
Cancer-Cell-Derived IgG and Its Potential Role in Tumor Development


Abstract
:1. Introduction
2. Current State of Knowledge
2.1. CIgs Use a Limited Repertoire
2.2. Ig Repertoire in Human Cancers
2.3. Regulation of IgG Expression in Tumors
2.4. Correlation of CIgG Expression with Clinicopathological Characteristics
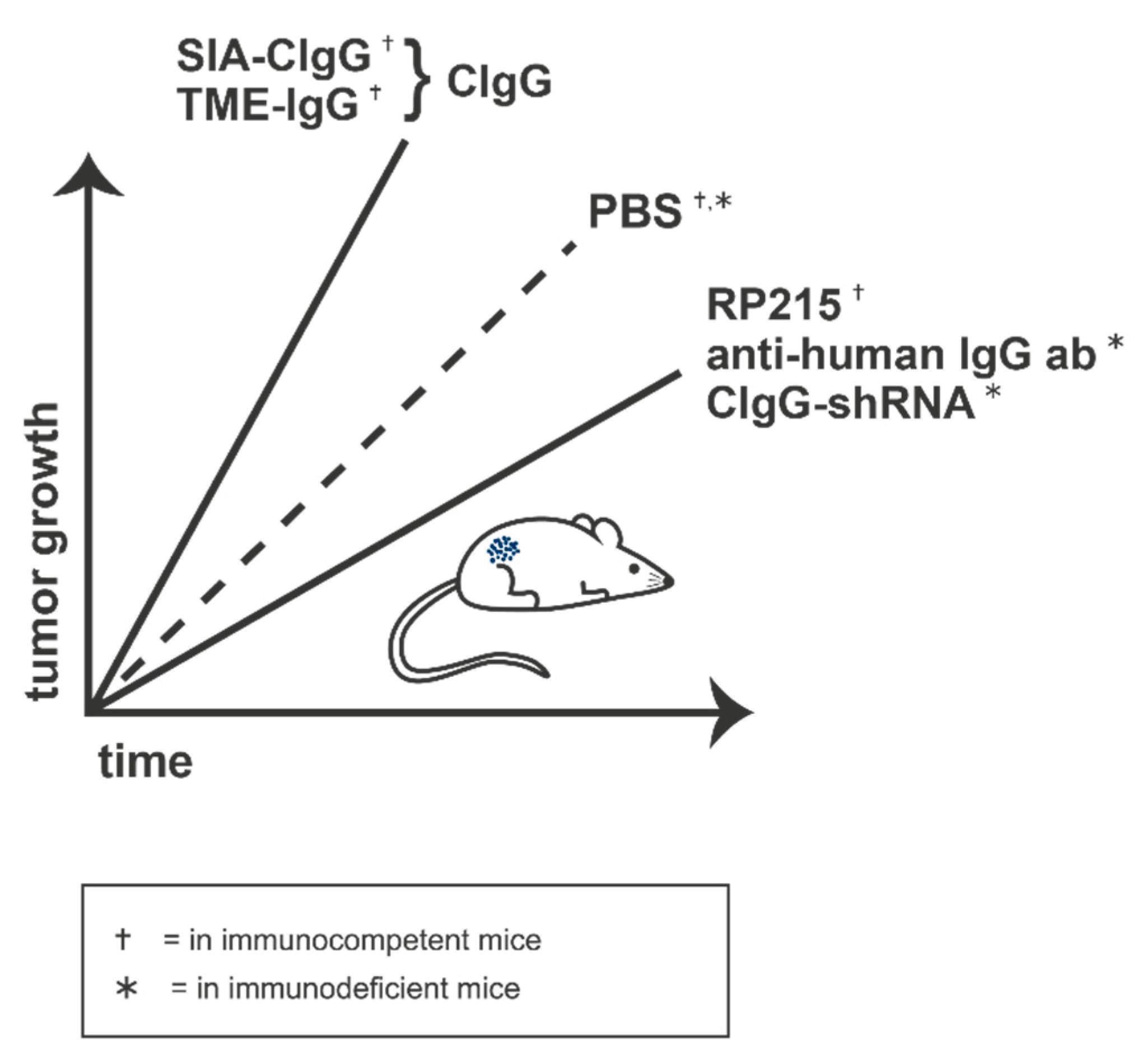
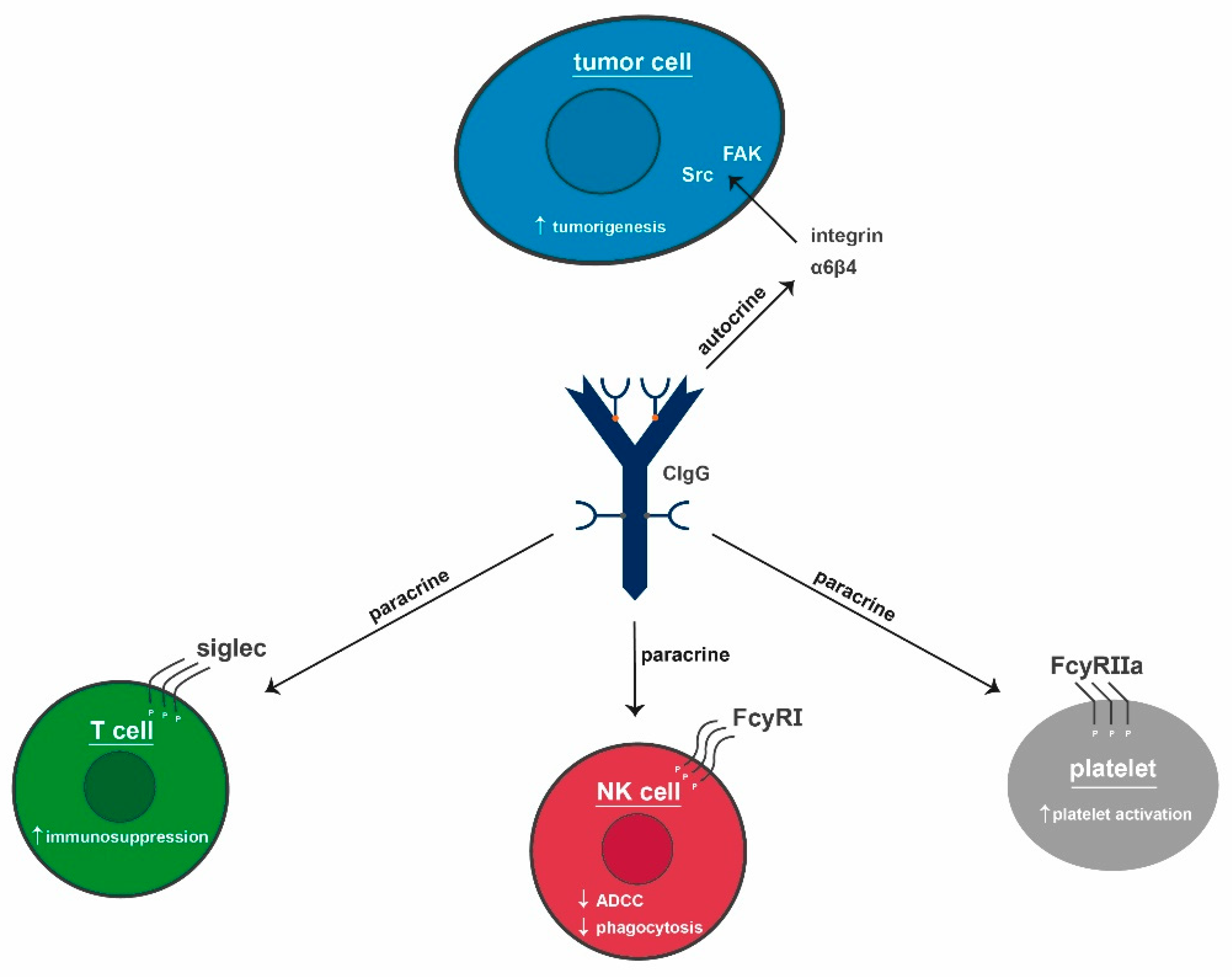
2.5. Effects of CIgG
2.6. IgG Signaling
2.6.1. Physiological
2.6.2. CIgG
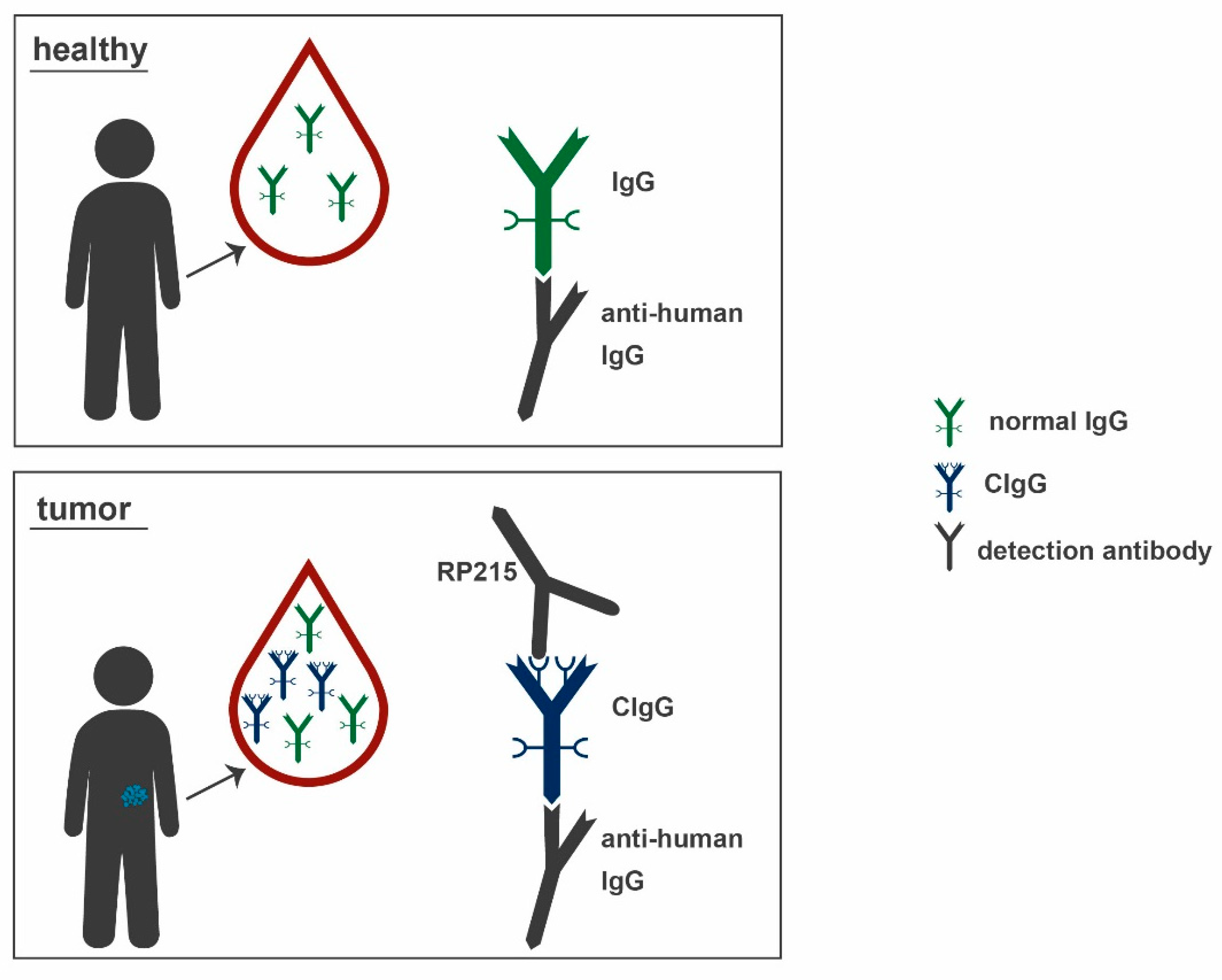
2.7. RP215
2.8. Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG subclasses and allotypes: From structure to effector functions. Front. Immunol. 2014, 5, 520. [Google Scholar] [CrossRef] [Green Version]
- Schur, P.H. IgG subclasses. A historical perspective. Monogr. Allergy 1988, 23, 1–11. [Google Scholar]
- Jung, D.; Giallourakis, C.; Mostoslavsky, R.; Alt, F.W. Mechanism and control of V(D)J recombination at the immunoglobulin heavy chain locus. Annu. Rev. Immunol. 2006, 24, 541–570. [Google Scholar] [CrossRef] [Green Version]
- Teng, G.; Papavasiliou, F.N. Immunoglobulin somatic hypermutation. Annu. Rev. Genet. 2007, 41, 107–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Picard, C.; Bobby Gaspar, H.; Al-Herz, W.; Bousfiha, A.; Casanova, J.-L.; Chatila, T.; Crow, Y.J.; Cunningham-Rundles, C.; Etzioni, A.; Franco, J.L.; et al. International Union of Immunological Societies: 2017 Primary Immunodeficiency Diseases Committee Report on Inborn Errors of Immunity. J. Clin. Immunol. 2018, 38, 96–128. [Google Scholar] [CrossRef] [Green Version]
- Pecoraro, A.; Crescenzi, L.; Granata, F.; Genovese, A.; Spadaro, G. Immunoglobulin replacement therapy in primary and secondary antibody deficiency: The correct clinical approach. Int. Immunopharmacol. 2017, 52, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Qin, Y.-H.; Zhou, T.-B.; Su, L.-N.; Lei, F.-Y.; Zhao, Y.-J.; Huang, W.-F. The efficacy of different dose intravenous immunoglobulin in treating acute idiopathic thrombocytopenic purpura: A meta-analysis of 13 randomized controlled trials. Blood Coagul. Fibrinolysis 2010, 21, 713–721. