
Protein Tyrosine Phosphatases: Mechanisms in Cancer

 ,
, 
 ,
,
Abstract
:1. Introduction
2. PTPs That Regulate the JAK-STAT Pathway
3. PTPs That Impact SFKs and PTEN
4. PTPs That Affect RTK-Associated PI3K-AKT and Ras-Raf-Mek-Erk Signaling
5. The Unique Cases of SHP-1 and PTPN12
6. PTPs That Influence Related Pathways
7. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Abbreviation | Meaning |
| PTP | Protein Tyrosine Phosphatase |
| PTK | Protein Tyrosine Kinase |
| RTK | Receptor Tyrosine Kinase |
| LMWPTP | Low Molecular Weight Protein Tyrosine Phosphatase |
| TNBC | Triple Negative Breast Cancer |
| ER | Estrogen Receptor |
| PR | Progesterone Receptor |
| MCF-7 | Human breast cancer cell line (ER+/PR+) |
| T47D | Human breast cancer cell line (ER+/PR+) |
| PC3 | Aggressive human prostate cancer cell line |
| DU145 | Aggressive human prostate cancer cell line |
| LNCaP | Moderately aggressive human prostate cancer cell line |
| MDA PCa2b | Moderately aggressive human prostate cancer cell line |
| JAK-STAT | Janus Kinase—Signal Transducer and Activator of Transcription |
| PI3K-AKT | Phosphoinositide 3 Kinase—Protein Kinase B |
| MAPK signaling pathway | Mitogen-Activated Protein Kinase (Ras-Raf-Mek-Erk pathway) |
References
- Regad, T. Targeting RTK Signaling Pathways in Cancer. Cancers 2015, 7, 1758–1784. [Google Scholar] [CrossRef]
- Lemmon, M.A.; Schlessinger, J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2010, 141, 1117–1134. [Google Scholar] [CrossRef] [Green Version]
- Iwakura, Y.; Nawa, H. ErbB1-4-dependent EGF/neuregulin signals and their cross talk in the central nervous system: Pathological implications in schizophrenia and Parkinson’s disease. Front. Cell. Neurosci. 2013, 7, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bollu, L.; Mazumdar, A.; Savage, M.I.; Brown, P.H. Molecular Pathways: Targeting Protein Tyrosine Phosphatases in Cancer. Clin. Cancer Res. 2017, 23, 2136–2142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motiwala, T.; Jacob, S.T. Role of Protein Tyrosine Phosphatases in Cancer. Prog. Nucleic Acid Res. Mol. Biol. 2006, 81, 297–329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alonso, A.; Sasin, J.; Bottini, N.; Friedberg, I.; Friedberg, I.; Osterman, A.; Godzik, A.; Hunter, T.; Dixon, J.; Mustelin, T. Protein Tyrosine Phosphatases in the Human Genome. Cell 2004, 117, 699–711. [Google Scholar] [CrossRef] [Green Version]
- Qu, C.K. The SHP-2 tyrosine phosphatase: Signaling mechanisms and biological functions. Cell Res. 2000, 10, 279–288. [Google Scholar] [CrossRef] [Green Version]
- Eck, M.J.; Pluskey, S.; Trüb, T.; Harrison, S.C.; Shoelson, S.E. Spatial constraints on the recognition of phosphoproteins by the tandem SH2 domains of the phosphatase SH-PTP2. Nat. Cell Biol. 1996, 379, 277–280. [Google Scholar] [CrossRef]
- Chan, R.J.; Feng, G.-S. PTPN11 is the first identified proto-oncogene that encodes a tyrosine phosphatase. Blood 2006, 109, 862–867. [Google Scholar] [CrossRef] [Green Version]
- Hof, P.; Pluskey, S.; Dhe-Paganon, S.; Eck, M.J.; Shoelson, S.E. Crystal Structure of the Tyrosine Phosphatase SHP-2. Cell 1998, 92, 441–450. [Google Scholar] [CrossRef] [Green Version]
- Matozaki, T.; Murata, Y.; Saito, Y.; Okazawa, H.; Ohnishi, H. Protein tyrosine phosphatase SHP-2: A proto-oncogene product that promotes Ras activation. Cancer Sci. 2009, 100, 1786–1793. [Google Scholar] [CrossRef]
- Zhang, S.Q.; Yang, W.; Kontaridis, M.I.; Bivona, T.G.; Wen, G.; Araki, T.; Luo, J.; Thompson, J.A.; Schraven, B.L.; Philips, M.R.; et al. Shp2 Regulates Src Family Kinase Activity and Ras/Erk Activation by Controlling Csk Recruitment. Mol. Cell 2004, 13, 341–355. [Google Scholar] [CrossRef]
- Hanafusa, H.; Torii, S.; Yasunaga, T.; Nishida, E. Sprouty1 and Sprouty2 provide a control mechanism for the Ras/MAPK signalling pathway. Nat. Cell Biol. 2002, 4, 850–858. [Google Scholar] [CrossRef] [PubMed]
- Agazie, Y.M.; Hayman, M.J. Molecular Mechanism for a Role of SHP2 in Epidermal Growth Factor Receptor Signaling. Mol. Cell. Biol. 2003, 23, 7875–7886. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Jang, H.; Nussinov, R. The structural basis for Ras activation of PI3Kα lipid kinase. Phys. Chem. Chem. Phys. 2019, 21, 12021–12028. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Yu, Q.; Liu, J.H.; Zhang, J.; Wang, H.; Koul, D.; McMurray, J.S.; Fang, X.; Yung, W.A.; Siminovitch, K.A.; et al. Src Family Protein-tyrosine Kinases Alter the Function of PTEN to Regulate Phosphatidylinositol 3-Kinase/AKT Cascades. J. Biol. Chem. 2003, 278, 40057–40066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irby, R.B.; Yeatman, T.J. Role of Src expression and activation in human cancer. Oncogene 2000, 19, 5636–5642. [Google Scholar] [CrossRef] [Green Version]
- Jiao, Q.; Bi, L.; Ren, Y.; Song, S.; Wang, Q.; Wang, Y.-S. Advances in studies of tyrosine kinase inhibitors and their acquired resistance. Mol. Cancer 2018, 17, 36. [Google Scholar] [CrossRef] [PubMed]
