Summary of the Available Molecular Methods for Detection of SARS-CoV-2 during the Ongoing Pandemic


Abstract
:1. Background
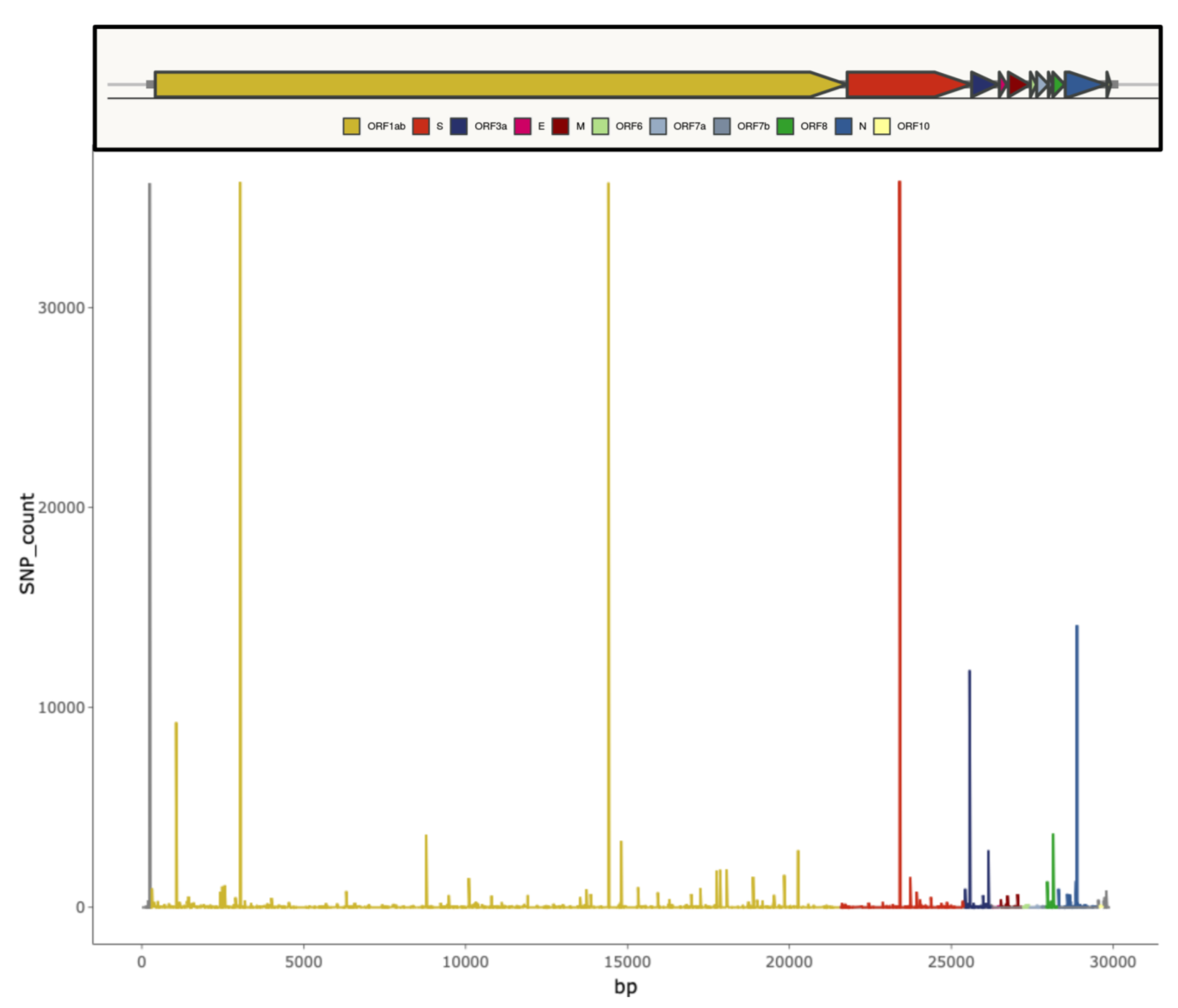
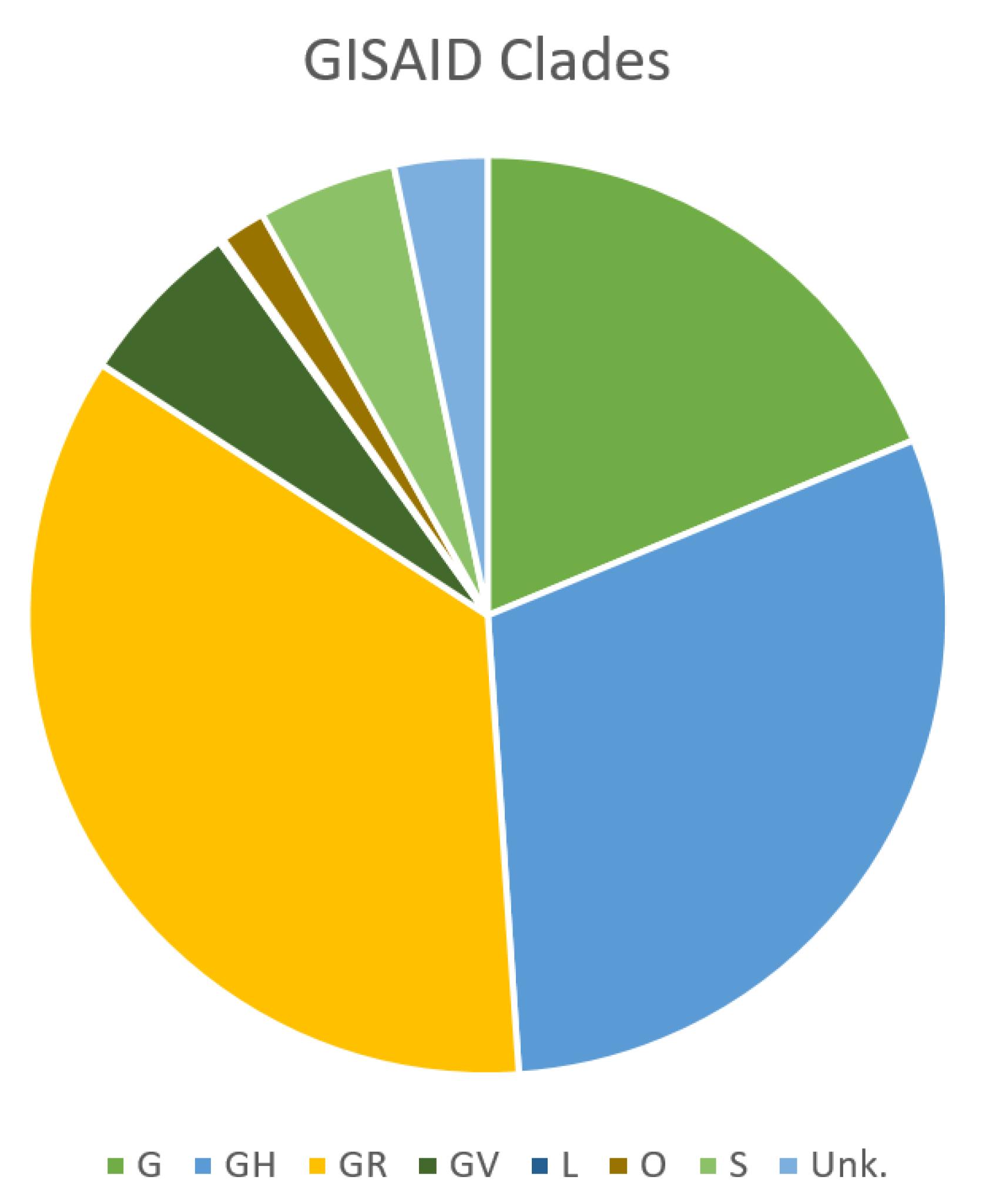
2. SARS-CoV-2 Genomic Features and Variability
3. Diagnostic Tests for SARS-CoV-2 Infection
4. Influence of SARS-CoV-2 Genetic Variability on Molecular Diagnostic Protocols
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Coronaviridae Study Group of the International Committee on Taxonomy of Viruses. The species Severe acute respiratory syndrome-related coronavirus: Classifying 2019-nCoV and naming it SARS-CoV-2. Nat. Microbiol. 2020, 5, 536–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, J.; Li, F.; Shi, Z.L. Origin and evolution of pathogenic coronaviruses. Nat. Rev. Microbiol. 2019, 17, 181–192. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sola, I.; Almazán, F.; Zúñiga, S.; Enjuanes, L. Continuous and Discontinuous RNA Synthesis in Coronaviruses. Annu. Rev. Virol. 2015, 2, 265–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, F.; Zhao, S.; Yu, B.; Chen, Y.M.; Wang, W.; Song, Z.G.; Hu, Y.; Tao, Z.W.; Tian, J.H.; Pei, Y.Y.; et al. A new coronavirus associated with human respiratory disease in China. Nature 2020, 579, 265–269. [Google Scholar] [CrossRef] [Green Version]
- Ji, W.; Wang, W.; Zhao, X.; Zai, J.; Li, X. Cross-species transmission of the newly identified coronavirus 2019-nCoV. J. Med. Virol. 2020, 92, 433–440. [Google Scholar] [CrossRef]
