
Metformin Treatment Attenuates Brain Inflammation and Rescues PACAP/VIP Neuropeptide Alterations in Mice Fed a High-Fat Diet

 , ,
, ,  , ,
, , 
Abstract
:1. Introduction
- (1)
- HFD mice show signatures of neuroinflammation in the hypothalamus and other vulnerable extra-hypothalamic CNS sites;
- (2)
- The PACAP/VIP neuropeptide system is dysregulated by HFD in brain regions showing HFD-triggered neuroinflammations; and
- (3)
- The antidiabetic drug metformin ameliorates neuroinflammation by rescuing HFD-induced dysregulation of the PACAP/VIP system and/or the PI3K/AKT pathways.
2. Results
2.1. Metformin Treatment Attenuated HFD-Induced Glucose Tolerance, Insulin Resistance, and Weight Gain
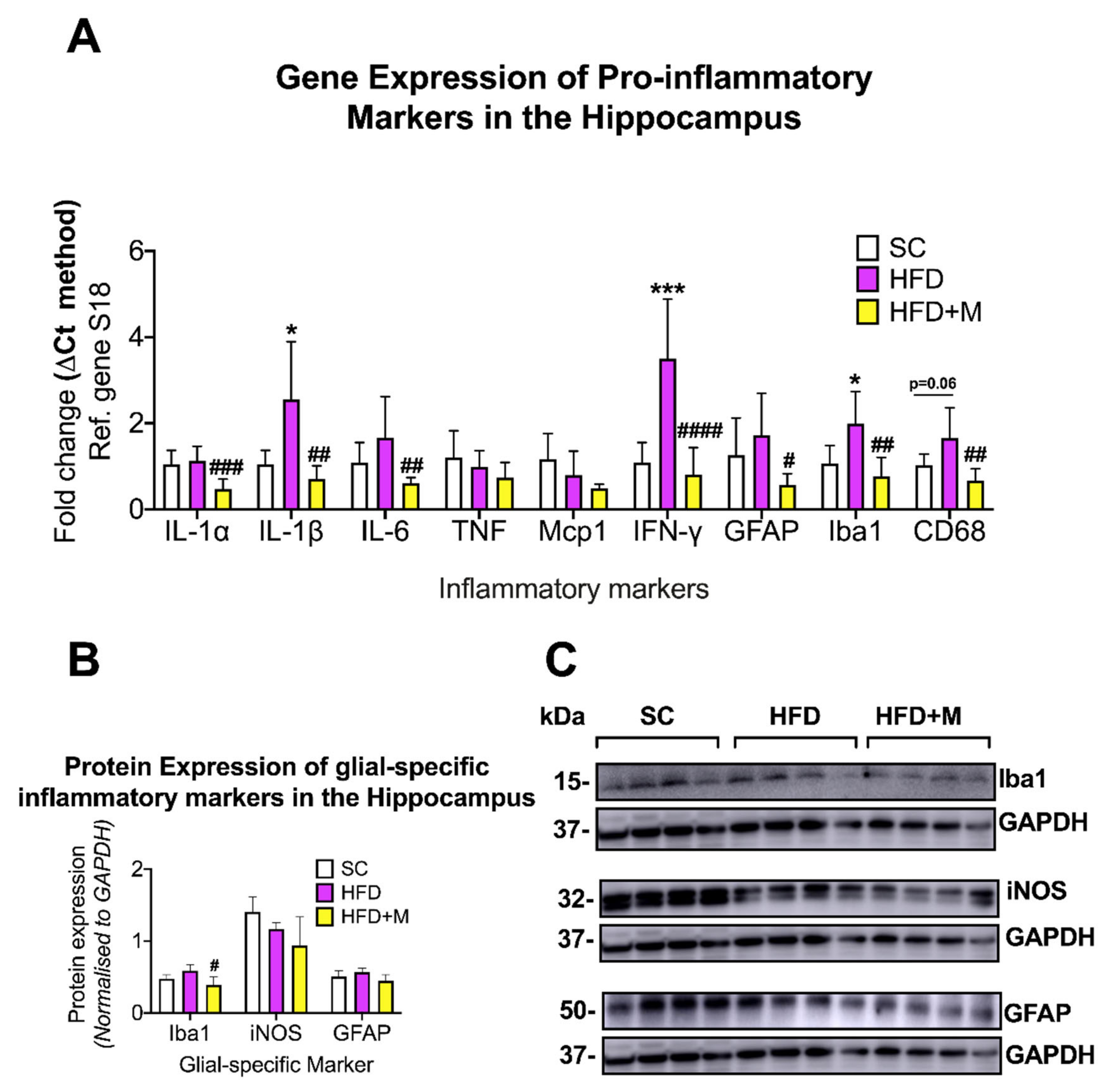
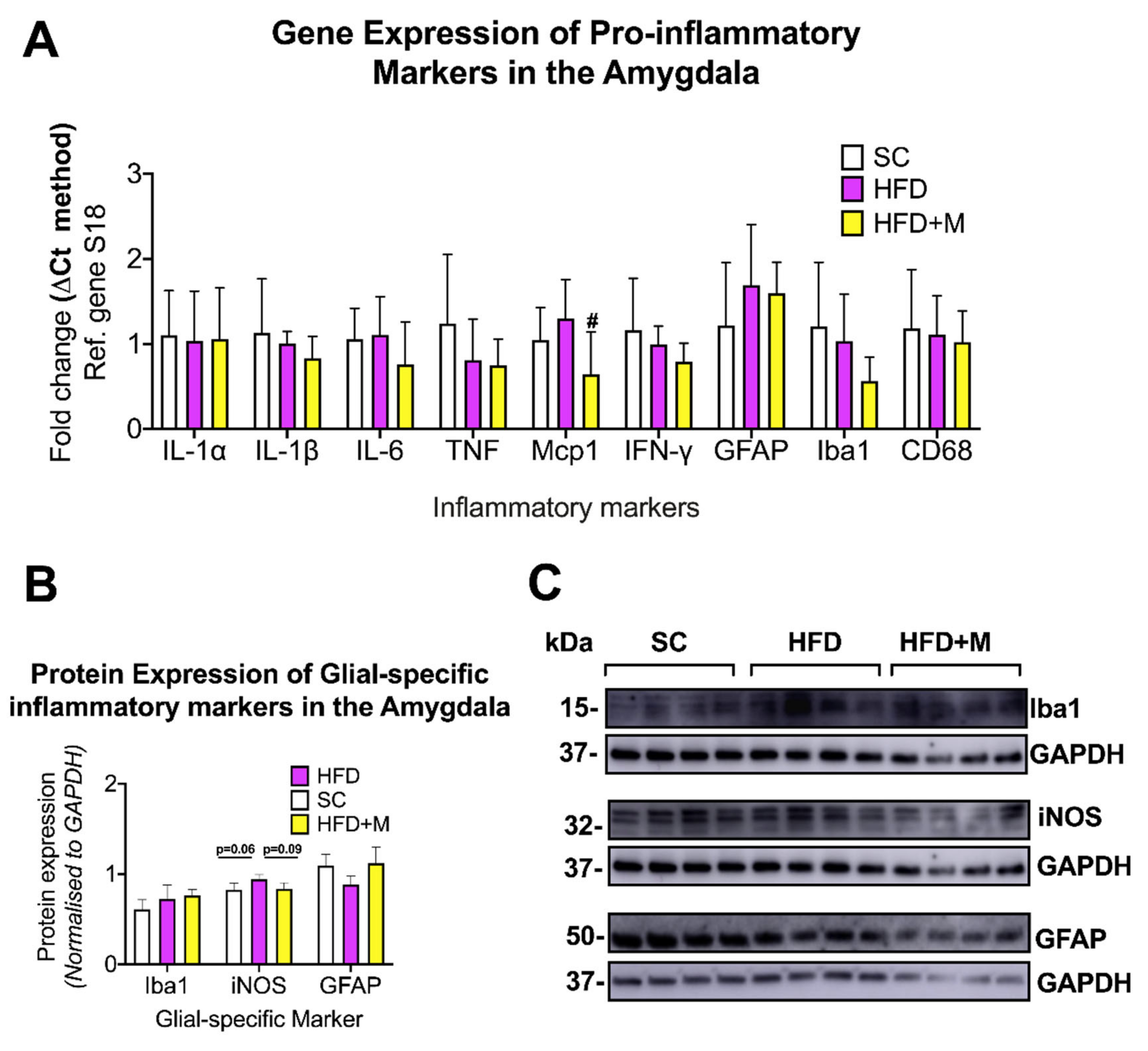
2.2. Metformin Treatment Rescues HFD-Induced Neuroinflammation and Glial Cell Activation in a Region-Specific Manner
2.3. Diet and Metformin Treatment Have No Effect on the mRNA Expression of the Anti-Inflammatory Cytokine IL-10
2.4. Metformin Treatment Triggers the Increase of Phospho-Akt(Ser473) Protein Levels in the Brain of Mice Fed with a HFD
2.5. Metformin Administration Rescues HFD-Induced Dysregulations of the PACAP/VIP Neuropeptide System
3. Discussion
3.1. Establishing a Suitable Animal Model of HFD-Induced Obesity and Assessing the Hypoglycemic Effects of Metformin
3.2. High-Fat Diet Triggers Distinct and Regional-Specific Neuroinflammatory Changes and Promotes Glial Activation
3.2.1. Hypothalamus
3.2.2. Hippocampus
3.2.3. PFC
3.2.4. Amygdala
3.3. Metformin Ameliorates HFD-Induced Neuroinflammation
3.4. Metformin Activates the PI3K/AKT Pathway in the Brain of HFD Mice
3.5. PACAP/VIP System in Response to Chronic HFD and Metformin Administration
3.5.1. Hypothalamus
3.5.2. Hippocampus
3.5.3. PFC
3.5.4. Amygdala
4. Materials and Methods
4.1. Animals
4.2. Drug Administration
4.3. Fasting Glucose and Insulin Measurements
4.4. Method of Euthanasia
4.5. Tissue Collection and Microdissections
4.6. RNA Extraction, cDNA Synthesis, and Real-Time Quantitative Polymerase Chain Reaction
4.7. BCA Protein Quantification Assay
4.8. Western Blot Analyses
4.9. Statistical Analyses
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dalvi, P.S.; Chalmers, J.A.; Luo, V.; Han, D.Y.; Wellhauser, L.; Liu, Y.; Tran, D.Q.; Castel, J.; Luquet, S.; Wheeler, M.B.; et al. High fat induces acute and chronic inflammation in the hypothalamus: Effect of high-fat diet, palmitate and tnf-alpha on appetite-regulating npy neurons. Int. J. Obes. 2017, 41, 149–158. [Google Scholar] [CrossRef] [PubMed]
- Mazon, J.N.; de Mello, A.H.; Ferreira, G.K.; Rezin, G.T. The impact of obesity on neurodegenerative diseases. Life Sci. 2017, 182, 22–28. [Google Scholar] [CrossRef] [PubMed]
- Hotamisligil, G.S. Inflammation and metabolic disorders. Nature 2006, 444, 860–867. [Google Scholar] [CrossRef] [PubMed]
- De Souza, C.T.; Araujo, E.P.; Bordin, S.; Ashimine, R.; Zollner, R.L.; Boschero, A.C.; Saad, M.J.; Velloso, L.A. Consumption of a fat-rich diet activates a proinflammatory response and induces insulin resistance in the hypothalamus. Endocrinology 2005, 146, 4192–4199. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Zhang, G.; Zhang, H.; Karin, M.; Bai, H.; Cai, D. Hypothalamic ikkbeta/nf-kappab and er stress link overnutrition to energy imbalance and obesity. Cell 2008, 135, 61–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Posey, K.A.; Clegg, D.J.; Printz, R.L.; Byun, J.; Morton, G.J.; Vivekanandan-Giri, A.; Pennathur, S.; Baskin, D.G.; Heinecke, J.W.; Woods, S.C.; et al. Hypothalamic proinflammatory lipid accumulation, inflammation, and insulin resistance in rats fed a high-fat diet. Am. J. Physiol. Endocrinol. Metab. 2009, 296, E1003–E1012. [Google Scholar] [CrossRef] [PubMed]
- Elias, M.F.; Elias, P.K.; Sullivan, L.M.; Wolf, P.A.; D’Agostino, R.B. Lower cognitive function in the presence of obesity and hypertension: The framingham heart study. Int. J. Obes. Relat. Metab. Disord. J. Int. Assoc. Study Obes. 2003, 27, 260–268. [Google Scholar] [CrossRef] [Green Version]
- Cournot, M.; Marquie, J.C.; Ansiau, D.; Martinaud, C.; Fonds, H.; Ferrieres, J.; Ruidavets, J.B. Relation between body mass index and cognitive function in healthy middle-aged men and women. Neurology 2006, 67, 1208–1214. [Google Scholar] [CrossRef]
- Kerwin, D.R.; Gaussoin, S.A.; Chlebowski, R.T.; Kuller, L.H.; Vitolins, M.; Coker, L.H.; Kotchen, J.M.; Nicklas, B.J.; Wassertheil-Smoller, S.; Hoffmann, R.G.; et al. Interaction between body mass index and central adiposity and risk of incident cognitive impairment and dementia: Results from the women’s health initiative memory study. J. Am. Geriatr. Soc. 2011, 59, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Elias, M.F.; Elias, P.K.; Sullivan, L.M.; Wolf, P.A.; D’Agostino, R.B. Obesity, diabetes and cognitive deficit: The framingham heart study. Neurobiol. Aging 2005, 26 (Suppl. 1), 11–16. [Google Scholar] [CrossRef]
