
A Lumenal Loop Associated with Catalytic Asymmetry in Plant Vacuolar H+-Translocating Pyrophosphatase


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
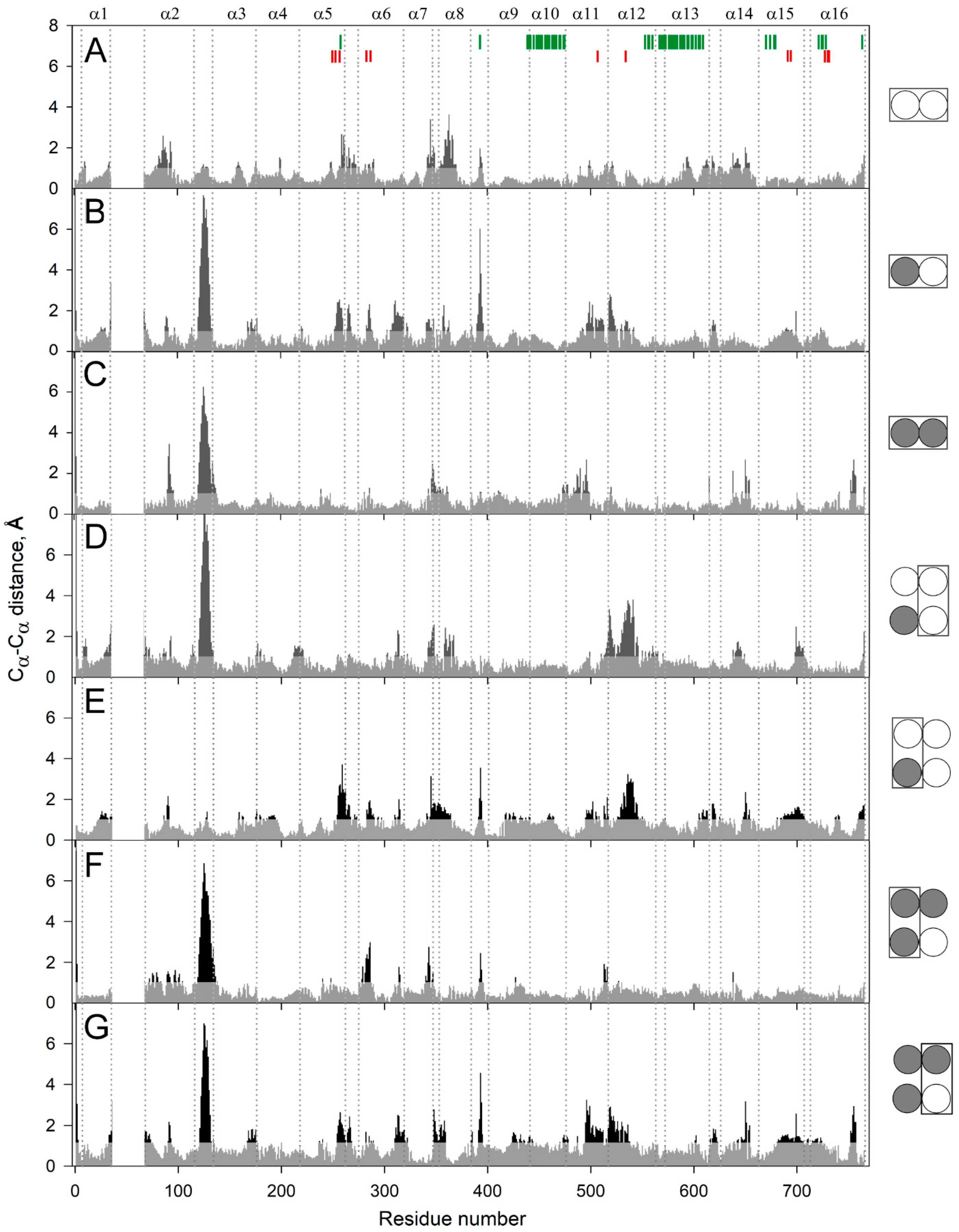
2.1. Molecular Dynamics Simulations of mPPase Dimer
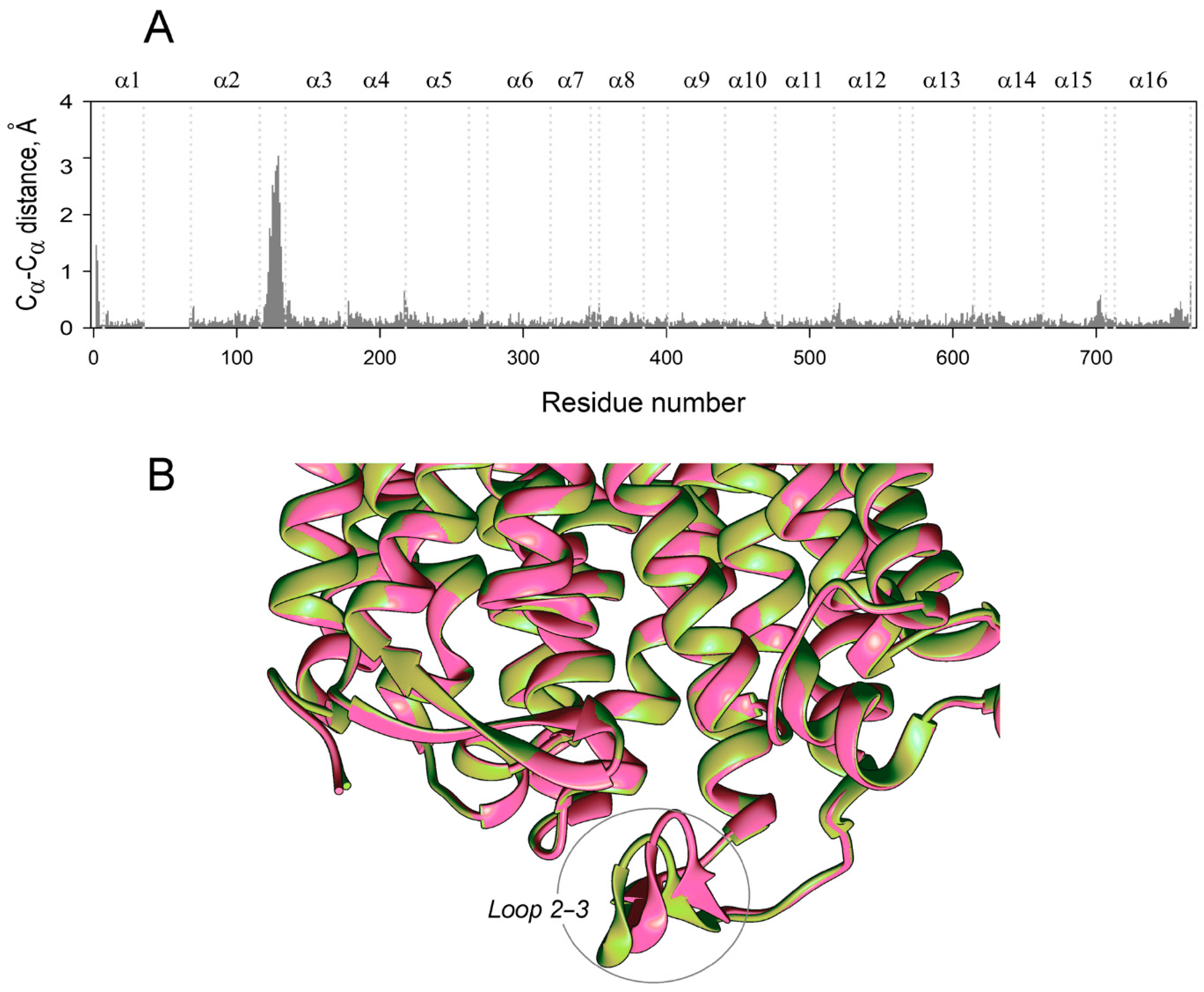
2.2. The Structural Elements Demonstrating Asymmetry in Vr-mPPase
2.3. Loop 2–3 Structures in Various Vr-mPPase Complexes
2.4. The Interactions Stabilizing Loop 2–3 Positions
2.5. Loop 2–3 Conservation in mPPases
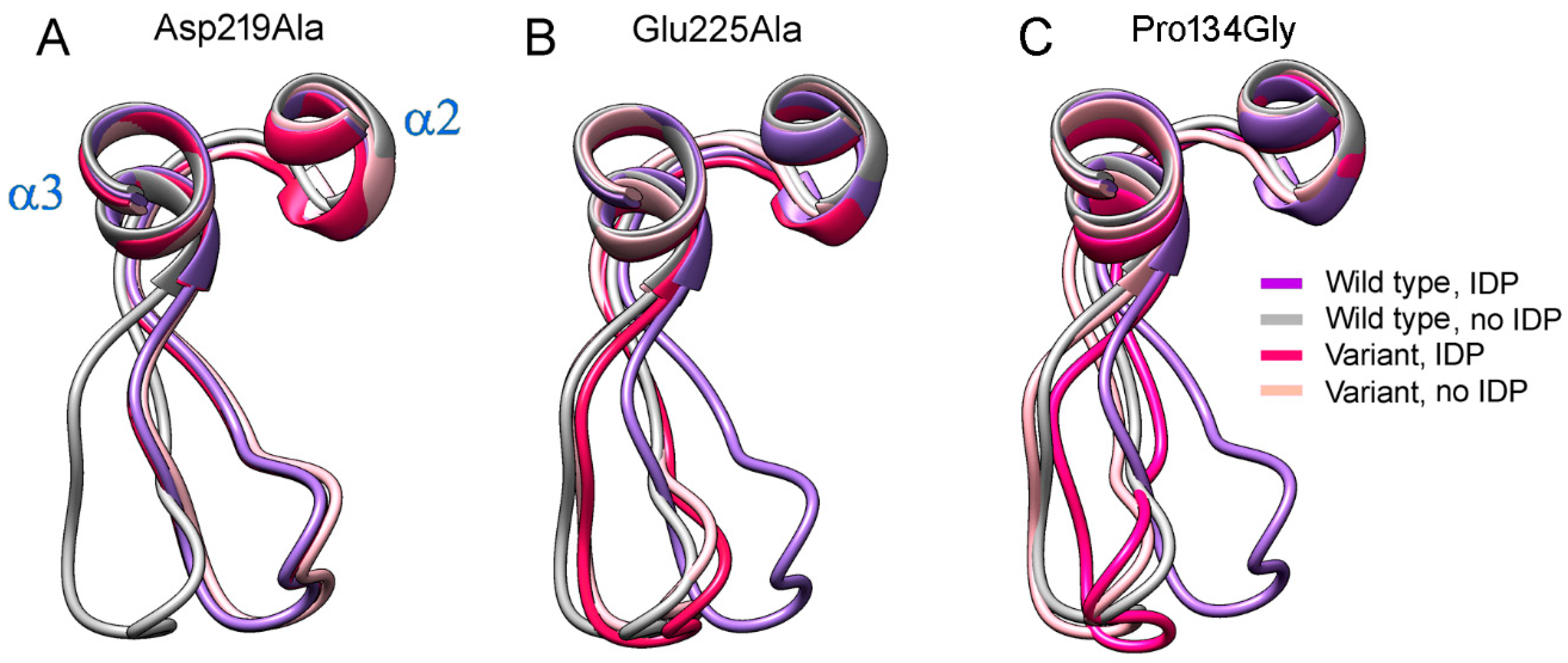
2.6. Mutations in Loop 2–3 and Its Interacting Partners
2.7. Water Accessibility in Structures with Different Loop 2-3 Conformations
3. Discussion
3.1. Structural Asymmetry in Vr-mPPase Dimer—A Myth or Reality?
3.2. Loop 2–3 and Other Asymmetry-Related Elements in mPPase
3.3. A Possible Role of Asymmetry in the Coupling Mechanism of mPPase
3.4. Loop 2–3 Role in mPPase Asymmetry and Functioning
4. Methods
4.1. Structure Preparation for Calculations
4.2. Molecular Dynamics Simulations
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Baltscheffsky, H.; Von Stedingk, L.V.; Heldt, H.W.; Klingenberg, M. Inorganic pyrophosphate: Formation in bacterial photophosphorylation. Science 1966, 153, 1120–1122. [Google Scholar] [CrossRef] [PubMed]
- Drozdowicz, Y.M.; Rea, P.A. Vacuolar H+ pyrophosphatases: From the evolutionary backwaters into the mainstream. Trends Plant Sci. 2001, 6, 206–211. [Google Scholar] [CrossRef]
- Serrano, A.; Pérez-Castiñeira, J.; Baltscheffsky, M.; Baltscheffsky, H. H+-PPases: Yesterday, today and tomorrow. IUBMB Life 2007, 59, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Baykov, A.A.; Malinen, A.M.; Luoto, H.H.; Lahti, R. Pyrophosphate-fueled Na+ and H+ transport in prokaryotes. Microbiol. Mol. Biol. Rev. 2013, 77, 267–276. [Google Scholar] [CrossRef] [Green Version]
- Tsai, J.Y.; Kellosalo, J.; Sun, Y.J.; Goldman, A. Proton/sodium pumping pyrophosphatases: The last of the primary ion pumps. Curr. Opin. Struct. Biol. 2014, 27, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Segami, S.; Asaoka, M.; Kinoshita, S.; Fukuda, M.; Nakanishi, Y.; Maeshima, M. Biochemical, structural and physiological characteristics of vacuolar H+-pyrophosphatase. Plant Cell. Physiol. 2018, 59, 1300–1308. [Google Scholar] [CrossRef]