- Fiordalisi, J.J.; Dewar, B.J.; Graves, L.M.; Madigan, J.P.; Cox, A.D. Src-Mediated Phosphorylation of the Tyrosine Phosphatase PRL-3 Is Required for PRL-3 Promotion of Rho Activation, Motility and Invasion. PLoS ONE 2013, 8, e64309. [Google Scholar] [CrossRef] [Green Version]
- Liang, F.; Liang, J.; Wang, W.-Q.; Sun, J.-P.; Udho, E.; Zhang, Z.-Y. PRL3 Promotes Cell Invasion and Proliferation by Down-regulation of Csk Leading to Src Activation. J. Biol. Chem. 2007, 282, 5413–5419. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Yan, F.; Ye, Q.; Wu, X.; Jiang, F. PTP1B inhibitor promotes endothelial cell motility by activating the DOCK180/Rac1 pathway. Sci. Rep. 2016, 6, 24111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, F.; Sells, M.A.; Chernoff, J. Protein tyrosine phosphatase 1B negatively regulates integrin signaling. Curr. Biol. 1998, 8, 173–S2. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Guan, J.-L. Focal adhesion kinase and its signaling pathways in cell migration and angiogenesis. Adv. Drug Deliv. Rev. 2011, 63, 610–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Defilippi, P.; Di Stefano, P.; Cabodi, S. p130Cas: A versatile scaffold in signaling networks. Trends Cell Biol. 2006, 16, 257–263. [Google Scholar] [CrossRef]
- Liu, F.; Hill, D.E.; Chernoff, J. Direct Binding of the Proline-rich Region of Protein Tyrosine Phosphatase 1B to the Src Homology 3 Domain of p130Cas. J. Biol. Chem. 1996, 271, 31290–31295. [Google Scholar] [CrossRef] [Green Version]
- Burridge, K.; Wennerberg, K. Rho and Rac Take Center Stage. Cell 2004, 116, 167–179. [Google Scholar] [CrossRef] [Green Version]
- Brábek, J.; Constancio, S.S.; Shin, N.-Y.; Pozzi, A.; Weaver, A.M.; Hanks, S.K. CAS promotes invasiveness of Src-transformed cells. Oncogene 2004, 23, 7406–7415. [Google Scholar] [CrossRef] [Green Version]
- Liao, S.-C.; Li, J.-X.; Yu, L.; Sun, S.-R. Protein tyrosine phosphatase 1B expression contributes to the development of breast cancer. J. Zhejiang Univ. Sci. B 2017, 18, 334–342. [Google Scholar] [CrossRef] [Green Version]
- Xu, Q.; Wu, N.; Li, X.; Guo, C.; Li, C.; Jiang, B.; Wang, H.; Shi, D. Inhibition of PTP1B blocks pancreatic cancer progression by targeting the PKM2/AMPK/mTOC1 pathway. Cell Death Dis. 2019, 10, 874. [Google Scholar] [CrossRef] [Green Version]
- Zahra, K.; Dey, T.; Ashish, A.; Mishra, S.P.; Pandey, U. Pyruvate Kinase M2 and Cancer: The Role of PKM2 in Promoting Tumorigenesis. Front. Oncol. 2020, 10, 159. [Google Scholar] [CrossRef] [Green Version]
- Bartolomé, R.A.; Martín-Regalado, Á.; Jaén, M.; Zannikou, M.; Zhang, P.; de los Ríos, V.; Balyasnikova, I.V.; Casal, J.I. Protein Tyrosine Phosphatase-1B Inhibition Disrupts IL13Rα2-Promoted Invasion and Metastasis in Cancer Cells. Cancers 2020, 12, 500. [Google Scholar] [CrossRef] [Green Version]
- Bjorge, J.D.; Jakymiw, A.; Fujita, D.J. Selected glimpses into the activation and function of Src kinase. Oncogene 2000, 19, 5620–5635. [Google Scholar] [CrossRef] [Green Version]
- Aleshin, A.; Finn, R.S. SRC: A Century of Science Brought to the Clinic. Neoplasia 2010, 12, 599–607. [Google Scholar] [CrossRef] [Green Version]
- Fan, G.; Lin, G.; Lucito, R.; Tonks, N.K. Protein-tyrosine Phosphatase 1B Antagonized Signaling by Insulin-like Growth Factor-1 Receptor and Kinase BRK/PTK6 in Ovarian Cancer Cells. J. Biol. Chem. 2013, 288, 24923–24934. [Google Scholar] [CrossRef] [Green Version]
- Alho, I.; Costa, L.; Bicho, M.; Coelho, C. The role of low-molecular-weight protein tyrosine phosphatase (LMW-PTP ACP1) in oncogenesis. Tumor Biol. 2013, 34, 1979–1989. [Google Scholar] [CrossRef]
- Nikolaienko, R.M.; Agyekum, B.; Bouyain, S. Receptor protein tyrosine phosphatases and cancer. Cell Adhes. Migr. 2012, 6, 356–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Wu, H.; Yi, B.; Zhou, J.; Wei, L.; Chen, Y.; Zhang, L. RING finger protein 38 induces gastric cancer cell growth by decreasing the stability of the protein tyrosine phosphatase SHP-1. FEBS Lett. 2018, 592, 3092–3100. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joo, M.K.; Park, J.-J.; Yoo, H.S.; Lee, B.J.; Chun, H.J.; Lee, S.W.; Bak, Y.-T. Epigenetic regulation and anti-tumorigenic effects of SH2-containing protein tyrosine phosphatase 1 (SHP1) in human gastric cancer cells. Tumor Biol. 2015, 37, 4603–4612. [Google Scholar] [CrossRef]
- Bentires-Alj, M.; Neel, B.G. Protein-Tyrosine Phosphatase 1B Is Required for HER2/Neu–Induced Breast Cancer. Cancer Res. 2007, 67, 2420–2424. [Google Scholar] [CrossRef] [Green Version]
- Karlsson, E.; Veenstra, C.; Gårsjö, J.; Nordenskjöld, B.; Fornander, T.; Stål, O. PTPN2 deficiency along with activation of nuclear Akt predict endocrine resistance in breast cancer. J. Cancer Res. Clin. Oncol. 2019, 145, 599–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musgrove, E.A.; Sutherland, R.L. Biological determinants of endocrine resistance in breast cancer. Nat. Rev. Cancer 2009, 9, 631–643. [Google Scholar] [CrossRef]