- Benvenuto, D.; Giovanetti, M.; Salemi, M.; Prosperi, M.; De Flora, C.; Junior Alcantara, L.C.; Angeletti, S.; Ciccozzi, M. The global spread of 2019-nCoV: A molecular evolutionary analysis. Pathog. Glob. Health 2020, 114, 64–67. [Google Scholar] [CrossRef] [Green Version]
- Lu, R.; Zhao, X.; Li, J.; Niu, P.; Yang, B.; Wu, H.; Wang, W.; Song, H.; Huang, B.; Zhu, N.; et al. Genomic characterisation and epidemiology of 2019 novel coronavirus: Implications for virus origins and receptor binding. Lancet 2020, 395, 565–574. [Google Scholar] [CrossRef] [Green Version]
- Lam, T.T.; Jia, N.; Zhang, Y.W.; Shum, M.H.; Jiang, J.F.; Zhu, H.C.; Tong, Y.G.; Shi, Y.X.; Ni, X.B.; Liao, Y.S.; et al. Identifying SARS-CoV-2-related coronaviruses in Malayan pangolins. Nature 2020, 583, 282–285. [Google Scholar] [CrossRef] [Green Version]
- Giovanetti, M.; Angeletti, S.; Benvenuto, D.; Ciccozzi, M. A doubt of multiple introduction of SARS-CoV-2 in Italy: A preliminary overview. J. Med. Virol. 2020, 92, 1634–1636. [Google Scholar] [CrossRef]
- Gonzalez-Reiche, A.S.; Hernandez, M.M.; Sullivan, M.J.; Ciferri, B.; Alshammary, H.; Obla, A.; Fabre, S.; Kleiner, G.; Polanco, J.; Khan, Z.; et al. Introductions and early spread of SARS-CoV-2 in the New York City area. Science 2020, 369, 297–301. [Google Scholar] [CrossRef]
- Stefanelli, P.; Faggioni, G.; Lo Presti, A.; Fiore, S.; Marchi, A.; Benedetti, E.; Fabiani, C.; Anselmo, A.; Ciammaruconi, A.; Fortunato, A.; et al. On Behalf of Iss Covid-Study Group. Whole genome and phylogenetic analysis of two SARS-CoV-2 strains isolated in Italy in January and February 2020: Additional clues on multiple introductions and further circulation in Europe. Eurosurveillance 2020, 25, 2000305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Snijder, E.J.; Bredenbeek, P.J.; Dobbe, J.C.; Thiel, V.; Ziebuhr, J.; Poon, L.L.; Guan, Y.; Rozanov, M.; Spaan, W.J.; Gorbalenya, A.E. Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J. Mol. Biol. 2003, 331, 991–1004. [Google Scholar] [CrossRef]
- van Dorp, L.; Acmana, M.; Richardb, D.; Shawd, L.P.; Forda, C.E.; Ormonda, L.; Owena, C.J.; Panga, J.; Tana, C.C.S.; Boshiere, F.A.T.; et al. Emergence of genomic diversity and recurrent mutations in SARS-CoV-2. Infect. Genet. Evol. 2020. [Google Scholar] [CrossRef] [PubMed]
- van Dorp, L.; Richard, D.; Tan, C.C.S.; Shaw, L.P.; Acman, M.; Balloux, F. No evidence for increased transmissibility from recurrent mutations in SARS-CoV-2. Nat. Commun. 2020, 11, 5986. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Wang, W.; Zhao, X.; Zai, J.; Zhao, Q.; Li, Y.; Chaillon, A. Transmission dynamics evolutionary history f 2019-nCoV. J. Med. Virol. 2020, 92, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Giovanetti, M.; Benvenuto, D.; Angeletti, S.; Ciccozzi, M. The first two cases of 2019-nCoV in Italy: Where they come from? J. Med. Virol. 2020, 92, 518–521. [Google Scholar] [CrossRef] [Green Version]
- Duchene, S.; Featherstone, L.; Haritopoulou-Sinanidou, M.; Rambaut, A.; Lemey, P.; Baele, G. Temporal Signal and the Phylodynamic Threshold of SARS-CoV-2. Virus Evol. 2020, 6, veaa061. [Google Scholar] [CrossRef]
- SARS-CoV-2 Alignment Screen. Available online: https://macman123.shinyapps.io/ugi-scov2-alignment-screen/ (accessed on 9 December 2020).