- Gutiérrez-Luna, F.M.; Hernandez-Dominguez, E.E.; Gabriela Valencia-Turcotte, L.G.; Rodríguez-Sotres, R. Pyrophosphate and pyrophosphatases in plants, their involvement in stress responses and their possible relationship to secondary metabolism. Plant Sci. 2018, 267, 11–19. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.M.; Tsai, J.Y.; Hsiao, C.D.; Huang, Y.T.; Chiu, C.L.; Liu, M.H.; Tung, J.Y.; Liu, T.H.; Pan, R.L.; Sun, Y.J. Crystal structure of a membrane-embedded H+-translocating pyrophosphatase. Nature 2012, 484, 399–403. [Google Scholar] [CrossRef] [PubMed]
- Kellosalo, J.; Kajander, T.; Kogan, K.; Pokharel, K.; Goldman, A. The structure and catalytic cycle of a sodium-pumping pyrophosphatase. Science 2012, 337, 473–476. [Google Scholar] [CrossRef] [PubMed]
- Calisto, F.; Sousa, F.M.; Sena, F.V.; Refojo, P.N.; Pereira, M.M. Mechanisms of energy transduction by charge translocating membrane proteins. Chem. Rev. 2021, 121, 1804–1844. [Google Scholar] [CrossRef]
- Li, K.M.; Wilkinson, C.; Kellosalo, J.; Tsai, J.Y.; Kajander, T.; Jeuken, L.J.C.; Sun, Y.J.; Goldman, A. Membrane pyrophosphatases from Thermotoga maritima and Vigna radiata suggest a conserved coupling mechanism. Nat. Commun. 2016, 7, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baykov, A.A. Energy coupling in cation-pumping pyrophosphatase—Back to Mitchell. Front. Plant Sci. 2020, 11, 107. [Google Scholar] [CrossRef] [Green Version]
- Luoto, H.H.; Nordbom, E.; Malinen, A.M.; Baykov, A.A.; Lahti, R. Evolutionarily divergent, Na+-regulated H+-transporting membrane-bound pyrophosphatases. Biochem. J. 2015, 467, 281–291. [Google Scholar] [CrossRef] [PubMed]
- Artukka, E.; Luoto, H.H.; Baykov, A.A.; Lahti, R.; Malinen, A.M. Role of the potassium/lysine cationic center in catalysis and functional asymmetry in membrane-bound pyrophosphatases. Biochem. J. 2018, 475, 1141–1158. [Google Scholar] [CrossRef]