- Miller, T.W. Endocrine Resistance: What Do We Know? Am. Soc. Clin. Oncol. Educ. Book 2013, e37–e42. [Google Scholar] [CrossRef]
- Klingler-Hoffmann, M.; Fodero-Tavoletti, M.T.; Mishima, K.; Narita, Y.; Cavenee, W.K.; Furnari, F.; Huang, H.-J.S.; Tiganis, T. The Protein Tyrosine Phosphatase TCPTP Suppresses the Tumorigenicity of Glioblastoma Cells Expressing a Mutant Epidermal Growth Factor Receptor. J. Biol. Chem. 2001, 276, 46313–46318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiganis, T.; Kemp, B.E.; Tonks, N.K. The Protein-tyrosine Phosphatase TCPTP Regulates Epidermal Growth Factor Receptor-mediated and Phosphatidylinositol 3-Kinase-dependent Signaling. J. Biol. Chem. 1999, 274, 27768–27775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shields, B.J.; Wiede, F.; Gurzov, E.N.; Wee, K.; Hauser, C.; Zhu, H.-J.; Molloy, T.J.; O’Toole, S.A.; Daly, R.J.; Sutherland, R.L.; et al. TCPTP Regulates SFK and STAT3 Signaling and Is Lost in Triple-Negative Breast Cancers. Mol. Cell. Biol. 2013, 33, 557–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiganis, T.; Bennett, A.M. Protein tyrosine phosphatase function: The substrate perspective. Biochem. J. 2007, 402, 1–15. [Google Scholar] [CrossRef]
- Shields, B.J.; Hauser, C.; Bukczynska, P.E.; Court, N.W.; Tiganis, T. DNA Replication Stalling Attenuates Tyrosine Kinase Signaling to Suppress S Phase Progression. Cancer Cell 2008, 14, 166–179. [Google Scholar] [CrossRef] [Green Version]
- Simoncic, P.D.; Lee-Loy, A.; Barber, D.L.; Tremblay, M.L.; McGlade, C. The T Cell Protein Tyrosine Phosphatase Is a Negative Regulator of Janus Family Kinases 1 and 3. Curr. Biol. 2002, 12, 446–453. [Google Scholar] [CrossRef] [Green Version]
- Tiganis, T.; Bennett, A.M.; Ravichandran, K.S.; Tonks, N.K. Epidermal Growth Factor Receptor and the Adaptor Protein p52 Shc Are Specific Substrates of T-Cell Protein Tyrosine Phosphatase. Mol. Cell. Biol. 1998, 18, 1622–1634. [Google Scholar] [CrossRef] [Green Version]
- Van Vliet, C.; Bukczynska, P.E.; Puryer, M.A.; Sadek, C.M.; Shields, B.J.; Tremblay, M.L.; Tiganis, T. Selective regulation of tumor necrosis factor–induced Erk signaling by Src family kinases and the T cell protein tyrosine phosphatase. Nat. Immunol. 2005, 6, 253–260. [Google Scholar] [CrossRef]
- Yamada, O.; Ozaki, K.; Akiyama, M.; Kawauchi, K. JAK–STAT and JAK–PI3K–mTORC1 Pathways Regulate Telomerase Transcriptionally and Posttranslationally in ATL Cells. Mol. Cancer Ther. 2012, 11, 1112–1121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.; Gao, L.; Kong, D.; Xue, H. Loss of Tyrosine Phosphatase Delta Promotes Gastric Cancer Progression via Signal Transducer and Activator of Transcription 3 Pathways. Dig. Dis. Sci. 2019, 64, 3164–3172. [Google Scholar] [CrossRef] [PubMed]
- Arias-Romero, L.E.; Saha, S.; Villamar-Cruz, O.; Yip, S.-C.; Ethier, S.P.; Zhang, Z.-Y.; Chernoff, J. Activation of Src by Protein Tyrosine Phosphatase 1B Is Required for ErbB2 Transformation of Human Breast Epithelial Cells. Cancer Res. 2009, 69, 4582–4588. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Chen, Q.; Hu, X.-G.; Zhang, X.-C.; Fu, T.-W.; Liu, Q.; Liang, Y.; Zhao, X.-L.; Zhang, X.; Ping, Y.-F.; et al. PTP1B promotes aggressiveness of breast cancer cells by regulating PTEN but not EMT. Tumor Biol. 2016, 37, 13479–13487. [Google Scholar] [CrossRef]
- Wu, C.; Zhang, L.; Bourne, P.A.; Reeder, J.E.; di Sant’Agnese, P.A.; Yao, J.L.; Na, Y.; Huang, J. Protein tyrosine phosphatase PTP1B is involved in neuroendocrine differentiation of prostate cancer. Prostate 2006, 66, 1125–1135. [Google Scholar] [CrossRef]
- Hu, C.-D.; Choo, R.; Huang, J. Neuroendocrine Differentiation in Prostate Cancer: A Mechanism of Radioresistance and Treatment Failure. Front. Oncol. 2015, 5, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahlgren, G.; Pedersen, K.; Lundberg, S.; Aus, G.; Hugosson, J.; Abrahamsson, P.-A. Regressive changes and neuroendocrine differentiation in prostate cancer after neoadjuvant hormonal treatment. Prostate 2000, 42, 274–279. [Google Scholar] [CrossRef]
- Jiborn, T.; Bjartell, A.; Abrahamsson, P.-A. Neuroendocrine Differentiation in Prostatic Carcinoma During Hormonal Treatment. Urology 1998, 51, 585–589. [Google Scholar] [CrossRef]
- Di Sant’Agnese, P.A.; de Mesy Jensen, K.L.; Churukian, C.J.; Agarwal, M.M. Human prostatic endocrine-paracrine (APUD) cells. Distributional analysis with a comparison of serotonin and neuron-specific enolase immunoreactivity and silver stains. Arch. Pathol. Lab. Med. 1985, 109, 607–612. [Google Scholar]
- Abrahamsson, P.-A.; Wadström, L.B.; Alumets, J.; Falkmer, S.; Grimelius, L. Peptide-Hormone- and Serotonin-Immunoreactive Tumour Cells in Carcinoma of the Prostate. Pathol.-Res. Pr. 1987, 182, 298–307. [Google Scholar] [CrossRef]
- Lee, L.-F.; Guan, J.; Qiu, Y.; Kung, H.-J. Neuropeptide-Induced Androgen Independence in Prostate Cancer Cells: Roles of Nonreceptor Tyrosine Kinases Etk/Bmx, Src, and Focal Adhesion Kinase. Mol. Cell. Biol. 2001, 21, 8385–8397. [Google Scholar] [CrossRef] [Green Version]