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.; Dong, Y.; Yin, Y.; Krucoff, M.W. Intravenous immunoglobulin plus corticosteroid to prevent coronary artery abnormalities in Kawasaki disease: A meta-analysis. Heart 2013, 99, 76–82. [Google Scholar] [CrossRef] [Green Version]
- Oaklander, A.L.; Lunn, M.P.; Hughes, R.A.; van Schaik, I.N.; Frost, C.; Chalk, C.H. Treatments for chronic inflammatory demyelinating polyradiculoneuropathy (CIDP): An overview of systematic reviews. Cochrane Database Syst. Rev. 2017, 1, CD010369. [Google Scholar] [CrossRef]
- Iro, M.A.; Martin, N.G.; Absoud, M.; Pollard, A.J. Intravenous immunoglobulin for the treatment of childhood encephalitis. Cochrane Database Syst. Rev. 2017, 10, CD011367. [Google Scholar] [CrossRef] [PubMed]
- Berger, M. Subcutaneous administration of IgG. Immunol. Allergy Clin. N. Am. 2008, 28, 779–802. [Google Scholar] [CrossRef] [PubMed]
- Qiu, X.; Zhu, X.; Zhang, L.; Mao, Y.; Zhang, J.; Hao, P.; Li, G.; Lv, P.; Li, Z.; Sun, X.; et al. Human epithelial cancers secrete immunoglobulin g with unidentified specificity to promote growth and survival of tumor cells. Cancer Res. 2003, 63, 6488–6495. [Google Scholar]
- Liang, P.-Y.; Li, H.-Y.; Zhou, Z.-Y.; Jin, Y.-X.; Wang, S.-X.; Peng, X.-H.; Ou, S.-J. Overexpression of immunoglobulin G prompts cell proliferation and inhibits cell apoptosis in human urothelial carcinoma. Tumour Biol. 2013, 34, 1783–1791. [Google Scholar] [CrossRef] [Green Version]
- Geng, Z.-H.; Ye, C.-X.; Huang, Y.; Jiang, H.-P.; Ye, Y.-J.; Wang, S.; Zhou, Y.; Shen, Z.-L.; Qiu, X.-Y. Human colorectal cancer cells frequently express IgG and display unique Ig repertoire. World J. Gastrointest. Oncol. 2019, 11, 195–207. [Google Scholar] [CrossRef]
- Ma, C.; Wang, Y.; Zhang, G.; Chen, Z.; Qiu, Y.; Li, J.; Luo, J.; Huang, B.; Jiang, C.; Huang, G.; et al. Immunoglobulin G expression and its potential role in primary and metastatic breast cancers. Curr. Mol. Med. 2013, 13, 429–437. [Google Scholar] [PubMed]