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-time tracking of pathogen evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Han, A.X.; Parker, E.; Scholer, F.; Maurer-Stroh, S.; Russell, C.A. Phylogenetic Clustering by Linear Integer Programming (PhyCLIP). Mol. Biol. Evol. 2019, 36, 1580–1595. [Google Scholar] [CrossRef]
- Alm, E.; Broberg, E.K.; Connor, T.; Hodcroft, E.B.; Komissarov, A.B.; Maurer-Stroh, S.; Melidou, A.; Neher, R.A.; O’Toole, Á.; Pereyaslov, D.; et al. Geographical and temporal distribution of SARS-CoV-2 clades in the WHO European Region, January to June 2020. Eurosurveillance 2020, 25, 2001410. [Google Scholar] [CrossRef]
- Mercatelli, D.; Giorgi, F.M. Geographic and Genomic Distribution of SARS-CoV-2 Mutations. Front. Microbiol. 2020, 11, 1800. [Google Scholar] [CrossRef]
- Influenza Genomic Epidemiology—GISAID. Available online: https://www.gisaid.org/epiflu-applications/influenza-genomic-epidemiology/ (accessed on 9 December 2020).
- Kemp, S.A.; Datir, R.P.; Collier, D.A.; Ferreira, I.A.T.M.; Carabelli, A.; Harvey, W.; Robertson, D.L.; Gupta, R.K. Recurrent emergence and transmission of a SARS-CoV-2 Spike deletion DeltaH69/DeltaV70. bioRxiv 2020. [Google Scholar] [CrossRef]
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N. Emergence and rapid spread of a new severe acute respiratory syndrome-related coronavirus 2 (SARS-CoV-2) lineage with multiple spike mutations in South Africa. medRxiv 2020. [Google Scholar] [CrossRef]
- World Health Organization. Coronavirus Disease (COVID-19) Technical Guidance: Laboratory Testing for 2019-nCoV in Humans (19 March 2020). Available online: https://www.who.int/emergencies/diseases/novel-coronavirus-2019/technical-guidance-publications (accessed on 9 December 2020).
- Corman, V.M.; Landt, O.; Kaiser, M.; Molenkamp, R.; Meijer, A.; Chu, D.K.; Bleicker, T.; Brünink, S.; Schneider, J.; Schmidt, M.L.; et al. 2020 Detection of 2019 novel coronavirus (2019-nCoV) by real-time RT-PCR. Eurosurveillance 2020, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holshue, M.L.; DeBolt, C.; Lindquist, S.; Lofy, K.H.; Wiesman, J.; Bruce, H.; Spitters, C.; Ericson, K.; Wilkerson, S.; Tural, A.; et al. Washington State 2019-nCoV Case Investigation Team. 2020. First case of 2019 novel coronavirus in the United States. N. Engl. J. Med. 2020, 382, 929–936. [Google Scholar] [CrossRef] [PubMed]
- Esbin, M.N.; Whitney, O.N.; Chong, S.; Maurer, A.; Darzacq, X.; Tjian, R. Overcoming the bottleneck to widespread testing: A rapid review of nucleic acid testing approaches for COVID-19 detection. RNA 2020, 26, 771–783. [Google Scholar] [CrossRef] [PubMed]
- James, A.S.; Alwneh, J.I. COVID-19 Infection Diagnosis: Potential Impact of Isothermal Amplification Technology to Reduce Community Transmission of SARS-CoV-2. Diagnostics 2020, 10, 399. [Google Scholar] [CrossRef]
- Shen, M.; Zhou, Y.; Ye, J.; Abdullah Al-Maskri, A.A.; Kang, Y.; Zeng, S.; Cai, S. Recent advances and perspectives of nucleic acid detection for coronavirus. J. Pharm. Anal. 2020, 10, 97–101. [Google Scholar] [CrossRef]
- World Health Organization. Diagnostic Testing for SARS-CoV-2, Interim Guidance. 11 September 2020. Available online: https://wwwwhoint/emergencies/diseases/novel-coronavirus-2019/technical-guidance-publications (accessed on 9 December 2020).