- Lessard, L.; Labbé, D.; Deblois, G.; Bégin, L.R.; Hardy, S.; Mes-Masson, A.-M.; Saad, F.; Trotman, L.C.; Giguère, V.; Tremblay, M.L. PTP1B Is an Androgen Receptor–Regulated Phosphatase That Promotes the Progression of Prostate Cancer. Cancer Res. 2012, 72, 1529–1537. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Li, Z.; Fan, X.; Xiong, J.; Zhang, G.; Luo, X.; Li, K.; Jie, Z.; Cao, Y.; Huang, Z.; et al. PRL-3 promotes gastric cancer peritoneal metastasis via the PI3K/AKT signaling pathway in�vitro and in�vivo. Oncol. Lett. 2018, 15, 9069–9074. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Quah, S.Y.; Dong, J.M.; Manser, E.; Tang, J.P.; Zeng, Q. PRL-3 Down-regulates PTEN Expression and Signals through PI3K to Promote Epithelial-Mesenchymal Transition. Cancer Res. 2007, 67, 2922–2926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, J.; Li, Z.; Zhang, Y.; Li, D.; Zhang, G.; Luo, X.; Jie, Z.; Liu, Y.; Cao, Y.; Le, Z.; et al. PRL-3 promotes the peritoneal metastasis of gastric cancer through the PI3K/Akt signaling pathway by regulating PTEN. Oncol. Rep. 2016, 36, 1819–1828. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hollander, P.D.; Rawls, K.; Tsimelzon, A.; Shepherd, J.; Mazumdar, A.; Hill, J.; Fuqua, S.A.W.; Chang, J.C.; Osborne, C.K.; Hilsenbeck, S.G.; et al. Phosphatase PTP4A3 Promotes Triple-Negative Breast Cancer Growth and Predicts Poor Patient Survival. Cancer Res. 2016, 76, 1942–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diamantopoulou, Z.; Kitsou, P.; Menashi, S.; Courty, J.; Katsoris, P. Loss of Receptor Protein Tyrosine Phosphatase β/ζ (RPTPβ/ζ) Promotes Prostate Cancer Metastasis. J. Biol. Chem. 2012, 287, 40339–40349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maeda, N.; Nishiwaki, T.; Shintani, T.; Hamanaka, H.; Noda, M. 6B4 Proteoglycan/Phosphacan, an Extracellular Variant of Receptor-like Protein-tyrosine Phosphatase ζ/RPTPβ, Binds Pleiotrophin/Heparin-binding Growth-associated Molecule (HB-GAM). J. Biol. Chem. 1996, 271, 21446–21452. [Google Scholar] [CrossRef] [Green Version]
- Meng, K.; Rodriguez-Pena, A.; Dimitrov, T.; Chen, W.; Yamin, M.; Noda, M.; Deuel, T.F. Pleiotrophin signals increased tyrosine phosphorylation of beta-catenin through inactivation of the intrinsic catalytic activity of the receptor-type protein tyrosine phosphatase beta/zeta. Proc. Natl. Acad. Sci. USA 2000, 97, 2603–2608. [Google Scholar] [CrossRef] [Green Version]
- Raulo, E.; Chernousov, M.; Carey, D.; Nolo, R.; Rauvala, H. Isolation of a neuronal cell surface receptor of heparin binding growth-associated molecule (HB-GAM). Identification as N-syndecan (syndecan-3). J. Biol. Chem. 1994, 269, 12999–13004. [Google Scholar] [CrossRef]
- Perez-Pinera, P.; Alcántara, S.; Dimitrov, T.; Vega, J.A.; Deuel, T.F. Pleiotrophin disrupts calcium-dependent homophilic cell-cell adhesion and initiates an epithelial-mesenchymal transition. Proc. Natl. Acad. Sci. USA 2006, 103, 17795–17800. [Google Scholar] [CrossRef] [Green Version]
- Keane, M.M.; Lowrey, G.A.; Ettenberg, S.A.; Dayton, M.A.; Lipkowitz, S. The protein tyrosine phosphatase DEP-1 is induced during differentiation and inhibits growth of breast cancer cells. Cancer Res. 1996, 56, 4236–4243. [Google Scholar]
- Tonks, N.K. Protein tyrosine phosphatases—From housekeeping enzymes to master regulators of signal transduction. FEBS J. 2012, 280, 346–378. [Google Scholar] [CrossRef] [Green Version]
- Spring, K.; Fournier, P.; Lapointe, L.; Chabot, C.; Roussy, J.; Pommey, S.; Stagg, J.; Royal, I. The protein tyrosine phosphatase DEP-1/PTPRJ promotes breast cancer cell invasion and metastasis. Oncogene 2015, 34, 5536–5547. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Zhang, R.; Xu, R.; Shang, J.; He, H.; Yang, Q. MicroRNA-574-5p in gastric cancer cells promotes angiogenesis by targeting protein tyrosine phosphatase non-receptor type 3 (PTPN3). Gene 2020, 733, 144383. [Google Scholar] [CrossRef]
- Li, M.-Y.; Peng, W.-H.; Wu, C.-H.; Chang, Y.-M.; Lin, Y.-L.; Chang, G.-D.; Wu, H.-C.; Chen, G.-C. PTPN3 suppresses lung cancer cell invasiveness by counteracting Src-mediated DAAM1 activation and actin polymerization. Oncogene 2019, 38, 7002–7016. [Google Scholar] [CrossRef]
- Aspenström, P.; Richnau, N.; Johansson, A.-S. The diaphanous-related formin DAAM1 collaborates with the Rho GTPases RhoA and Cdc42, CIP4 and Src in regulating cell morphogenesis and actin dynamics. Exp. Cell Res. 2006, 312, 2180–2194. [Google Scholar] [CrossRef]
- Wong, G.S.; Zhou, J.; Bin Liu, J.; Wu, Z.; Xu, X.; Li, T.; Xu, D.; Schumacher, S.E.; Puschhof, J.; McFarland, J.; et al. Targeting wild-type KRAS-amplified gastroesophageal cancer through combined MEK and SHP2 inhibition. Nat. Med. 2018, 24, 968–977. [Google Scholar] [CrossRef] [PubMed]