- Jiang, H.; Kang, B.; Huang, X.; Yan, Y.; Wang, S.; Ye, Y.; Shen, Z. Cancer IgG, a potential prognostic marker, promotes colorectal cancer progression. Chin. J. Cancer Res. 2019, 31, 499–510. [Google Scholar] [CrossRef]
- Niu, N.; Zhang, J.; Huang, T.; Sun, Y.; Chen, Z.; Yi, W.; Korteweg, C.; Wang, J.; Gu, J. IgG expression in human colorectal cancer and its relationship to cancer cell behaviors. PLoS ONE 2012, 7, e47362. [Google Scholar] [CrossRef] [Green Version]
- Qiu, Y.; Korteweg, C.; Chen, Z.; Li, J.; Luo, J.; Huang, G.; Gu, J. Immunoglobulin G expression and its colocalization with complement proteins in papillary thyroid cancer. Mod. Pathol. 2012, 25, 36–45. [Google Scholar] [CrossRef]
- Hu, J.-B.; Zheng, S.; Deng, Y.-C. Expression of a novel immunoglobulin gene SNC73 in human cancer and non-cancerous tissues. WJG 2003, 9, 1054–1057. [Google Scholar] [CrossRef] [PubMed]
- Lee, G. Cancer cell-expressed immunoglobulins: CA215 as a pan cancer marker and its diagnostic applications. Cancer Biomark. 2009, 5, 137–142. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Geng, Z.; Shao, W.; Liu, E.; Zhang, J.; Tang, J.; Wang, P.; Sun, X.; Xiao, L.; Xu, W.; et al. Cancer-derived sialylated IgG promotes tumor immune escape by binding to Siglecs on effector T cells. Cell. Mol. Immunol. 2019, 17, 1148–1162. [Google Scholar] [CrossRef] [PubMed]
- National Cancer Institute (US). PDQ Cancer Information Summaries: Plasma Cell Neoplasms (Including Multiple Myeloma) Treatment (PDQ®): Patient Version; National Cancer Institute (US): Bethesda, MD, USA, 2002.
- Cohen, H.J.; Crawford, J.; Rao, M.K.; Pieper, C.F.; Currie, M.S. Racial Differences in the Prevalence of Monoclonal Gammopathy in a Community-based Sample of the Elderly. Am. J. Med. 1998, 104, 439–444. [Google Scholar] [CrossRef]
- Kyle, R.A.; Beard, C.M.; O’Fallon, W.M.; Kurland, L.T. Incidence of multiple myeloma in Olmsted County, Minnesota: 1978 through 1990, with a review of the trend since 1945. JCO 1994, 12, 1577–1583. [Google Scholar] [CrossRef] [PubMed]