- Gariepy, G.; Nitka, D.; Schmitz, N. The association between obesity and anxiety disorders in the population: A systematic review and meta-analysis. Int. J. Obes. 2010, 34, 407–419. [Google Scholar] [CrossRef] [Green Version]
- Lykouras, L.; Michopoulos, J. Anxiety disorders and obesity. Psychiatrike Psychiatriki 2011, 22, 307–313. [Google Scholar] [PubMed]
- Gariepy, G.; Wang, J.; Lesage, A.D.; Schmitz, N. The longitudinal association from obesity to depression: Results from the 12-year national population health survey. Obesity 2010, 18, 1033–1038. [Google Scholar] [CrossRef] [PubMed]
- Stunkard, A.J.; Faith, M.S.; Allison, K.C. Depression and obesity. Biol. Psychiatry 2003, 54, 330–337. [Google Scholar] [CrossRef]
- Theriault, P.; ElAli, A.; Rivest, S. High fat diet exacerbates alzheimer’s disease-related pathology in appswe/ps1 mice. Oncotarget 2016, 7, 67808–67827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nam, K.N.; Mounier, A.; Wolfe, C.M.; Fitz, N.F.; Carter, A.Y.; Castranio, E.L.; Kamboh, H.I.; Reeves, V.L.; Wang, J.; Han, X.; et al. Effect of high fat diet on phenotype, brain transcriptome and lipidome in alzheimer’s model mice. Sci. Rep. 2017, 7, 4307. [Google Scholar] [CrossRef]
- Cai, D. Neuroinflammation and neurodegeneration in overnutrition-induced diseases. Trends Endocrinol. Metab. 2013, 24, 40–47. [Google Scholar] [CrossRef] [Green Version]
- Winzell, M.S.; Ahren, B. The high-fat diet-fed mouse: A model for studying mechanisms and treatment of impaired glucose tolerance and type 2 diabetes. Diabetes 2004, 53 (Suppl. 3), S215–S219. [Google Scholar] [CrossRef] [Green Version]
- Fraulob, J.C.; Ogg-Diamantino, R.; Fernandes-Santos, C.; Aguila, M.B.; Mandarim-de-Lacerda, C.A. A mouse model of metabolic syndrome: Insulin resistance, fatty liver and non-alcoholic fatty pancreas disease (nafpd) in c57bl/6 mice fed a high fat diet. J. Clin. Biochem. Nutr. 2010, 46, 212–223. [Google Scholar] [CrossRef] [Green Version]
- Buettner, R.; Scholmerich, J.; Bollheimer, L.C. High-fat diets: Modeling the metabolic disorders of human obesity in rodents. Obesity 2007, 15, 798–808. [Google Scholar] [CrossRef] [PubMed]
- Cordner, Z.A.; Tamashiro, K.L.K. Effects of high-fat diet exposure on learning & memory. Physiol. Behav. 2015, 152, 363–371. [Google Scholar]
- Sivanathan, S.; Thavartnam, K.; Arif, S.; Elegino, T.; McGowan, P.O. Chronic high fat feeding increases anxiety-like behaviour and reduces transcript abundance of glucocorticoid signalling genes in the hippocampus of female rats. Behav. Brain Res. 2015, 286, 265–270. [Google Scholar] [CrossRef] [PubMed]
- Guillemot-Legris, O.; Muccioli, G.G. Obesity-induced neuroinflammation: Beyond the hypothalamus. Trends Neurosci. 2017, 40, 237–253. [Google Scholar] [CrossRef] [PubMed]
- Milanski, M.; Degasperi, G.; Coope, A.; Morari, J.; Denis, R.; Cintra, D.E.; Tsukumo, D.M.; Anhe, G.; Amaral, M.E.; Takahashi, H.K.; et al. Saturated fatty acids produce an inflammatory response predominantly through the activation of tlr4 signaling in hypothalamus: Implications for the pathogenesis of obesity. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 359–370. [Google Scholar] [CrossRef]
- Yarlagadda, A.; Alfson, E.; Clayton, A.H. The blood brain barrier and the role of cytokines in neuropsychiatry. Psychiatry 2009, 6, 18–22. [Google Scholar]
- Selmaj, K.W. The role of cytokines in inflammatory conditions of the central nervous system. Semin. Neurosci. 1992, 4, 221–229. [Google Scholar] [CrossRef]
- Val-Laillet, D.; Layec, S.; Guérin, S.; Meurice, P.; Malbert, C.-H. Changes in brain activity after a diet-induced obesity. Obesity 2011, 19, 749–756. [Google Scholar] [CrossRef] [PubMed]
- Molteni, R.; Barnard, R.J.; Ying, Z.; Roberts, C.K.; Gómez-Pinilla, F. A high-fat, refined sugar diet reduces hippocampal brain-derived neurotrophic factor, neuronal plasticity, and learning. Neuroscience 2002, 112, 803–814. [Google Scholar] [CrossRef] [Green Version]
- Wu, A.; Molteni, R.; Ying, Z.; Gomez-Pinilla, F. A saturated-fat diet aggravates the outcome of traumatic brain injury on hippocampal plasticity and cognitive function by reducing brain-derived neurotrophic factor. Neuroscience 2003, 119, 365–375. [Google Scholar] [CrossRef]
- Laroche, S.; Davis, S.; Jay, T.M. Plasticity at hippocampal to prefrontal cortex synapses: Dual roles in working memory and consolidation. Hippocampus 2000, 10, 438–446. [Google Scholar] [CrossRef]
- Cazettes, F.; Cohen, J.I.; Yau, P.L.; Talbot, H.; Convit, A. Obesity-mediated inflammation may damage the brain circuit that regulates food intake. Brain Res. 2011, 1373, 101–109. [Google Scholar] [CrossRef] [Green Version]
- Miller, A.A.; Spencer, S.J. Obesity and neuroinflammation: A pathway to cognitive impairment. Brain Behav. Immun. 2014, 42, 10–21. [Google Scholar] [CrossRef]