- Vigil, D.; Cherfils, J.; Rossman, K.L.; Der, C.J. Ras superfamily GEFs and GAPs: Validated and tractable targets for cancer therapy? Nat. Rev. Cancer 2010, 10, 842–857. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buday, L.; Downward, J. Epidermal growth factor regulates p21ras through the formation of a complex of receptor, Grb2 adapter protein, and Sos nucleotide exchange factor. Cell 1993, 73, 611–620. [Google Scholar] [CrossRef]
- Castellano, E.; Downward, J. RAS Interaction with PI3K: More Than Just Another Effector Pathway. Genes Cancer 2011, 2, 261–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.S.; Shin, O.R.; Kim, H.K.; Cho, Y.S.; An, C.H.; Lim, K.W.; Kim, S.S. Overexpression of Protein Phosphatase Non-receptor Type 11 (PTPN11) in Gastric Carcinomas. Dig. Dis. Sci. 2009, 55, 1565–1569. [Google Scholar] [CrossRef]
- Matalkah, F.; Martin, E.; Zhao, H.; Agazie, Y.M. SHP2 acts both upstream and downstream of multiple receptor tyrosine kinases to promote basal-like and triple-negative breast cancer. Breast Cancer Res. 2016, 18, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, K.; Zhao, H.; Ji, Z.; Zhang, C.; Zhou, P.; Wang, L.; Chen, Q.; Wang, J.; Zhang, P.; Chen, Z.; et al. Shp2 promotes metastasis of prostate cancer by attenuating the PAR3/PAR6/aPKC polarity protein complex and enhancing epithelial-to-mesenchymal transition. Oncogene 2016, 35, 1271–1282. [Google Scholar] [CrossRef]
- Etienne-Manneville, S. Polarity proteins in migration and invasion. Oncogene 2008, 27, 6970–6980. [Google Scholar] [CrossRef] [Green Version]
- Sun, T.; Aceto, N.; Meerbrey, K.L.; Kessler, J.D.; Zhou, C.; Migliaccio, I.; Nguyen, D.X.; Pavlova, N.N.; Botero, M.; Huang, J.; et al. Activation of Multiple Proto-oncogenic Tyrosine Kinases in Breast Cancer via Loss of the PTPN12 Phosphatase. Cell 2011, 144, 703–718. [Google Scholar] [CrossRef] [Green Version]
- Veeramani, S.; Lee, M.-S.; Lin, M.-F. Revisiting histidine-dependent acid phosphatases: A distinct group of tyrosine phosphatases. Trends Biochem. Sci. 2009, 34, 273–278. [Google Scholar] [CrossRef] [Green Version]
- Veeramani, S.; Yuan, T.-C.; Chen, S.-J.; Lin, F.-F.; E Petersen, J.; Shaheduzzaman, S.; Srivastava, S.; Macdonald, R.G.; Lin, M.-F. Cellular prostatic acid phosphatase: A protein tyrosine phosphatase involved in androgen-independent proliferation of prostate cancer. Endocr.-Relat. Cancer 2005, 12, 805–822. [Google Scholar] [CrossRef]
- Reif, A.E.; Schlesinger, R.M.; Fish, C.A.; Robinson, C.M. Acid phosphatase isozymes in cancer of the prostate. Cancer 1973, 31, 689–699. [Google Scholar] [CrossRef]
- Foti, A.G.; Cooper, J.F.; Herschman, H.; Malvaez, R.R. Detection of Prostatic Cancer by Solid-Phase Radioimmunoassay of Serum Prostatic Acid Phosphatase. N. Engl. J. Med. 1977, 297, 1357–1361. [Google Scholar] [CrossRef] [PubMed]
- Vihko, P.; Kostama, A.; Jänne, O.; Sajanti, E.; Vihko, R. Rapid radioimmunoassay for prostate-specific acid phosphatase in human serum. Clin. Chem. 1980, 26, 1544–1547. [Google Scholar] [CrossRef] [PubMed]
- Vihko, P.; Lukkarinen, O.; Kontturi, M.; Vihko, R. Effectiveness of radioimmunoassay of human prostate-specific acid phosphatase in the diagnosis and follow-up of therapy in prostatic carcinoma. Cancer Res. 1981, 41, 1180–1183. [Google Scholar] [CrossRef]
- Loor, R.; Wang, M.C.; Valenzuela, L.; Chu, T. Expression of prostatic acid phosphatase in human prostate cancer. Cancer Lett. 1981, 14, 63–69. [Google Scholar] [CrossRef]
- Hakalahti, L.; Vihko, P.; Henttu, P.; Vihko, R.; Autio-Harmainen, H.; Soini, Y. Evaluation of PAP and PSA gene expression in prostatic hyperplasia and prostatic carcinoma using northern-blot analyses, in situ hybridization and immunohistochemical stainings with monoclonal and bispecific antibodies. Int. J. Cancer 1993, 55, 590–597. [Google Scholar] [CrossRef]
- Lin, M.F.; Lee, M.S.; Zhou, X.W.; Andressen, J.C.; Meng, T.C.; Johansson, S.L.; West, W.W.; Taylor, R.J.; Anderson, J.R.; Lin, F.F. Decreased Expression of Cellular Prostatic Acid Phosphatase Increases Tumorigenicity of Human Prostate Cancer Cells. J. Urol. 2001, 166, 1943–1950. [Google Scholar] [CrossRef]
- Lin, M.F.; Davolio, J.; Garcia-Arenas, R. Expression of human prostatic acid phosphatase activity and the growth of prostate carcinoma cells. Cancer Res. 1992, 52, 4600–4607. [Google Scholar]
- Meng, T.-C.; Lin, M.-F. Tyrosine Phosphorylation of c-ErbB-2 Is Regulated by the Cellular Form of Prostatic Acid Phosphatase in Human Prostate Cancer Cells. J. Biol. Chem. 1998, 273, 22096–22104. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.-F.; Meng, T.-C.; Rao, P.S.; Chang, C.; Schönthal, A.H.; Lin, F.-F. Expression of Human Prostatic Acid Phosphatase Correlates with Androgen-stimulated Cell Proliferation in Prostate Cancer Cell Lines. J. Biol. Chem. 1998, 273, 5939–5947. [Google Scholar] [CrossRef] [Green Version]
- Igawa, T.; Lin, F.-F.; Lee, M.-S.; Karan, D.; Batra, S.K.; Lin, M.-F. Establishment and characterization of androgen-independent human prostate cancer LNCaP cell model. Prostate 2002, 50, 222–235. [Google Scholar] [CrossRef]