- Kyle, R.A.; Rajkumar, S.V. Monoclonal gammopathies of undetermined significance. Hematol. Oncol. Clin. N. Am. 1999, 13, 1181–1202. [Google Scholar] [CrossRef]
- Kyle, R.A.; Gertz, M.A.; Witzig, T.E.; Lust, J.A.; Lacy, M.Q.; Dispenzieri, A.; Fonseca, R.; Rajkumar, S.V.; Offord, J.R.; Larson, D.R.; et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin. Proc. 2003, 78, 21–33. [Google Scholar] [CrossRef]
- Bladé, J.; Lust, J.A.; Kyle, R.A. Immunoglobulin D multiple myeloma: Presenting features, response to therapy, and survival in a series of 53 cases. JCO 1994, 12, 2398–2404. [Google Scholar] [CrossRef]
- Perri, R.T.; Hebbel, R.P.; Oken, M.M. Influence of treatment and response status on infection risk in multiple myeloma. Am. J. Med. 1981, 71, 935–940. [Google Scholar] [CrossRef]
- Qin, C.; Sheng, Z.; Huang, X.; Tang, J.; Liu, Y.; Xu, T.; Qiu, X. Cancer-driven IgG promotes the development of prostate cancer though the SOX2-CIgG pathway. Prostate 2020, 80, 1134–1144. [Google Scholar] [CrossRef]
- Lee, Y.N.; Frugoni, F.; Dobbs, K.; Tirosh, I.; Du, L.; Ververs, F.A.; Ru, H.; Ott de Bruin, L.; Adeli, M.; Bleesing, J.H.; et al. Characterization of T and B cell repertoire diversity in patients with RAG deficiency. Sci. Immunol. 2016, 1, eaah6109. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Huang, X.; Ye, J.; Pan, P.; Cao, Q.; Yang, B.; Li, Z.; Su, M.; Huang, C.; Gu, J. Immunoglobulin G is present in a wide variety of soft tissue tumors and correlates well with proliferation markers and tumor grades. Cancer 2010, 116, 1953–1963. [Google Scholar] [CrossRef]
- Lee, G.; Ge, B.; Huang, T.-K.; Zheng, G.; Duan, J.; Wang, I.H.Y. Positive identification of CA215 pan cancer biomarker from serum specimens of cancer patients. Cancer Biomark. 2010, 6, 111–117. [Google Scholar] [CrossRef]