- Anashkin, V.A.; Malinen, A.M.; Bogachev, A.V.; Baykov, A.A. Catalytic asymmetry in homodimeric H+-pumping membrane pyrophosphatase demonstrated by non-hydrolyzable pyrophosphate analogs. Int. J. Mol. Sci. 2021, 22, 9820. [Google Scholar] [CrossRef] [PubMed]
- Leigh, R.A.; Pope, A.J.; Jennings, I.R.; Sanders, D. Kinetics of the vacuolar H+-pyrophosphatase. The roles of magnesium, pyrophosphate, and their complexes as substrates, activators, and inhibitors. Plant Physiol. 1992, 100, 1698–1705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidilaseris, K.; Kiriazis, A.; Turku, A.; Khattab, A.; Johansson, N.G.; Leino, T.O.; Kiuru, P.S.; Boije af Gennäs, G.; Meri, S.; Yli-Kauhaluoma, J.; et al. Asymmetry in catalysis by Thermotoga maritima membrane bound pyrophosphatase demonstrated by a nonphosphorus allosteric inhibitor. Sci. Adv. 2019, 5, eaav7574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmes, A.O.M.; Kalli, A.C.; Goldman, A. The function of membrane integral pyrophosphatases from whole organism to single molecule. Front. Mol. Biosci. 2019, 6, 132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shah, N.R.; Wilkinson, C.; Harborne, S.P.; Turku, A.; Li, K.M.; Sun, Y.J.; Sarah Harris, S.; Goldman, A. Insights into the mechanism of membrane pyrophosphatases by combining experiment and computer simulation. Struct. Dyn. 2017, 4, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belogurov, G.A.; Lahti, R. A lysine substitute for K+: A460K mutation eliminates K+ dependence in H+-pyrophosphatase of Carboxydothermus hydrogenoformans. J. Biol. Chem. 2002, 277, 49651–49654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Drozdowicz, Y.M.; Kissinger, J.C.; Rea, P.A. AVP2, a sequence-divergent, K+-insensitive H+-translocating inorganic pyrophosphatase from Arabidopsis. Plant Physiol. 2000, 123, 353–362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fuglsang, A.T.; Paez-Valencia, J.; Gaxiola, R.A. Plant proton pumps: Regulatory circuits involving H+-ATPase and H+-PPase. In Transporters and Pumps in Plant Signaling; Geisler, M., Venema, K., Eds.; Springer: Berlin/Heidelberg, Germany, 2011; pp. 39–64. [Google Scholar]