- Pannacciulli, N.; Del Parigi, A.; Chen, K.; Le, D.S.N.T.; Reiman, E.M.; Tataranni, P.A. Brain abnormalities in human obesity: A voxel-based morphometric study. NeuroImage 2006, 31, 1419–1425. [Google Scholar] [CrossRef] [PubMed]
- Tataranni, P.A.; Gautier, J.F.; Chen, K.; Uecker, A.; Bandy, D.; Salbe, A.D.; Pratley, R.E.; Lawson, M.; Reiman, E.M.; Ravussin, E. Neuroanatomical correlates of hunger and satiation in humans using positron emission tomography. Proc. Natl. Acad. Sci. USA 1999, 96, 4569–4574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adler, A.I.; Shaw, E.J.; Stokes, T.; Ruiz, F. Newer agents for blood glucose control in type 2 diabetes: Summary of nice guidance. BMJ Clin. Res. Ed. 2009, 338, b1668. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Gallagher, D.; DeVito, L.M.; Cancino, G.I.; Tsui, D.; He, L.; Keller, G.M.; Frankland, P.W.; Kaplan, D.R.; Miller, F.D. Metformin activates an atypical pkc-cbp pathway to promote neurogenesis and enhance spatial memory formation. Cell Stem Cell 2012, 11, 23–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, T.; Yu, J.T.; Zhu, X.C.; Wang, H.F.; Tan, M.S.; Cao, L.; Zhang, Q.Q.; Gao, L.; Shi, J.Q.; Zhang, Y.D.; et al. Acute metformin preconditioning confers neuroprotection against focal cerebral ischaemia by pre-activation of ampk-dependent autophagy. Br. J. Pharmacol. 2014, 171, 3146–3157. [Google Scholar] [CrossRef] [PubMed]
- Ashabi, G.; Khalaj, L.; Khodagholi, F.; Goudarzvand, M.; Sarkaki, A. Pre-treatment with metformin activates nrf2 antioxidant pathways and inhibits inflammatory responses through induction of ampk after transient global cerebral ischemia. Metab. Brain Dis. 2015, 30, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Afshari, K.; Dehdashtian, A.; Haddadi, N.S.; Haj-Mirzaian, A.; Iranmehr, A.; Ebrahimi, M.A.; Tavangar, S.M.; Faghir-Ghanesefat, H.; Mohammadi, F.; Rahimi, N.; et al. Anti-inflammatory effects of metformin improve the neuropathic pain and locomotor activity in spinal cord injured rats: Introduction of an alternative therapy. Spinal Cord 2018, 302, 8–20. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Tennekoon, G.I.; Birnbaum, M.; Marchionni, M.A.; Rutkowski, J.L. Neuregulin signaling through a pi3k/akt/bad pathway in schwann cell survival. Mol. Cell. Neurosci. 2001, 17, 761–767. [Google Scholar] [CrossRef]
- Battu, C.E.; Rieger, D.; Loureiro, S.; Furtado, G.V.; Bock, H.; Saraiva-Pereira, M.L.; Pessoa-Pureur, R.; Goncalves, C.A.; Perry, M.L. Alterations of pi3k and akt signaling pathways in the hippocampus and hypothalamus of wistar rats treated with highly palatable food. Nutr. Neurosci. 2012, 15, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Sokolowska, P.; Dejda, A.; Nowak, J.Z. Neuroprotective role of pacap, vip, and phi in the central nervous system. Postepy Hig. Med. Dosw. 2004, 58, 416–427. [Google Scholar]
- Yang, S.; Yang, J.; Yang, Z.; Chen, P.; Fraser, A.; Zhang, W.; Pang, H.; Gao, X.; Wilson, B.; Hong, J.-S.; et al. Pituitary adenylate cyclase-activating polypeptide (pacap) 38 and pacap4–6 are neuroprotective through inhibition of nadph oxidase: Potent regulators of microglia-mediated oxidative stress. J. Pharmacol. Exp. Ther. 2006, 319, 595–603. [Google Scholar] [CrossRef] [Green Version]
- Waschek, J.A. Vip and pacap: Neuropeptide modulators of cns inflammation, injury, and repair. Br. J. Pharmacol. 2013, 169, 512–523. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marzagalli, R.; Leggio, G.M.; Bucolo, C.; Pricoco, E.; Keay, K.A.; Cardile, V.; Castorina, S.; Salomone, S.; Drago, F.; Castorina, A. Genetic blockade of the dopamine d3 receptor enhances hippocampal expression of pacap and receptors and alters their cortical distribution. Neuroscience 2016, 316, 279–295. [Google Scholar] [CrossRef] [PubMed]
- Musumeci, G.; Leggio, G.M.; Marzagalli, R.; Al-Badri, G.; Drago, F.; Castorina, A. Identification of dysregulated microrna networks in schwann cell-like cultures exposed to immune challenge: Potential crosstalk with the protective vip/pacap neuropeptide system. Int. J. Mol. Sci. 2018, 19, 981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nussdorfer, G.G.; Malendowicz, L.K. Role of vip, pacap, and related peptides in the regulation of the hypothalamo-pituitary-adrenal axis. Peptides 1998, 19, 1443–1467. [Google Scholar] [CrossRef]
- Giunta, S.; Castorina, A.; Bucolo, C.; Magro, G.; Drago, F.; D’Agata, V. Early changes in pituitary adenylate cyclase-activating peptide, vasoactive intestinal peptide and related receptors expression in retina of streptozotocin-induced diabetic rats. Peptides 2012, 37, 32–39. [Google Scholar] [CrossRef] [PubMed]
- Castorina, A.; Vogiatzis, M.; Kang, J.W.M.; Keay, K.A. Pacap and vip expression in the periaqueductal grey of the rat following sciatic nerve constriction injury. Neuropeptides 2019, 74, 60–69. [Google Scholar] [CrossRef]
- Sekar, R.; Wang, L.; Chow, B.K. Central control of feeding behavior by the secretin, pacap, and glucagon family of peptides. Front. Endocrinol. 2017, 8, 18. [Google Scholar] [CrossRef] [Green Version]