- Babbage, G.; Ottensmeier, C.H.; Blaydes, J.; Stevenson, F.K.; Sahota, S.S. Immunoglobulin heavy chain locus events and expression of activation-induced cytidine deaminase in epithelial breast cancer cell lines. Cancer Res. 2006, 66, 3996–4000. [Google Scholar] [CrossRef] [Green Version]
- JI, F.; Chang, X.; LIU, C.; Meng, L.; QU, L.; WU, J.; LIU, C.; Cui, H.; SHOU, C. Prognostic value and characterization of the ovarian cancer-specific antigen CA166-9. Int. J. Oncol. 2015, 47, 1405–1415. [Google Scholar] [CrossRef]
- Chen, Z.; Gu, J. Immunoglobulin G expression in carcinomas and cancer cell lines. FASEB J. 2007, 21, 2931–2938. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Hu, S.; Korteweg, C.; Chen, Z.; Qiu, Y.; Su, M.; Gu, J. Expression of immunoglobulin G in esophageal squamous cell carcinomas and its association with tumor grade and Ki67. Hum. Pathol. 2012, 43, 423–434. [Google Scholar] [CrossRef]
- Li, X.; Ni, R.; Chen, J.; Liu, Z.; Xiao, M.; Jiang, F.; Lu, C. The presence of IGHG1 in human pancreatic carcinomas is associated with immune evasion mechanisms. Pancreas 2011, 40, 753–761. [Google Scholar] [CrossRef] [PubMed]
- Cui, M.; You, L.; Zheng, B.; Huang, X.; Liu, Q.; Huang, J.; Pan, B.; Qiu, X.; Liao, Q.; Zhao, Y. High Expression of Cancer-Derived Glycosylated Immunoglobulin G Predicts Poor Prognosis in Pancreatic Ductal Adenocarcinoma. J. Cancer 2020, 11, 2213–2221. [Google Scholar] [CrossRef]
- Liu, Y.; Chen, Z.; Niu, N.; Chang, Q.; Deng, R.; Korteweg, C.; Gu, J. IgG gene expression and its possible significance in prostate cancers. Prostate 2012, 72, 690–701. [Google Scholar] [CrossRef]
- Li, L.; Cheng, X.; Zheng, Z.; Xing, X.; Wang, X.; Zhang, L.; Guo, T.; Du, H.; Hu, Y.; Qiu, X.; et al. Overexpression of cancer cell-derived immunoglobulin G correlates with poor prognosis in gastric cancer patients. Transl. Cancer Res. 2016, 5, 285–293. [Google Scholar] [CrossRef]