- Asaoka, M.; Segami, S.; Maeshima, M. Identification of the critical residues for the function of vacuolar H+-pyrophosphatase by mutational analysis based on the 3D structure. J. Biochem. 2014, 156, 333–344. [Google Scholar] [CrossRef]
- Baykov, A.A.; Bakuleva, N.P.; Rea, P.A. Steady-state kinetics of substrate hydrolysis by vacuolar H+-pyrophosphatase. A simple three-state model. Eur. J. Biochem. 1993, 217, 755–762. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.Y.; Tang, K.Z.; Li, K.M.; Hsu, B.L.; Chiang, Y.W.; Goldman, A.; Sun, Y.-J. Roles of the hydrophobic gate and exit channel in Vigna radiata pyrophosphatase ion translocation. J. Mol. Biol. 2019, 431, 1619–1632. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsiao, Y.Y.; Pan, Y.J.; Hsu, S.H.; Huang, Y.T.; Liu, T.H.; Lee, C.H.; Liu, P.F.; Chang, W.C.; Wang, Y.K.; Chien, L.F.; et al. Functional roles of arginine residues in mung bean vacuolar H+-pyrophosphatase. Biochim. Biophys. Acta 2007, 1767, 965–973. [Google Scholar] [CrossRef] [PubMed]
- Van, R.C.; Pan, Y.J.; Hsu, S.H.; Huang, Y.T.; Hsiao, Y.Y.; Pan, R.L. Role of transmembrane segment 5 of the plant vacuolar H+-pyrophosphatase. Biochim. Biophys. Acta 2005, 1709, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Pellegrini-Calace, M.; Maiwald, T.; Thornton, J.M. PoreWalker: A novel tool for the identification and characterization of channels in transmembrane proteins from their three-dimensional structure. PLoS Comput. Biol. 2009, 5, e1000440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, Y.D.; Huang, Y.F.; Pan, Y.J.; Huang, L.K.; Liao, Y.Y.; Lin, W.H.; Liu, T.Y.; Lee, C.H.; Pan, R.L. Regulation of H+-pyrophosphatase by 14-3-3 proteins from Arabidopsis thaliana. J. Membr. Biol. 2018, 251, 263–276. [Google Scholar] [CrossRef]
- Swapna, L.S.; Srikeerthana, K.; Srinivasan, N. Extent of structural asymmetry in homodimeric proteins: Prevalence and relevance. PLoS ONE 2012, 7, e36688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaber, A.; Pavšič, M. Modeling and structure determination of homo-oligomeric proteins: An overview of challenges and current approaches. Int. J. Mol. Sci. 2021, 22, 9081. [Google Scholar] [CrossRef]