- Hawke, Z.; Ivanov, T.R.; Bechtold, D.A.; Dhillon, H.; Lowell, B.B.; Luckman, S.M. Pacap neurons in the hypothalamic ventromedial nucleus are targets of central leptin signaling. J. Neurosci. Off. J. Soc. Neurosci. 2009, 29, 14828–14835. [Google Scholar] [CrossRef] [Green Version]
- Castorina, A.; Scuderi, S.; D’Amico, A.G.; Drago, F.; D’Agata, V. Pacap and vip increase the expression of myelin-related proteins in rat schwannoma cells: Involvement of pac1/vpac2 receptor-mediated activation of pi3k/akt signaling pathways. Exp. Cell Res. 2014, 322, 108–121. [Google Scholar] [CrossRef]
- Castorina, A.; Waschek, J.A.; Marzagalli, R.; Cardile, V.; Drago, F. Pacap interacts with pac1 receptors to induce tissue plasminogen activator (tpa) expression and activity in schwann cell-like cultures. PLoS ONE 2015, 10, e0117799. [Google Scholar] [CrossRef]
- Vaudry, D.; Falluel-Morel, A.; Bourgault, S.; Basille, M.; Burel, D.; Wurtz, O.; Fournier, A.; Chow, B.K.C.; Hashimoto, H.; Galas, L.; et al. Pituitary adenylate cyclase-activating polypeptide and its receptors: 20 years after the discovery. Pharmacol. Rev. 2009, 61, 283–357. [Google Scholar] [CrossRef] [PubMed]
- Dore, R.; Iemolo, A.; Smith, K.L.; Wang, X.; Cottone, P.; Sabino, V. Crf mediates the anxiogenic and anti-rewarding, but not the anorectic effects of pacap. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2013, 38, 2160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mabuchi, T.; Shintani, N.; Matsumura, S.; Okuda-Ashitaka, E.; Hashimoto, H.; Muratani, T.; Minami, T.; Baba, A.; Ito, S. Pituitary adenylate cyclase-activating polypeptide is required for the development of spinal sensitization and induction of neuropathic pain. J. Neurosci. 2004, 24, 7283–7291. [Google Scholar] [CrossRef]
- Dejda, A.; Jolivel, V.; Bourgault, S.; Seaborn, T.; Fournier, A.; Vaudry, H.; Vaudry, D. Inhibitory effect of pacap on caspase activity in neuronal apoptosis: A better understanding towards therapeutic applications in neurodegenerative diseases. J. Mol. Neurosci. 2008, 36, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Dejda, A.; Sokolowska, P.; Nowak, J.Z. Neuroprotective potential of three neuropeptides pacap, vip and phi. Pharmacol. Rep. PR 2005, 57, 307–320. [Google Scholar] [PubMed]
- Farnham, M.M.J.; Inglott, M.A.; Pilowsky, P.M. Intrathecal pacap-38 causes increases in sympathetic nerve activity and heart rate but not blood pressure in the spontaneously hypertensive rat. Am. J. Physiol. Heart Circ. Physiol. 2011, 300, H214–H222. [Google Scholar] [CrossRef] [Green Version]
- Said, S.I.; Mutt, V. Isolation from porcine-intestinal wall of a vasoactive octacosapeptide related to secretin and to glucagon. Eur. J. Biochem. 1972, 28, 199–204. [Google Scholar] [CrossRef]
- Said, S.I.; Mutt, V. Polypeptide with broad biological activity: Isolation from small intestine. Science 1970, 169, 1217–1218. [Google Scholar] [CrossRef]
- Besson, J.; Sarrieau, A.; Vial, M.; Marie, J.-C.; Rosselin, G.; Rostene, W. Characterization and autoradiographic distribution of vasoactive intestinal peptide binding sites in the rat central nervous system. Brain Res. 1986, 398, 329–336. [Google Scholar] [CrossRef]
- Martin, J.L.; Dietl, M.M.; Hof, P.R.; Palacios, J.M.; Magistretti, P.J. Autoradiographic mapping of [mono[125i]iodo-tyr10, meto17]vasoactive intestinal peptide binding sites in the rat brain. Neuroscience 1987, 23, 539–565. [Google Scholar] [CrossRef]
- Brenneman, D.E.; Eiden, L.E. Vasoactive intestinal peptide and electrical activity influence neuronal survival. Proc. Natl. Acad. Sci. USA 1986, 83, 1159–1162. [Google Scholar] [CrossRef] [Green Version]
- Yu, R.; Zhang, H.; Huang, L.; Liu, X.; Chen, J. Anti-hyperglycemic, antioxidant and anti-inflammatory effects of vip and a vpac1 agonist on streptozotocin-induced diabetic mice. Peptides 2011, 32, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Verma, A.K.; Manohar, M.; Upparahalli Venkateshaiah, S.; Mishra, A. Neuroendocrine cells derived chemokine vasoactive intestinal polypeptide (vip) in allergic diseases. Cytokine Growth Factor Rev. 2017, 38, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Liao, J.K. A mouse model of diet-induced obesity and insulin resistance. Methods Mol. Biol. 2012, 821, 421–433. [Google Scholar]
- Sclafani, A. Animal models of obesity: Classification and characterization. Int. J. Obes. 1984, 8, 491–508. [Google Scholar]
- Guyenet, S.J.; Nguyen, H.T.; Hwang, B.H.; Schwartz, M.W.; Baskin, D.G.; Thaler, J.P. High-fat diet feeding causes rapid, non-apoptotic cleavage of caspase-3 in astrocytes. Brain Res. 2013, 1512, 97–105. [Google Scholar] [CrossRef] [Green Version]