- Li, M.; Zheng, H.; Duan, Z.; Liu, H.; Hu, D.; Bode, A.; Dong, Z.; Cao, Y. Promotion of cell proliferation and inhibition of ADCC by cancerous immunoglobulin expressed in cancer cell lines. Cell. Mol. Immunol. 2012, 9, 54–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheng, Z.; Liu, Y.; Qin, C.; Liu, Z.; Yuan, Y.; Yin, H.; Qiu, X.; Xu, T. Involvement of cancer-derived IgG in the proliferation, migration and invasion of bladder cancer cells. Oncol. Lett. 2016, 12, 5113–5121. [Google Scholar] [CrossRef] [PubMed]
- Sheng, Z.; Liu, Y.; Qin, C.; Liu, Z.; Yuan, Y.; Hu, F.; Du, Y.; Yin, H.; Qiu, X.; Xu, T. IgG is involved in the migration and invasion of clear cell renal cell carcinoma. J. Clin. Pathol. 2016, 69, 497–504. [Google Scholar] [CrossRef] [Green Version]
- Lv, W.-Q.; Peng, J.; Wang, H.-C.; Chen, D.; Yang, Y.; Zhao, Y.; Qiu, X.-Y.; Jiang, J.-H.; Li, C.-Y. Expression of cancer cell-derived IgG and extra domain A-containing fibronectin in salivary adenoid cystic carcinoma. Arch. Oral Biol. 2017, 81, 15–20. [Google Scholar] [CrossRef]
- Zheng, H.; Li, M.; Ren, W.; Zeng, L.; Liu, H.; Hu, D.; Deng, X.; Tang, M.; Shi, Y.; Gong, J.; et al. Expression and secretion of immunoglobulin alpha heavy chain with diverse VDJ recombinations by human epithelial cancer cells. Mol. Immunol. 2007, 44, 2221–2227. [Google Scholar] [CrossRef]
- Hu, D.; Duan, Z.; Li, M.; Jiang, Y.; Liu, H.; Zheng, H.; Li, L.; Bode, A.M.; Dong, Z.; Cao, Y. Heterogeneity of aberrant immunoglobulin expression in cancer cells. Cell Mol. Immunol. 2011, 8, 479–485. [Google Scholar] [CrossRef] [Green Version]
- Kimoto, Y. Expression of heavy-chain constant region of immunoglobulin and T-cell receptor gene transcripts in human non-hematopoietic tumor cell lines. Genes Chromosomes Cancer 1998, 22, 83–86. [Google Scholar] [CrossRef]
- Wang, H.; Cao, X.; Liu, E.-C.; He, D.; Ma, Y.; Zhang, T.; Feng, Y.; Qin, G. Prognostic significance of immunoglobulin M overexpression in laryngeal squamous cell carcinoma. Acta Oto-Laryngol. 2013, 133, 1080–1087. [Google Scholar] [CrossRef]
