derived Superoxide Skews Macrophage Phenotypes to Delay Type 1 Diabetes. Diabetes 2015, 64, 937–946. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-E.; Choi, S.-E.; Lee, S.-J.; Lee, J.-H.; Lee, Y.-J.; Kang, S.S.; Chun, J.; Kang, Y. Tumour Necrosis Factor-Induced Glucose-Stimulated Insulin Secretion Inhibition in INS-1 Cells Is Ascribed to a Reduction of the Glucose-Stimulated Ca2+ Influx. J. Endocrinol. 2008, 198, 549–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardozo, A.K.; Ortis, F.; Storling, J.; Feng, Y.-M.; Rasschaert, J.; Tonnesen, M.; Van Eylen, F.; Mandrup-Poulsen, T.; Herchuelz, A.; Eizirik, D.L. Cytokines Downregulate the Sarcoendoplasmic Reticulum Pump Ca2+ ATPase 2b and Deplete Endoplasmic Reticulum Ca2+, Leading to Induction of Endoplasmic Reticulum Stress in Pancreatic-Cells. Diabetes 2005, 54, 452–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Zhang, H.; Zhao, B.; Fei, H. IL-1beta Caused Pancreatic Beta-Cells Apoptosis Is Mediated in Part by Endoplasmic Reticulum Stress via the Induction of Endoplasmic Reticulum ca2+ Release through the C-Jun N-Terminal Kinase Pathway. Mol. Cell. Biochem. 2009, 324, 183–190. [Google Scholar] [CrossRef] [PubMed]
- Kawamori, D.; Kaneto, H.; Nakatani, Y.; Matsuoka, T.-A.; Matsuhisa, M.; Hori, M.; Yamasaki, Y. The Forkhead Transcription Factor Foxo1 Bridges the JNK Pathway and the Transcription Factor PDX-1 through Its Intracellular Translocation. J. Biol. Chem. 2006, 281, 1091–1098. [Google Scholar] [CrossRef] [Green Version]
- Sun, Y.; Mao, Q.; Shen, C.; Wang, C.; Jia, W. Exosomes from β-Cells Alleviated Hyperglycemia and Enhanced Angiogenesis in Islets of Streptozotocin-Induced Diabetic Mice. Diabetes Metab. Syndr. Obes. Targets Ther. 2019, 12, 2053–2064. [Google Scholar] [CrossRef] [Green Version]
- Tang, S.; Luo, F.; Feng, Y.M.; Wei, X.; Miao, H.; Lu, Y.B.; Tang, Y.; Ding, D.F.; Jin, J.F.; Zhu, Q. Neutral Ceramidase Secreted Via Exosome Protects Against Palmitate-Induced Apoptosis in INS-1 Cells. Exp. Clin. Endocrinol. Diabetes 2016, 125, 130–135. [Google Scholar] [CrossRef] [PubMed]
- Guay, C.; Menoud, V.; Rome, S.; Regazzi, R. Horizontal Transfer of Exosomal MicroRNAs Transduce Apoptotic Signals between Pancreatic Beta-Cells. Cell Commun. Signal. 2015, 13, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robbins, P.D.; Morelli, A.E. Regulation of Immune Responses by Extracellular Vesicles. Nat. Rev. Immunol. 2014, 14, 195–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guay, C.; Kruit, J.K.; Rome, S.; Menoud, V.; Mulder, N.L.; Jurdzinski, A.; Mancarella, F.; Sebastiani, G.; Donda, A.; Gonzalez, B.J.; et al. Lymphocyte-Derived Exosomal MicroRNAs Promote Pancreatic β Cell Death and May Contribute to Type 1 Diabetes Development. Cell Metab. 2019, 29, 348–361.e6. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Islet Resident Macrophages | Monocyte Derived Macrophages | Dendritic Cells | Ref. | |
|---|---|---|---|---|
| MHCII | +++ | +++ | +++ | [71,80] |
| F4/80 | +++ | +++ | − | [71,81] |
| CD11c | ++ | ++ | ++ | [61,78,82] |
| CD11b | ++ | ++ | + | [82,83] |
| Ly6C | − | ++ | − | [83] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cosentino, C.; Regazzi, R. Crosstalk between Macrophages and Pancreatic β-Cells in Islet Development, Homeostasis and Disease. Int. J. Mol. Sci. 2021, 22, 1765. https://doi.org/10.3390/ijms22041765
Cosentino C, Regazzi R. Crosstalk between Macrophages and Pancreatic β-Cells in Islet Development, Homeostasis and Disease. International Journal of Molecular Sciences. 2021; 22(4):1765. https://doi.org/10.3390/ijms22041765
Chicago/Turabian StyleCosentino, Cristina, and Romano Regazzi. 2021. "Crosstalk between Macrophages and Pancreatic β-Cells in Islet Development, Homeostasis and Disease" International Journal of Molecular Sciences 22, no. 4: 1765. https://doi.org/10.3390/ijms22041765