- Veeramani, S.; Igawa, T.; Yuan, T.-C.; Lin, F.-F.; Lee, M.-S.; Lin, J.S.; Johansson, S.L.; Lin, M.-F. Expression of p66Shc protein correlates with proliferation of human prostate cancer cells. Oncogene 2005, 24, 7203–7212. [Google Scholar] [CrossRef] [Green Version]
- Tassidis, H.; Culig, Z.; Wingren, A.G.; Härkönen, P. Role of the protein tyrosine phosphatase SHP-1 in Interleukin-6 regulation of prostate cancer cells. Prostate 2010, 70, 1491–1500. [Google Scholar] [CrossRef]
- Rodriguez-Ubreva, F.J.; Cariaga-Martinez, A.E.; Cortés, M.A.; Romero-De Pablos, M.; Ropero, S.; López-Ruiz, P.; Colas, B. Knockdown of protein tyrosine phosphatase SHP-1 inhibits G1/S progression in prostate cancer cells through the regulation of components of the cell-cycle machinery. Oncogene 2009, 29, 345–355. [Google Scholar] [CrossRef] [Green Version]
- Wander, S.A.; Zhao, D.; Slingerland, J.M. p27: A Barometer of Signaling Deregulation and Potential Predictor of Response to Targeted Therapies: Figure 1. Clin. Cancer Res. 2011, 17, 12–18. [Google Scholar] [CrossRef] [Green Version]
- Liang, J.; Zubovitz, J.; Petrocelli, T.; Kotchetkov, R.; Connor, M.K.; Han, K.; Lee, J.-H.; Ciarallo, S.; Catzavelos, C.; Beniston, R.; et al. PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat. Med. 2002, 8, 1153–1160. [Google Scholar] [CrossRef]
- Fujita, N.; Sato, S.; Katayama, K.; Tsuruo, T. Akt-dependent Phosphorylation of p27Kip1Promotes Binding to 14-3-3 and Cytoplasmic Localization. J. Biol. Chem. 2002, 277, 28706–28713. [Google Scholar] [CrossRef] [Green Version]
- Weidemann, S.A.; Sauer, C.; Luebke, A.M.; Möller-Koop, C.; Steurer, S.; Hube-Magg, C.; Büscheck, F.; Höflmayer, D.; Tsourlakis, M.C.; Clauditz, T.S.; et al. High-level expression of protein tyrosine phosphatase non-receptor 12 is a strong and independent predictor of poor prognosis in prostate cancer. BMC Cancer 2019, 19, 944. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.-H.; Ye, L.; Mason, M.D.; Jiang, W.G. Receptor-like protein tyrosine phosphatase κ negatively regulates the apoptosis of prostate cancer cells via the JNK pathway. Int. J. Oncol. 2013, 43, 1560–1568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohtaka, M.; Miyoshi, Y.; Kawahara, T.; Ohtake, S.; Yasui, M.; Uemura, K.; Yoneyama, S.; Hattori, Y.; Teranishi, J.-I.; Yokomizo, Y.; et al. Low-molecular-weight protein tyrosine phosphatase expression as a prognostic factor for men with metastatic hormone-naïve prostate cancer. Urol. Oncol. Semin. Orig. Investig. 2017, 35, 607.e9–607.e14. [Google Scholar] [CrossRef] [PubMed]
- Ruela-De-Sousa, R.R.; Hoekstra, E.; Hoogland, A.M.; Queiroz, K.C.S.; Peppelenbosch, M.P.; Stubbs, A.P.; Pelizzaro-Rocha, K.; van Leenders, G.J.; Jenster, G.; Aoyama, H.; et al. Low-Molecular-Weight Protein Tyrosine Phosphatase Predicts Prostate Cancer Outcome by Increasing the Metastatic Potential. Eur. Urol. 2016, 69, 710–719. [Google Scholar] [CrossRef]
- Anderton, M.; Van Der Meulen, E.; Blumenthal, M.J.; Schäfer, G. The Role of the Eph Receptor Family in Tumorigenesis. Cancers 2021, 13, 206. [Google Scholar] [CrossRef]
- Tan, Y.-H.C.; Srivastava, S.; Won, B.M.; Kanteti, R.; Arif, Q.; Husain, A.N.; Li, H.; Vigneswaran, W.T.; Pang, K.-M.; Kulkarni, P.; et al. EPHA2 mutations with oncogenic characteristics in squamous cell lung cancer and malignant pleural mesothelioma. Oncogenesis 2019, 8, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhi, H.-Y.; Hou, S.-W.; Li, R.-S.; Basir, Z.; Xiang, Q.; Szabo, A.; Chen, G. PTPH1 cooperates with vitamin D receptor to stimulate breast cancer growth through their mutual stabilization. Oncogene 2010, 30, 1706–1715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suresh, P.S.; Ma, S.; Migliaccio, A.; Chen, G. Protein-Tyrosine Phosphatase H1 Increases Breast Cancer Sensitivity to Antiestrogens by Dephosphorylating Estrogen Receptor at Tyr537. Mol. Cancer Ther. 2014, 13, 230–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivaganesh, V.; Promi, N.; Maher, S.; Peethambaran, B. Emerging Immunotherapies against Novel Molecular Targets in Breast Cancer. Int. J. Mol. Sci. 2021, 22, 2433. [Google Scholar] [CrossRef]
- Kostrzewa, T.; Styszko, J.; Gorska-Ponikowska, M.; Sledzinski, T.; Kuban-Jankowska, A. Inhibitors of Protein Tyrosine Phosphatase PTP1B With Anticancer Potential. Anticancer. Res. 2019, 39, 3379–3384. [Google Scholar] [CrossRef]
- Le, H.; Cho, Y.-C.; Cho, A.S. Inhibition of protein tyrosine phosphatase non-receptor type 2 by PTP inhibitor XIX: Its role as a multiphosphatase inhibitor. BMB Rep. 2017, 50, 329–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frankson, R.; Ruo-Yu, Z.; Bai, Y.; Li, Q.; Zhang, R.-Y.; Zhang, Z.-Y. Therapeutic Targeting of Oncogenic Tyrosine Phosphatases. Cancer Res. 2017, 77, 5701–5705. [Google Scholar] [CrossRef] [Green Version]
- Navire Pharma Inc., A BridgeBio Company. A Phase 1/1B First-in-Human Study of the SHP2 Inhibitor BBP-398 (Formerly Known as IACS-15509) in Patients with Advanced Solid Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT04528836 (accessed on 22 November 2021).