- Martinoia, E. Vacuolar transporters—Companions on a longtime journey. Plant Physiol. 2018, 176, 1384–1407. [Google Scholar] [CrossRef] [PubMed]
- Rea, P.A.; Sanders, D. Tonoplast energization—2 H+ pumps, one membrane. Physiol. Plant. 1987, 71, 131–141. [Google Scholar] [CrossRef]
- Gaxiola, R.A.; Palmgren, M.G.; Schumacher, K. Plant proton pumps. FEBS Lett. 2007, 581, 2204–2214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holzheu, P.; Krebs, M.; Larasati, C.; Schumacher, K.; Kummer, U. An integrative view on vacuolar pH homeostasis in Arabidopsis thaliana: Combining mathematical modeling and Experimentation. Plant J. 2021, 106, 1541–1556. [Google Scholar] [CrossRef] [PubMed]
- Johansson, N.G.; Turku, A.; Vidilaseris, K.; Dreano, L.; Khattab, A.; Pérez, D.A.; Wilkinson, A.; Zhang, Y.; Tamminen, M.; Grazhdankin, E.; et al. Discovery of membrane-bound pyrophosphatase inhibitors derived from an isoxazole fragment. ACS Med. Chem. Lett. 2020, 11, 605–610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scholz-Starke, J.; Primo, C.; Yang, J.; Kandel, R.; Gaxiola, R.A.; Hirschi, K.D. The flip side of the Arabidopsis type I proton-pumping pyrophosphatase (AVP1): Using a transmembrane H+ gradient to synthesize pyrophosphate. J. Biol. Chem. 2019, 294, 1290–1299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Primo, C.; Pizzio, G.A.; Yang, J.; Gaxiola, R.A.; Scholz-Starke, J.; Hirschi, K.D. Plant proton pumping pyrophosphatase: The potential for its pyrophosphate synthesis activity to modulate plant growth. Plant Biol. 2019, 21, 989–996. [Google Scholar] [CrossRef] [PubMed]
- Igamberdiev, A.U.; Kleczkowski, L.A. Pyrophosphate as an alternative energy currency in plants. Biochem. J. 2021, 478, 1515–1524. [Google Scholar] [CrossRef]
- Segami, S.; Tomoyama, T.; Sakamoto, S.; Gunji, S.; Fukuda, F.; Kinoshita, S.; Mitsuda, N.; Ferjani, A.; Maeshima, M. Vacuolar H+-pyrophosphatase and cytosolic soluble pyrophosphatases cooperatively regulate pyrophosphate levels in Arabidopsis thaliana. Plant Cell 2018, 30, 1040–1061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Webb, B.; Sali, A. Comparative protein structure modeling using Modeller. Curr. Protoc. Bioinform. 