- Dinnes, J.; Deeks, J.J.; Adriano, A.; Berhane, S.; Davenport, C.; Dittrich, S.; Emperador, D.; Takwoingi, Y.; Cunningham, J.; Beese, S.; et al. Rapid, point-of-care antigen and molecular-based tests for diagnosis of SARS-CoV-2 infection. Cochrane COVID-19 Diagnostic Test Accuracy Group. Cochrane Database Syst. Rev. 2020, 8, CD013705. [Google Scholar] [CrossRef]
- Peñarrubia, L.; Ruiz, M.; Porco, R.; Rao, S.N.; Juanola-Falgarona, M.; Manissero, D.; López-Fontanals, M.; Pareja, J. Multiple assays in a real-time RT-PCR SARS-CoV-2 panel can mitigate the risk of loss of sensitivity by new genomic variants during the COVID-19 outbreak. Int. J. Infect. Dis. 2020, 97, 225–229. [Google Scholar] [CrossRef]
- Wolfel, R.; Corman, V.M.; Guggemos, W.; Seilmaier, M.; Zange, S.; Müller, M.A.; Niemeyer, D.; Jones, T.C.; Vollmar, P.; Rothe, C.; et al. Virological assessment of hospitalized patients with COVID-19. Nature 2020, 581, 465–469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- La Scola, B.; Le Bideau, M.; Andreani, J.; Hoang, V.T.; Grimaldier, C.; Colson, P.; Colson, P.; Raoult, D. Viral RNA load as determined by cell culture as a management tool for discharge of SARS-CoV-2 from infectious disease wards. Eur. J. Clin. Microbiol. Infect. Dis. 2020, 39, 1059–1061. [Google Scholar] [CrossRef] [PubMed]
- Khan, K.A.; Cheung, P. Presence of mismatches between diagnostic PCR assays and coronavirus SARS-CoV-2 genome. R. Soc. Open Sci. 2020, 7, 200636. [Google Scholar] [CrossRef] [PubMed]
- Vogels, C.B.F.; Brito, A.F.; Wyllie, A.L.; Fauver, J.R.; Ott, I.M.; Kalinich, C.C.; Petrone, M.E.; Casanovas-Massana, A.; Catherine Muenker, M.; Moore, A.J.; et al. Analytical sensitivity and efficiency comparisons of SARS-CoV-2 RT-qPCR primer-probe sets. Nat. Microbiol. 2020, 5, 1299–1305. [Google Scholar] [CrossRef] [PubMed]
- Gand, M.; Vanneste, K.; Thomas, I.; Van Gucht, S.; Capron, A.; Herman, P.; Roosens, N.H.C.; De Keersmaecker, S.C.J. Use of Whole Genome Sequencing Data for a First in Silico Specificity Evaluation of the RT-qPCR Assays Used for SARS-CoV-2 Detection. Int. J. Mol. Sci. 2020, 21, 5585. [Google Scholar] [CrossRef]
- Álvarez-Díaz, D.A.; Franco-Muñoz, C.; Laiton-Donato, K.; Usme-Ciro, J.A.; Franco-Sierra, N.D.; Flórez-Sánchez, A.C.; Gómez-Rangel, S.; Rodríguez-Calderon, L.D.; Barbosa-Ramirez, J.; Ospitia-Baez, E.; et al. Molecular analysis of several in-house rRT-PCR protocols for SARS-CoV-2 detection in the context of genetic variability of the virus in Colombia. Infect. Genet. Evol. 2020, 84, 104390. [Google Scholar] [CrossRef]
- Kuchinski, K.S.; Jassem, A.N.; Prystajecky, N.A. Assessing oligonucleotide designs from early lab developed PCR diagnostic tests for SARS-CoV-2 using the PCR_strainer pipeline. J. Clin. Virol. 2020, 131, 104581. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Assay Type | Principle of the Assay | Intended Use |
|---|---|---|
| Nucleic acid tests | detect the presence of viral RNA, generally by RT-PCR | decision making for clinical, infection control, or public health management (screening close contacts, outbreak investigations, or surveillance programs) |