- Protein Phosphatase 2A Inhibitor, in Recurrent Glioblastoma—Tabular View—ClinicalTrials.Gov. Available online: https://clinicaltrials.gov/ct2/show/record/NCT03027388 (accessed on 22 November 2021).
- Intra-IMMUSG Pte Ltd. An Open Label, Multicenter, Safety and Efficacy Phase 2 Study of PRL3-Zumab in Solid Tumors. Available online: https://clinicaltrials.gov/ct2/show/NCT04452955 (accessed on 22 November 2021).





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PTP | Classification | Cellular/Molecular Function | Oncogene (O)/Tumor Suppressor (TS) | Figure Illustration |
|---|---|---|---|---|
| Src homology region 2 domain containing phosphatase 1 (SHP-1)/Tyrosine protein phosphatase non-receptor type 6 (PTPN6) | Non-receptor, tyrosine-specific | Downregulation of JAK-STAT, XIAP, Cyclin D1, MMP-9, VEGF1 | TS in gastric cancer | Figure 1 |
| Tyrosine protein phosphatase non-receptor type 1 (PTPN1)/Protein Tyrosine Phosphatase 1B (PTP1B) | Non-receptor, tyrosine-specific | Dephosphorylates STAT3, increases CCL5; | O in breast cancer | Figure 1 |
| Tyrosine protein phosphatase non-receptor type 2 (PTPN2) | Non-receptor, tyrosine-specific | Dephosphorylates ErbB1 (HER1), p-JAK, p-STAT | TS in breast cancer | Figure 1 |
| Protein Tyrosine Phosphatase Receptor Type D (PTPRD) | Receptor-type, tyrosine-specific | Dephosphorylates STAT3 | TS in gastric cancer | Figure 1 |
| PTP | Classification | Cellular/Molecular Function | Oncogene (O)/Tumor Suppressor (TS) | Figure Illustration |
|---|---|---|---|---|
| Tyrosine protein phosphatase non-receptor type 1 (PTPN1)/Protein Tyrosine Phosphatase 1B (PTP1B) | Non-receptor, tyrosine-specific | Dephosphorylates Tyr527 residue of Src (activation); Inhibits PTEN expression | O in breast cancer | Figure 2 |
| PTPN1/PTP1B | Non-receptor, tyrosine-specific | Exact mechanism is unknown. May dephosphorylate STAT3, increases CCL5; Dephosphorylates Tyr527 residue of Src (activation); Inhibits PTEN expression | O in prostate cancer | Figure 1 and Figure 2 |
| Phosphatase of Regenerating Liver 3 (PRL-3)/Protein Tyrosine Phosphatase 4A3 (PTP4A3) | Non-receptor, tyrosine-specific | Inhibits PTEN expression, which heightens PI3K-AKT signaling | O in gastric cancer | Figure 2 |
| PRL-3/PTP4A3 | Non-receptor, tyrosine-specific | Not well understood. May heighten Src activity; Inhibits PTEN expression, which heightens PI3K-AKT signaling | O in breast cancer | Figure 2 |
| Receptor protein tyrosine phosphatase beta/zeta (RPTPβ/ζ) | Receptor-type, tyrosine-specific | Reduces Tyr416 phosphorylation of Src and inactivates it; Reduces phosphorylation of and activates PTEN | TS in prostate cancer | Figure 2 |
| Receptor-type tyrosine protein phosphatase eta (PTPRJ)/Density Enhanced Phosphatase 1 (DEP-1) | Receptor-type, tyrosine-specific | Dephosphorylates Src at Tyr529, which increases Src Tyr418 and subsequent Cortactin phosphorylation | O in breast cancer | Figure 2 |
| Protein Tyrosine Phosphatase H1 (PTPH1)/Tyrosine protein phosphatase non-receptor type 3 (PTPN3) | Non-receptor, tyrosine-specific | Dephosphorylates and inhibits Src mediated DAAM1 phosphorylation; Directly inhibits DAAM1 phosphorylation | TS in gastric cancer | Figure 2 |
| PTP | Classification | Cellular/Molecular Function | Oncogene (O)/Tumor Suppressor (TS) | Figure Illustration |