- Buckman, L.B.; Thompson, M.M.; Moreno, H.N.; Ellacott, K.L. Regional astrogliosis in the mouse hypothalamus in response to obesity. J. Comp. Neurol. 2013, 521, 1322–1333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thaler, J.P.; Yi, C.X.; Schur, E.A.; Guyenet, S.J.; Hwang, B.H.; Dietrich, M.O.; Zhao, X.; Sarruf, D.A.; Izgur, V.; Maravilla, K.R.; et al. Obesity is associated with hypothalamic injury in rodents and humans. J. Clin. Investig. 2012, 122, 153–162. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osborn, L.M.; Kamphuis, W.; Wadman, W.J.; Hol, E.M. Astrogliosis: An integral player in the pathogenesis of Alzheimer’s disease. Prog. Neurobiol. 2016, 144, 121–141. [Google Scholar] [CrossRef] [PubMed]
- Codeluppi, S.; Fernandez-Zafra, T.; Sandor, K.; Kjell, J.; Liu, Q.; Abrams, M.; Olson, L.; Gray, N.S.; Svensson, C.I.; Uhlén, P. Interleukin-6 secretion by astrocytes is dynamically regulated by pi3k-mtor-calcium signaling. PLoS ONE 2014, 9, e92649. [Google Scholar]
- Gao, Y.; Bielohuby, M.; Fleming, T.; Grabner, G.F.; Foppen, E.; Bernhard, W.; Guzmán-Ruiz, M.; Layritz, C.; Legutko, B.; Zinser, E.; et al. Dietary sugars, not lipids, drive hypothalamic inflammation. Mol. Metab. 2017, 6, 897–908. [Google Scholar] [CrossRef] [PubMed]
- Kopelman, P.G. Obesity as a medical problem. Nature 2000, 404, 635. [Google Scholar] [CrossRef]
- Du Plessis, S.S.; Cabler, S.; McAlister, D.A.; Sabanegh, E.; Agarwal, A. The effect of obesity on sperm disorders and male infertility. Nat. Rev. Urol. 2010, 7, 153. [Google Scholar] [CrossRef] [PubMed]
- Aslani, S.; Vieira, N.; Marques, F.; Costa, P.S.; Sousa, N.; Palha, J.A. The effect of high-fat diet on rat’s mood, feeding behavior and response to stress. Transl. Psychiatry 2015, 5, e684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutheil, S.; Ota, K.T.; Wohleb, E.S.; Rasmussen, K.; Duman, R.S. High-fat diet induced anxiety and anhedonia: Impact on brain homeostasis and inflammation. Neuropsychopharmacol. Off. Publ. Am. Coll. Neuropsychopharmacol. 2016, 41, 1874–1887. [Google Scholar] [CrossRef] [Green Version]
- Goshen, I.; Kreisel, T.; Ounallah-Saad, H.; Renbaum, P.; Zalzstein, Y.; Ben-Hur, T.; Levy-Lahad, E.; Yirmiya, R. A dual role for interleukin-1 in hippocampal-dependent memory processes. Psychoneuroendocrinology 2007, 32, 1106–1115. [Google Scholar] [CrossRef]
- Rachal Pugh, C.; Fleshner, M.; Watkins, L.R.; Maier, S.F.; Rudy, J.W. The immune system and memory consolidation: A role for the cytokine il-1β. Neurosci. Biobehav. Rev. 2001, 25, 29–41. [Google Scholar] [CrossRef]
- Hein, A.M.; Stasko, M.R.; Matousek, S.B.; Scott-McKean, J.J.; Maier, S.F.; Olschowka, J.A.; Costa, A.C.S.; O’Banion, M.K. Sustained hippocampal il-1β overexpression impairs contextual and spatial memory in transgenic mice. Brain Behav. Immun. 2010, 24, 243–253. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Kim, S.J.; Son, T.G.; Chan, S.L.; Mattson, M.P. Interferon-gamma is up-regulated in the hippocampus in response to intermittent fasting and protects hippocampal neurons against excitotoxicity. J. Neurosci. Res. 2006, 83, 1552–1557. [Google Scholar] [CrossRef] [PubMed]
- Monteiro, S.; Ferreira, F.M.; Pinto, V.; Roque, S.; Morais, M.; de Sá-Calçada, D.; Mota, C.; Correia-Neves, M.; Cerqueira, J.J. Absence of ifnγ promotes hippocampal plasticity and enhances cognitive performance. Transl. Psychiatry 2016, 6, e707. [Google Scholar] [CrossRef] [PubMed]
- Tsai, S.F.; Wu, H.T.; Chen, P.C.; Chen, Y.W.; Yu, M.; Tzeng, S.F.; Wu, P.H.; Chen, P.S.; Kuo, Y.M. Stress aggravates high-fat-diet-induced insulin resistance via a mechanism that involves the amygdala and is associated with changes in neuroplasticity. Neuroendocrinology 2018, 107, 147–157. [Google Scholar] [CrossRef]
- Anderson, R.J.; Freedland, K.E.; Clouse, R.E.; Lustman, P.J. The prevalence of comorbid depression in adults with diabetes: A meta-analysis. Diabetes Care 2001, 24, 1069–1078. [Google Scholar] [CrossRef] [Green Version]
- Arcego, D.M.; Krolow, R.; Lampert, C.; Toniazzo, A.P.; Berlitz, C.; Lazzaretti, C.; Schmitz, F.; Rodrigues, A.F.; Wyse, A.T.; Dalmaz, C. Early life adversities or high fat diet intake reduce cognitive function and alter bdnf signaling in adult rats: Interplay of these factors changes these effects. Int. J. Dev. Neurosci. Off. J. Int. Soc. Dev. Neurosci. 2016, 50, 16–25. [Google Scholar] [CrossRef] [PubMed]
- De Pablos, R.M.; Villaran, R.F.; Arguelles, S.; Herrera, A.J.; Venero, J.L.; Ayala, A.; Cano, J.; Machado, A. Stress increases vulnerability to inflammation in the rat prefrontal cortex. J. Neurosci. Off. J. Soc. Neurosci. 2006, 26, 5709–5719. [Google Scholar] [CrossRef]
- Prud’homme, G.; Glinka, Y.; Wang, Q. Gaba exerts anti-inflammatory and immunosuppressive effects (p5175). J. Immunol. 2013, 190, 68.15. [Google Scholar]
- McEwen, B.S.; Morrison, J.H. Brain on stress: Vulnerability and plasticity of the prefrontal cortex over the life course. Neuron 2013, 79, 16–29. [Google Scholar] [CrossRef] [Green Version]