| Antigen tests | detect the presence of a viral antigen, typically part of a surface protein, by lateral flow assays or chemiluminescence immunoassays | decision making for clinical, infection control, or public health management (screening close contacts, outbreak investigations, or surveillance programs) |
| Antibody tests | detect the presence of antibodies generated against SARS-CoV-2. The three most used assays are enzyme-linked immunosorbent assays, chemiluminescence assays, and lateral flow assays | sero-epidemiological surveys and studies; complement to the virus-detection tests |
| Source | Primer/Probe Name | Target Gene | Sequence | Lenght | Genomic Region * |
|---|---|---|---|---|---|
| China CDC, China | Forward (F) | ORF1ab | CCCTGTGGGTTTTACACTTAA | 21 | 13,342–13,362 |
| China CDC, China | Reverse (R) | ORF1ab | ACGATTGTGCATCAGCTGA | 19 | 13,442–13,460 |
| China CDC, China | Fluorescence probe (P) | ORF1ab | CCGTCTGCGGTATGTGGAAAGGTTATGG | 28 | 13,377–13,404 |
| China CDC, China | Forward (F) | N | GGGGAACTTCTCCTGCTAGAAT | 22 | 28,881–28,902 |
| China CDC, China | Reverse (R) | N | CAGACATTTTGCTCTCAAGCTG | 22 | 28,958–28,979 |
| China CDC, China | Fluorescence probe (P) | N | TTGCTGCTGCTTGACAGATT | 20 | 28,934–28,953 |
| Institut Pasteur, France | nCoV_IP2-12669Fw | RdRp | ATGAGCTTAGTCCTGTTG | 18 | 12,690–12,707 |
| Institut Pasteur, France | nCoV_IP2-12759Rv | RdRp | CTCCCTTTGTTGTGTTGT | 18 | 12,780–12,797 |
| Institut Pasteur, France | nCoV_IP2-12696bProbe(+) | RdRp | ATGTCTTGTGCTGCCGGTA | 19 | 12,719–12,737 |
| Institut Pasteur, France | nCoV_IP4-14059Fw | RdRp | GGTAACTGGTATGATTTCG | 19 | 14,080–14,098 |
| Institut Pasteur, France | nCoV_IP4-14146Rv | RdRp | CTGGTCAAGGTTAATATAGG | 20 | 14,167–14,186 |
| Institut Pasteur, France | nCoV_IP4-14084Probe(+) | RdRp | TCATACAAACCACGCCAGG | 19 | 14,105–14,123 |
| Institut Pasteur, France | E_Sarbeco_F1 | E | ACAGGTACGTTAATAGTTAATAGCGT | 26 | 26,269–26,294 |
| Institut Pasteur, France | E_Sar beco_R2 | E | ATATTGCAGCAGTACGCACACA | 22 | 26,360–26,381 |
| Institut Pasteur, France | E_Sarbeco_P1 | E | ACACTAGCCATCCTTACTGCGCTTCG | 26 | 26,332–26,357 |
| US CDC, USA | 2019-nCoV_N1-F | ORF9b | GACCCCAAAATCAGCGAAAT | 20 | 28,287–28,306 |
| US CDC, USA | 2019-nCoV_N1-R | ORF9b | TCTGGTTACTGCCAGTTGAATCTG | 24 | 28,335–28,358 |
| US CDC, USA | 2019-nCoV_N1-P | ORF9b | ACCCCGCATTACGTTTGGTGGACC | 24 | 28,309–28,332 |
| US CDC, USA | 2019-nCoV_N2-F | ORF9b | TTACAAACATTGGCCGCAAA | 20 | 29,164–29,183 |
| US CDC, USA | 2019-nCoV_N2-R | ORF9b | GCGCGACATTCCGAAGAA | 18 | 29,213–29,230 |
| US CDC, USA | 2019-nCoV_N2-P | ORF9b | ACAATTTGCCCCCAGCGCTTCAG | 23 | 29,188–29,210 |
| US CDC, USA | 2019-nCoV_N3-F | ORF9b | GGGAGCCTTGAATACACCAAAA | 22 | 28,681–28,702 |
| US CDC, USA | 2019-nCoV_N3-R | ORF9b | TGTAGCACGATTGCAGCATTG | 21 | 28,732–28,752 |
| US CDC, USA | 2019-nCoV_N3-P | ORF9b | ATCACATTGGCACCCGCAATCCTG | 24 | 28,704–28,727 |
| National Institute of Infectious Diseases, Japan | NIID_2019-nCOV_N_F2 | N | AAATTTTGGGGACCAGGAAC | 20 | 29,142–29,161 |
| National Institute of Infectious Diseases, Japan | NIID_2019-nCOV_N_R2 | N | TGGCAGCTGTGTAGGTCAAC | 20 | 29,280–29,299 |
| National Institute of Infectious Diseases, Japan | NIID_2019-nCOV_N_P2 | N | ATGTCGCGCATTGGCATGGA | 20 | 29,239–29,258 |