|---|---|---|---|---|
| Src homology region 2 domain containing phosphatase 2 (SHP-2)/Tyrosine protein phosphatase non-receptor type 11 (PTPN11) | Non-receptor, tyrosine-specific | Recruits Grb2-SOS, which catalyzes conversion of inactive Ras to active GTP-Ras; Recruits Grb2-Gab1, which heightens PI3K-AKT signaling | O in gastric cancer | Figure 3 |
| SHP-2/PTPN11 | Non-receptor, tyrosine-specific | Recruits Grb2-SOS, which catalyzes the conversion of inactive Ras to active GTP-Ras; Recruits Grb2-Gab1, which heightens PI3K-AKT signaling | O in breast cancer | Figure 3 |
| SHP-2/PTPN11 | Non-receptor, tyrosine-specific | Dephosphorylates PAR3, disrupts PAR3/PAR6/aPKC cell polarity/cell-to-cell adhesion complex | O in prostate cancer | N/A |
| Tyrosine protein phosphatase non-receptor type 12 (PTPN12) | Non-receptor, tyrosine-specific | Dephosphorylates EGFR and HER2 RTKs, which inhibits downstream MAPK signaling | TS in breast cancer | Figure 3 |
| Cellular Prostatic Acid Phosphatase (cPAcP) | Histidine-dependent acid phosphatase | Dephosphorylates HER2 RTKs, inhibits downstream MAPK signaling | TS in prostate cancer | Figure 3 |
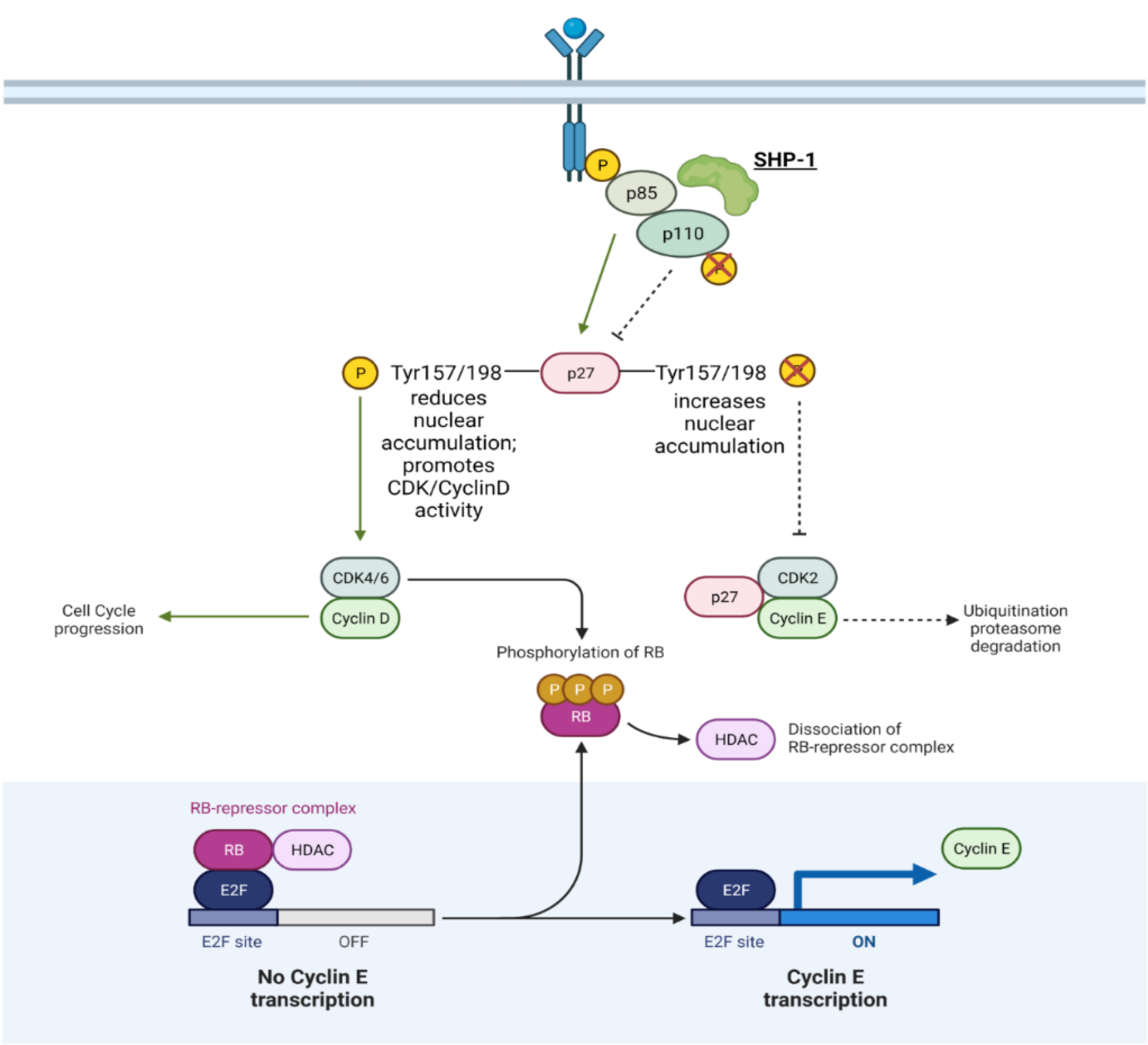
| SHP-1/PTPN6 | Non-receptor, tyrosine-specific | May function as a TS like in gastric cancer. Oncogenic activity as well: Reduces Cyclin/CDK degradation; Translocates CDK2 to the nucleus; Increases CDK6 expression and Rb phosphorylation → E2F→ Increases Cyclin E | TS/O in prostate cancer | Figure 4 |
| Tyrosine protein phosphatase non-receptor type 12 (PTPN12) | Non-receptor, tyrosine-specific | Unknown | O in prostate cancer | N/A |
| PTP | Classification | Cellular/Molecular Function | Oncogene (O)/Tumor Suppressor (TS) |
|---|---|---|---|
| Receptor-like protein tyrosine phosphatase K (PTPRK) | Non-receptor, tyrosine-specific | Inhibits JNK phosphorylation and subsequent apoptosis | O in prostate cancer |
| Low-molecular-weight protein tyrosine phosphatase (LMWPTP) | Non-receptor, low molecular weight | May dephosphorylate EphA2 at Tyr772 and upregulate FAK/AKT/ERK signaling | O in prostate cancer |
| Protein Tyrosine Phosphatase H1 (PTPH1)/Tyrosine protein phosphatase non-receptor type 3 (PTPN3) | Non-receptor, tyrosine-specific | Binds to VDR and inhibits nuclear localization and transcription, enhances tumor survival; Dephosphorylates ER causing accumulation/degradation | O in breast cancer, but sensitizes cancer to anti-hormone treatment |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sivaganesh, V.; Sivaganesh, V.; Scanlon, C.; Iskander, A.; Maher, S.; Lê, T.; Peethambaran, B. Protein Tyrosine Phosphatases: Mechanisms in Cancer. Int. J. Mol. Sci. 2021, 22, 12865. https://doi.org/10.3390/ijms222312865
Sivaganesh V, Sivaganesh V, Scanlon C, Iskander A, Maher S, Lê T, Peethambaran B. Protein Tyrosine Phosphatases: Mechanisms in Cancer. International Journal of Molecular Sciences. 2021; 22(23):12865. https://doi.org/10.3390/ijms222312865
Chicago/Turabian StyleSivaganesh, Vignesh, Varsha Sivaganesh, Christina Scanlon, Alexander Iskander, Salma Maher, Thư Lê, and Bela Peethambaran. 2021. "Protein Tyrosine Phosphatases: Mechanisms in Cancer" International Journal of Molecular Sciences 22, no. 23: 12865. https://doi.org/10.3390/ijms222312865