- Curtin, N.M.; Boyle, N.T.; Mills, K.H.; Connor, T.J. Psychological stress suppresses innate ifn-gamma production via glucocorticoid receptor activation: Reversal by the anxiolytic chlordiazepoxide. Brain Behav. Immun. 2009, 23, 535–547. [Google Scholar] [CrossRef]
- Vega-Torres, J.D.; Haddad, E.; Lee, J.B.; Kalyan-Masih, P.; Maldonado George, W.I.; López Pérez, L.; Piñero Vázquez, D.M.; Arroyo Torres, Y.; Santiago Santana, J.M.; Obenaus, A.; et al. Exposure to an obesogenic diet during adolescence leads to abnormal maturation of neural and behavioral substrates underpinning fear and anxiety. Brain Behav. Immun. 2018, 70, 96–117. [Google Scholar] [CrossRef]
- Duan, Y.; Zhang, R.; Zhang, M.; Sun, L.; Dong, S.; Wang, G.; Zhang, J.; Zhao, Z. Metformin inhibits food intake and neuropeptide y gene expression in the hypothalamus. Neural Regen. Res. 2013, 8, 2379–2388. [Google Scholar] [PubMed]
- Du, R.W.; Bu, W.G. Metformin improves depressive-like symptoms in mice via inhibition of peripheral and central nf-κb-nlrp3 inflammation activation. Exp. Brain Res. 2020, 238, 2549–2556. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.; Wang, K.; Ma, C.; Wang, X.; Gong, Z.; Zhang, R.; Zang, D.; Cheng, Y. Evaluation of metformin on cognitive improvement in patients with non-dementia vascular cognitive impairment and abnormal glucose metabolism. Front. Aging Neurosci. 2018, 10, 227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ying, M.A.; Maruschak, N.; Mansur, R.; Carvalho, A.F.; Cha, D.S.; McIntyre, R.S. Metformin: Repurposing opportunities for cognitive and mood dysfunction. CNS Neurol. Disord. Drug Targets 2014, 13, 1836–1845. [Google Scholar] [CrossRef] [PubMed]
- Cianciulli, A.; Calvello, R.; Porro, C.; Trotta, T.; Salvatore, R.; Panaro, M.A. Pi3k/akt signalling pathway plays a crucial role in the anti-inflammatory effects of curcumin in lps-activated microglia. Int. Immunopharmacol. 2016, 36, 282–290. [Google Scholar] [CrossRef]
- Vergadi, E.; Ieronymaki, E.; Lyroni, K.; Vaporidi, K.; Tsatsanis, C. Akt signaling pathway in macrophage activation and m1/m2 polarization. J. Immunol. 2017, 198, 1006–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kidd, L.B.; Schabbauer, G.A.; Luyendyk, J.P.; Holscher, T.D.; Tilley, R.E.; Tencati, M.; Mackma, N. Insulin activation of the phosphatidylinositol 3-kinase/protein kinase b (akt) pathway reduces lipopolysaccharide-induced inflammation in mice. J. Pharmacol. Exp. Ther. 2008, 326, 348–353. [Google Scholar] [CrossRef] [Green Version]
- Piggins, H.D.; Stamp, J.A.; Burns, J.; Rusak, B.; Semba, K. Distribution of pituitary adenylate cyclase activating polypeptide (pacap) immunoreactivity in the hypothalamus and extended amygdala of the rat. J. Comp. Neurol. 1996, 376, 278–294. [Google Scholar] [CrossRef]
- Rodriguez, E.M.; Blazquez, J.L.; Guerra, M. The design of barriers in the hypothalamus allows the median eminence and the arcuate nucleus to enjoy private milieus: The former opens to the portal blood and the latter to the cerebrospinal fluid. Peptides 2010, 31, 757–776. [Google Scholar] [CrossRef]
- Rostène, W.H.; Léránth, C.; Maletti, M.; Mezey, É.; Besson, J.; Eiden, L.E.; Rosselin, G.; Palkovits, M. Distribution of vasoactive intestinal peptide (vip) following various brain transections in the rat by radioimmunoassay and electronmicroscopic immunocytochemistry. Neuropeptides 1982, 2, 337–350. [Google Scholar] [CrossRef]
- Dalcik, H.; Phelps, C.J. Median eminence-afferent vasoactive intestinal peptide (vip) neurons in the hypothalamus: Localization by simultaneous tract tracing and immunocytochemistry. Peptides 1993, 14, 1059–1066. [Google Scholar] [CrossRef]
- Kingsbury, M.A. New perspectives on vasoactive intestinal polypeptide as a widespread modulator of social behavior. Curr. Opin. Behav. Sci. 2015, 6, 139–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammack, S.E.; Roman, C.W.; Lezak, K.R.; Kocho-Shellenberg, M.; Grimmig, B.; Falls, W.A.; Braas, K.; May, V. Roles for pituitary adenylate cyclase-activating peptide (pacap) expression and signaling in the bed nucleus of the stria terminalis (bnst) in mediating the behavioral consequences of chronic stress. J. Mol. Neurosci. 2010, 42, 327–340. [Google Scholar] [CrossRef] [Green Version]
- Smith, S.M.; Vale, W.W. The role of the hypothalamic-pituitary-adrenal axis in neuroendocrine responses to stress. Dialogues Clin. Neurosci. 2006, 8, 383–395. [Google Scholar] [PubMed]
- Matsuda, K.; Maruyama, K. Regulation of feeding behavior by pituitary adenylate cyclase-activating polypeptide (pacap) and vasoactive intestinal polypeptide (vip) in vertebrates. Peptides 2007, 28, 1761–1766. [Google Scholar] [CrossRef]
- Resch, J.M.; Maunze, B.; Gerhardt, A.K.; Magnuson, S.K.; Phillips, K.A.; Choi, S. Intrahypothalamic pituitary adenylate cyclase-activating polypeptide regulates energy balance via site-specific actions on feeding and metabolism. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E1452–E1463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carniglia, L.; Ramírez, D.; Durand, D.; Saba, J.; Turati, J.; Caruso, C.; Scimonelli, T.N.; Lasaga, M. Neuropeptides and microglial activation in inflammation, pain, and neurodegenerative diseases. Mediat. Inflamm. 2017, 2017, 5048616. [Google Scholar] [CrossRef] [PubMed]