| Charité, Germany | RdRP_SARSr-F2 | RdRp | GTGAAATGGTCATGTGTGGCGG | 22 | 15,431–15,452 |
| Charité, Germany | RdRP_SARSr-R1 | RdRp | CAAATGTTAAAAACACTATTAGCATA | 26 | 15,505–15,530 |
| Charité, Germany | RdRP_SARSr-P2 | RdRp | CAGGTGGAACCTCATCAGGAGATGC | 25 | 15,470–15,494 |
| Charité, Germany | E_Sarbeco_F1 | E | ACAGGTACGTTAATAGTTAATAGCGT | 26 | 26,269–26,294 |
| Charité, Germany | E_Sarbeco_R2 | E | ATATTGCAGCAGTACGCACACA | 22 | 26,360–26,381 |
| Charité, Germany | E_Sarbeco_P1 | E | ACACTAGCCATCCTTACTGCGCTTCG | 26 | 26,332–26,357 |
| HKU, HongKong SAR | HKU-ORF1b-nsp14F | ORF1b | TGGGGTTTTACAGGTAACCT | 20 | 18,778–18,797 |
| HKU, HongKong SAR | HKU-ORF1b-nsp14R | ORF1b | AACACGCTTAACAAAGCACTC | 21 | 18,889–18,909 |
| HKU, HongKong SAR | HKU-ORF1b-nsp141P | ORF1b | TAGTTGTGATGCAATCATGACTAG | 24 | 18,849–18,872 |
| HKU, HongKong SAR | HKU-NF | N | TAATCAGACAAGGAACTGATTA | 22 | 29,145–29,166 |
| HKU, HongKong SAR | HKU-NR | N | CGAAGGTGTGACTTCCATG | 19 | 29,236–29,254 |
| HKU, HongKong SAR | HKU-NP | N | GCAAATTGTGCAATTTGCGG | 20 | 29,177–29,196 |
| National Institute of Health, Thailand | WH-NICN-F | ORF9b | CGTTTGGTGGACCCTCAGAT | 20 | 28,320–28,339 |
| National Institute of Health, Thailand | WH-NICN-R | ORF9b | CCCCACTGCGTTCTCCATT | 19 | 28,358–28,376 |
| National Institute of Health, Thailand | WH-NICN-P | ORF9b | CAACTGGCAGTAACCA | 16 | 28,341–28,356 |
| Assay | Manufacturer | Viral Genes | Assay/Equipment Type | Approx. Time-to-Result |
|---|---|---|---|---|
| Xpert® Xpress SARS-CoV-2 | Cepheid | N, E | RT-PCR/single test, sample-to-result | 45 min. |
| Vivalytic analyzer/Vivalytic VRI test | BOSCH | Na a | RT-PCR/single test, sample-to-result | 39 min. |
| VitaPCRTM platform/VitaPCR™ SARS-CoV-2 assay | Menarini | N | RT-PCR/single test, sample-to-result | 20 min |
| GenMark ePlex instrument/ePlex® SARS-CoV-2 Test | GenMark | N | RT-PCR/single test, sample-to-result | 90 min. |
| ARIES® SARS-CoV-2 Assay | Luminex Corporation | Orf1ab, N | RT-PCR/single test, sample-to-result | 2 h |
| ID Now COVID-19 | Abbott | RdRp | Isothermal amplification/single test, sample-to-result | 13 min. |
| Simplexa COVID-19 Direct assay | DiaSorin | orf1ab, S | RT-PCR/batch testing, sample-to-result | 80 min. |
| ELITech InGenius platform/SARS-CoV-2 ELITe MGB® Kit | ELITech | RdRp, Orf8 | RT-PCR/batch testing, sample-to-result | 2 h 30 min. |
| Cobas 6800/8800/cobas SARS-CoV-2 | Roche | orf1ab, E | RT-PCR/batch testing, sample-to-result | 3 h 30 min. |
| Alinity m System/Alinity m SARS-CoV-2 assay | Abbott | RdRp, N | RT-PCR/batch testing, sample-to-result | 2 h |
| NeoMoDx™ molecular system/NeuMoDx™ SARS-CoV-2 Assay | QIAGEN | Nsp2, N | RT-PCR/batch testing, sample-to-result | 80 min. |
| BD MAX™ System/BD SARS-CoV-2 Reagents | Becton Dickinson | N | RT-PCR/batch testing, sample-to-result | 3 h |
| Panther/Aptima SARS-CoV-2 assay | Hologic | orf1ab | Isothermal amplification/batch testing, sample-to-result | 3 h 30 min. |
| Seegene NIMBUS/STARlet/Maelstrom 9600/Allplex™ SARS-CoV-2 Assay | Seegene | RdRp, N, S, E | RT-PCR/batch testing, integrated equipment for extraction and amplification b | From 3 h 20 min. to 4 h 40 min. |