2016, 54, 5.6.1–5.6.37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera: A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.; Hitzenberger, M.; Rieger, M.; Kern, N.R.; Zacharias, M.; Im, W. CHARMM-GUI supports the Amber force fields. J. Chem. Phys. 2020, 153, 035103. [Google Scholar] [CrossRef]
- Case, D.A.; Berryman, J.T.; Betz, R.M.; Cerutti, D.S.; Cheatham, T.E., III; Darden, T.A.; Duke, R.E.; Giese, T.J.; Gohlke, H.; Goetz, A.W.; et al. AMBER 2015; University of California: San Francisco, CA, USA, 2015. [Google Scholar]
- Larsen, M.; Willett, M.; Yount, R.G. Imidodiphosphate and pyrophosphate: Possible biological significance of similar structures. Science 1969, 166, 1510–1511. [Google Scholar] [CrossRef] [PubMed]
- Anashkin, V.A.; Orlov, V.N.; Lahti, R.; Baykov, A.A. An arginine residue involved in allosteric regulation of cystathionine β-synthase (CBS) domain-containing pyrophosphatase. Arch. Biochem. Biophys. 2019, 662, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD—Visual Molecular Dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Sindhikara, D.J.; Yoshida, N.; Hirata, F. Placevent: An algorithm for prediction of explicit solvent atom distribution—Application to HIV-1 protease and F-ATP synthase. J. Comput. Chem. 2012, 33, 1536–1543. [Google Scholar] [CrossRef] [PubMed]










Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anashkin, V.A.; Baykov, A.A. A Lumenal Loop Associated with Catalytic Asymmetry in Plant Vacuolar H+-Translocating Pyrophosphatase. Int. J. Mol. Sci. 2021, 22, 12902. https://doi.org/10.3390/ijms222312902
Anashkin VA, Baykov AA. A Lumenal Loop Associated with Catalytic Asymmetry in Plant Vacuolar H+-Translocating Pyrophosphatase. International Journal of Molecular Sciences. 2021; 22(23):12902. https://doi.org/10.3390/ijms222312902
Chicago/Turabian StyleAnashkin, Viktor A., and Alexander A. Baykov. 2021. "A Lumenal Loop Associated with Catalytic Asymmetry in Plant Vacuolar H+-Translocating Pyrophosphatase" International Journal of Molecular Sciences 22, no. 23: 12902. https://doi.org/10.3390/ijms222312902