- Matsuyama, S.; Matsumoto, A.; Hashimoto, H.; Shintani, N.; Baba, A. Impaired long-term potentiation in vivo in the dentate gyrus of pituitary adenylate cyclase-activating polypeptide (pacap) or pacap type 1 receptor-mutant mice. Neuroreport 2003, 14, 2095–2098. [Google Scholar] [CrossRef]
- Jinno, S.; Kosaka, T. Patterns of expression of neuropeptides in gabaergic nonprincipal neurons in the mouse hippocampus: Quantitative analysis with optical disector. J. Comp. Neurol. 2003, 461, 333–349. [Google Scholar] [CrossRef] [PubMed]
- Owens, D.F.; Kriegstein, A.R. Is there more to gaba than synaptic inhibition? Nat. Rev. Neurosci. 2002, 3, 715–727. [Google Scholar] [CrossRef]
- Sandoval-Salazar, C.; Ramírez-Emiliano, J.; Trejo-Bahena, A.; Oviedo-Solís, C.I.; Solís-Ortiz, M.S. A high-fat diet decreases gaba concentration in the frontal cortex and hippocampus of rats. Biol. Res. 2016, 49, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Restivo, M.R.; McKinnon, M.C.; Frey, B.N.; Hall, G.B.; Taylor, V.H. Effect of obesity on cognition in adults with and without a mood disorder: Study design and methods. BMJ Open 2016, 6, e009347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meloni, E.G.; Venkataraman, A.; Donahue, R.J.; Carlezon, W.A. Bi-directional effects of pituitary adenylate cyclase-activating polypeptide (pacap) on fear-related behavior and c-fos expression after fear conditioning in rats. Psychoneuroendocrinology 2016, 64, 12–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelley, A.E. Ventral striatal control of appetitive motivation: Role in ingestive behavior and reward-related learning. Neurosci. Biobehav. Rev. 2004, 27, 765–776. [Google Scholar] [CrossRef]
- Tachibana, T.; Saito, S.; Tomonaga, S.; Takagi, T.; Saito, E.S.; Boswell, T.; Furuse, M. Intracerebroventricular injection of vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide inhibits feeding in chicks. Neurosci. Lett. 2003, 339, 203–206. [Google Scholar] [CrossRef]
- Matsuda, K.; Maruyama, K.; Nakamachi, T.; Miura, T.; Uchiyama, M.; Shioda, S. Inhibitory effects of pituitary adenylate cyclase-activating polypeptide (pacap) and vasoactive intestinal peptide (vip) on food intake in the goldfish, carassius auratus. Peptides 2005, 26, 1611–1616. [Google Scholar] [CrossRef]
- Leceta, J.; Gomariz, R.P.; Martinez, C.; Abad, C.; Ganea, D.; Delgado, M. Receptors and transcriptional factors involved in the anti-inflammatory activity of vip and pacap. Ann. N. Y. Acad. Sci. 2000, 921, 92–102. [Google Scholar] [CrossRef] [PubMed]
- Yarchoan, M.; Arnold, S.E. Repurposing diabetes drugs for brain insulin resistance in alzheimer disease. Diabetes 2014, 63, 2253–2261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cetinkalp, S.; Simsir, I.Y.; Ertek, S. Insulin resistance in brain and possible therapeutic approaches. Curr. Vasc. Pharmacol. 2014, 12, 553–564. [Google Scholar] [CrossRef] [PubMed]
- Moody, T.W.; Ito, T.; Osefo, N.; Jensen, R.T. Vip and pacap. Recent insights into their functions/roles in physiology and disease from molecular and genetic studies. Curr. Opin. Endocrinol. Diabetes Obes. 2011, 18, 61–67. [Google Scholar] [CrossRef] [Green Version]
- Ashabi, G.; Khodagholi, F.; Khalaj, L.; Goudarzvand, M.; Nasiri, M. Activation of amp-activated protein kinase by metformin protects against global cerebral ischemia in male rats: Interference of ampk/pgc-1alpha pathway. Metab. Brain Dis. 2014, 29, 47–58. [Google Scholar] [CrossRef] [PubMed]
- Kappe, C.; Zhang, Q.; Nyström, T.; Sjöholm, Å. Effects of high-fat diet and the anti-diabetic drug metformin on circulating glp-1 and the relative number of intestinal l-cells. Diabetol. Metab. Syndr. 2014, 6, 70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karise, I.; Ornellas, F.; Barbosa-da-Silva, S.; Matsuura, C.; del Sol, M.; Aguila, M.B.; Mandarim-de-Lacerda, C.A. Liver and metformin: Lessons of a fructose diet in mice. Biochim. Open 2017, 4, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Martin-Montalvo, A.; Mercken, E.M.; Mitchell, S.J.; Palacios, H.H.; Mote, P.L.; Scheibye-Knudsen, M.; Gomes, A.P.; Ward, T.M.; Minor, R.K.; Blouin, M.-J.; et al. Metformin improves healthspan and lifespan in mice. Nat. Commun. 2013, 4, 2192. [Google Scholar] [CrossRef] [PubMed]
- Kita, Y.; Takamura, T.; Misu, H.; Ota, T.; Kurita, S.; Takeshita, Y.; Uno, M.; Matsuzawa-Nagata, N.; Kato, K.-i.; Ando, H.; et al. Metformin prevents and reverses inflammation in a non-diabetic mouse model of nonalcoholic steatohepatitis. PLoS ONE 2012, 7, e43056. [Google Scholar] [CrossRef] [PubMed]