| KingFisher Flex Purification system/TaqPath™ COVID-19 RT-PCR Kit | Life Technologies Corporation | orf1ab, N, S | RT-PCR/batch testing, integrated equipment for extraction and amplification | na |
| BIOFIRE® Respiratory Panel 2.1 | Biomérieux | S, M | RT-PCR/syndromic panel, sample-to-result | 45 min. |
| QIAstat-Dx Respiratory SARS-CoV-2 Panel | QIAGEN | RdRp, E | RT-PCR/syndromic panel, sample-to-result | 60 min. |
| No. of Genomes | No. of Primers/Probes Set Evaluated | Relevant Findings | Source | Period | Reference |
|---|---|---|---|---|---|
| 17,027 | 27 | 100% of mutation frequency in the Charité-ORF1b and 18% in the forward primer of CN-CDC-N | GISAID | Genomes sequenced before 7 May 2020 | [37] |
| 992 | 10 | mutations in the first 5′ three positions of the China CDC N forward primer, frequency 13% | GISAID | Genomes sequenced before 22 March 2020 | [38] |
| 2569 | 30 | mutations in the first 5′ three positions of the China CDC N forward primer, frequency 14% | GISAID | Genomes sequenced before 7 April 2020 | [39] |
| 30 | 13 | mutations in the China CDC N forward primer, frequency 16% | Locally sequenced genomes from Colombia | Period 6–24 March 2020 | [40] |
| 15,001 | 15 | A single mismatch in the Charité group’s RdRP gene assay and the Japan NIID’s N gene assay; AAC variant at the 5′ end of the China CDC N forward primer, frequency 18.8% | GISAID | Genomes sequenced before 8 June 2020 | [41] |
| 33,819 | 9 | AAC variant at the 5′ end of the China CDC N forward primer, frequency 24% | GISAID and GenBank | Genomes sequenced before June 2020 | [34] |
| Source | Primer/Probe Name | Target Gene | Sequence | Lenght | Genomic Region * | Mutation | Frequency (%) | Clade Nextstrain | Clade GISAID | Country |
|---|---|---|---|---|---|---|---|---|---|---|
| China CDC, China | Forward (F) | N | GGGGAACTTCTCCTGCTAGAAT | 22 | 28,881–28,902 | G28881A | 37.1 | 20A, 20B | G, GH, GR | Worldwide |
| G28882A | 36.9 | 20A, 20B | GH, GR | Worldwide | ||||||
| G28883C | 36.9 | 20A, 20B | GH, GR | Worldwide | ||||||
| C28887T | 2.9 | 19A, 20A, 20B, 20C | G, GH, GR, O | SriLanka | ||||||
| China CDC, China | Reverse (R) | N | CAGACATTTTGCTCTCAAGCTG | 22 | 28,958–28,979 | G28975C | 4.6 | 20A | GH | Europe |
| US CDC | 2019-nCoV_N3-P | ORF9b | ATCACATTGGCACCCGCAATCCTG | 24 | 28,704–28,727 | A28715T | 2.0 | 20A, 20B | GH, GR | Japan |
| HKU, HongKong, SAR | HKU-NR | N | CGAAGGTGTGACTTCCATG | 19 | 29,236–29,254 | G29254A | 1.0 | 20A, 20B, 20C | GH, GR | Latvia |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arena, F.; Pollini, S.; Rossolini, G.M.; Margaglione, M. Summary of the Available Molecular Methods for Detection of SARS-CoV-2 during the Ongoing Pandemic. Int. J. Mol. Sci. 2021, 22, 1298. https://doi.org/10.3390/ijms22031298
Arena F, Pollini S, Rossolini GM, Margaglione M. Summary of the Available Molecular Methods for Detection of SARS-CoV-2 during the Ongoing Pandemic. International Journal of Molecular Sciences. 2021; 22(3):1298. https://doi.org/10.3390/ijms22031298
Chicago/Turabian StyleArena, Fabio, Simona Pollini, Gian Maria Rossolini, and Maurizio Margaglione. 2021. "Summary of the Available Molecular Methods for Detection of SARS-CoV-2 during the Ongoing Pandemic" International Journal of Molecular Sciences 22, no. 3: 1298. https://doi.org/10.3390/ijms22031298