- Fu, L.; Bruckbauer, A.; Li, F.; Cao, Q.; Cui, X.; Wu, R.; Shi, H.; Zemel, M.B.; Xue, B. Interaction between metformin and leucine in reducing hyperlipidemia and hepatic lipid accumulation in diet-induced obese mice. Metabolism 2015, 64, 1426–1434. [Google Scholar] [CrossRef] [PubMed]
- McGrath, K.C.; Li, X.H.; Whitworth, P.T.; Kasz, R.; Tan, J.T.; McLennan, S.V.; Celermajer, D.S.; Barter, P.J.; Rye, K.A.; Heather, A.K. High density lipoproteins improve insulin sensitivity in high-fat diet-fed mice by suppressing hepatic inflammation. J. Lipid Res. 2014, 55, 421–430. [Google Scholar] [CrossRef] [Green Version]
- Franklin, K.; Paxinos, G. Atlas the Mouse Brain in Stereotaxic Coordinates; Elsevier: Amsterdam, The Netherlands, 2008. [Google Scholar]
- Schmittgen, T.D.; Livak, K.J. Analyzing real-time pcr data by the comparative c(t) method. Nat. Protoc. 2008, 3, 1101–1108. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene (Ref. Seq.) | Primers | Location of Primers | Tm (°C) | Length (bp) |
|---|---|---|---|---|
| PACAP peptide (NM_009625.2) | 5′-CTGCGTGACGCTTACGCCCT-3′ 3′-CCTAGGTTCTCCCCCGCGCC-5′ | 401 552 | 59.77 61.02 | 152 |
| VIP peptide (NM_011702.2) | 5′-TGGCAAACGAATCAGCAGCAGCA-3′ 3′-AGCCATTTGCTTTCTGAGGCGGG-5′ | 487 592 | 60.18 59.99 | 106 |
| PAC1 receptor (NM_007407.3) | 5′-CAGTCCCCAGACATGGGAGGCA-3′ 3′-AGCGGGCCAGCCGTAGAGTA-5′ | 1474 1612 | 59.92 59.40 | 139 |
| VPAC1 receptor (NM_011703.4) | 5′-TCAATGGCGAGGTGCAGGCAG-3′ 3′-TGTGTGCTGCACGAGACGCC-5′ | 1311 1437 | 59.71 60.25 | 127 |
| VPAC2 receptor (NM_009511.2) | 5′-GCGTCGGTGGTGCTGACCTG-3′ 3′-ACACCGCTGCAGGCTCTCTGAT-5′ | 127 281 | 60.32 60.24 | 155 |
| Interleukin 1-α (NM_010554.4) | 5′-ACGTCAAGCAACGGGAAGAT-3′ 3′- AAGGTGCTGATCTGGGTTGG-5′ | 229 352 | 59.97 59.96 | 124 |
| Interleukin 1-β (NM_008361.4) | 5′-GCTACCTGTGTCTTTCCCGT-3′ 3′- CATCTCGGAGCCTGTAGTGC-5′ | 293 456 | 59.68 60.25 | 164 |
| Interleukin 6 (NM_031168.2) | 5′-CCCCAATTTCCAATGCTCTCC-3′ 3′-CGCACTAGGTTTGCCGAGTA-5′ | 578 718 | 59.24 60.11 | 141 |
| Interleukin 10 (NM_010548.2) | 5′-GCATGGCCCAGAAATCAAGG-3′ 3′-GAGAAATCGATGACAGCGCC-5′ | 367 457 | 59.54 59.42 | 91 |
| Interferon-γ (NM_008337.4) | 5′-AGCAAGGCGAAAAAGGATGC-3′ 3′-TCATTGAATGCTTGGCGCTG-5′ | 425 507 | 59.76 59.83 | 83 |
| Tumor necrosis factor (NM_013693.3) | 5′-ATGGCCTCCCTCTCATCAGT-3′ 3′-TTTGCTACGACGTGGGCTAC-5′ | 364 460 | 60.03 60.39 | 97 |
| Monocyte chemoattractant protein 1 (NM_011333) | 5′-TGACCCCAAGAAGGAATGGG-3′ 3′-ACCTTAGGGCAGATGCAGTT-5′ | 313 416 | 59.30 59.00 | 104 |
| Ribosomal protein S18 (NM_011296.2) | 5′-CCCTGAGAAGTTCCAGCACA-3′ 3′-GGTGAGGTCGATGTCTGCTT-5′ | 36 180 | 59.60 59.75 | 145 |
| Glial Fibrillary Acidic Protein (NM_001131020.1) | 5’-GCGAAGAAAACCGCATCACC-3′ 3′-TCTGGTGAGCCTGTATTGGGA-5′ | 1189 1338 | 60 61 | 150 |
| Ionized calcium-Binding Adapter molecule 1 (NM_001361501.1) | 5′-GCTTTTGGACTGCTGAAGGC-3′ 3′- GTTTGGACGGCAGATCCTCA-5′ | 402 515 | 60.04 61.45 | 114 |
| Cluster of Differentiation 68 (NM_001291058.1) | 5′-CTCCCACCACAAATGGCACT-3′ 3′-CTTGGACCTTGGACTAGGCG-5′ | 386 480 | 60.54 60.11 | 95 |
| Antibody Name | Cat. # | Molecular Weight (kDa) | Dilution |
|---|---|---|---|
| Pituitary Adenylate Cyclase-Activating Peptide (PACAP) | GTX37576 | 18 | 1/500 |
| Vasoactive Intestinal Polypeptide (VIP) | GTX129461 | 18 | 1/300 |
| Pituitary Adenylate Cyclase 1 Receptor (PAC1) | GTX30026 | 53 | 1/1000 |
| Vasoactive Intestinal Polypeptide Type-1 Receptor (VPAC1) | LS-C177415 | 49 | 1/1000 |
| Vasoactive Intestinal Polypeptide Type-2 Receptor (VPAC2) | ab28624 | 50–60 | 1/1000 |
| Glial Fibrillary Acidic Protein (Iba1) | GTX100042 | 15 | 1/500 |
| inducible Nitric Oxide Synthase (iNOS) | GTX60599 | 32 | 1/500 |
| Glial Fibrillary Acidic Protein (GFAP) | G4546 | 50 | 1/500 |
| Phosphorylated protein kinase B (p-Akt) | 4060 Ser473 | 60 | 1/1000 |
| Protein Kinase B (Akt) | 4691 C67E7 | 60 | 1/1000 |
| Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) | VPA00187 | 37 | 1/1500 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mandwie, M.; Karunia, J.; Niaz, A.; Keay, K.A.; Musumeci, G.; Rennie, C.; McGrath, K.; Al-Badri, G.; Castorina, A. Metformin Treatment Attenuates Brain Inflammation and Rescues PACAP/VIP Neuropeptide Alterations in Mice Fed a High-Fat Diet. Int. J. Mol. Sci. 2021, 22, 13660. https://doi.org/10.3390/ijms222413660
Mandwie M, Karunia J, Niaz A, Keay KA, Musumeci G, Rennie C, McGrath K, Al-Badri G, Castorina A. Metformin Treatment Attenuates Brain Inflammation and Rescues PACAP/VIP Neuropeptide Alterations in Mice Fed a High-Fat Diet. International Journal of Molecular Sciences. 2021; 22(24):13660. https://doi.org/10.3390/ijms222413660
Chicago/Turabian StyleMandwie, Mawj, Jocelyn Karunia, Aram Niaz, Kevin A. Keay, Giuseppe Musumeci, Claire Rennie, Kristine McGrath, Ghaith Al-Badri, and Alessandro Castorina. 2021. "Metformin Treatment Attenuates Brain Inflammation and Rescues PACAP/VIP Neuropeptide Alterations in Mice Fed a High-Fat Diet" International Journal of Molecular Sciences 22, no. 24: 13660. https://doi.org/10.3390/ijms222413660