- Zhu, X.; Li, C.; Sun, X.; Mao, Y.; Li, G.; Liu, X.; Zhang, Y.; Qiu, X. Immunoglobulin mRNA and protein expression in human oral epithelial tumor cells. Appl. Immunohistochem. Mol. Morphol. 2008, 16, 232–238. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.-M.; Gan, Y.-H. Cancer-derived IgG involved in cisplatin resistance through PTP-BAS/Src/PDK1/AKT signaling pathway. Oral Dis. 2021, 27, 464–474. [Google Scholar] [CrossRef]
- Shang, Y.; Zhang, X.; Lu, L.; Jiang, K.; Krohn, M.; Matschos, S.; Mullins, C.S.; Vollmar, B.; Zechner, D.; Gong, P.; et al. Pharmaceutical immunoglobulin G impairs anti-carcinoma activity of oxaliplatin in colon cancer cells. Br. J. Cancer 2021, 124, 1411–1420. [Google Scholar] [CrossRef] [PubMed]
- Laumen, H.; Nielsen, P.J.; Wirth, T. The BOB.1/OBF.1 co-activator is essential for octamer-dependent transcription in B cells. Eur. J. Immunol. 2000, 30, 458–469. [Google Scholar] [CrossRef]
- Poellinger, L.; Yoza, B.K.; Roeder, R.G. Functional cooperativity between protein molecules bound at two distinct sequence elements of the immunoglobulin heavy-chain promoter. Nature 1989, 337, 573–576. [Google Scholar] [CrossRef]
- Zhu, X.; Wu, L.; Zhang, L.; Hao, P.; Zhang, S.; Huang, J.; Zheng, J.; Liu, Y.; Li, W.; Zhang, Y.; et al. Distinct regulatory mechanism of immunoglobulin gene transcription in epithelial cancer cells. Cell. Mol. Immunol. 2010, 7, 279–286. [Google Scholar] [CrossRef] [PubMed]
- Nieswandt, B.; Hafner, M.; Echtenacher, B.; Männel, D.N. Lysis of tumor cells by natural killer cells in mice is impeded by platelets. Cancer Res. 1999, 59, 1295–1300. [Google Scholar] [PubMed]
- Zucchella, M.; Dezza, L.; Pacchiarini, L.; Meloni, F.; Tacconi, F.; Bonomi, E.; Grignani, G.; Notario, A. Human tumor cells cultured “in vitro” activate platelet function by producing ADP or thrombin. Haematologica 1989, 74, 541–545. [Google Scholar] [PubMed]
- Yu, L.-X.; Yan, L.; Yang, W.; Wu, F.-Q.; Ling, Y.; Chen, S.-Z.; Tang, L.; Tan, Y.-X.; Cao, D.; Wu, M.-C.; et al. Platelets promote tumour metastasis via interaction between TLR4 and tumour cell-released high-mobility group box1 protein. Nat. Commun. 2014, 5, 5256. [Google Scholar] [CrossRef]
- Gay, L.J.; Felding-Habermann, B. Platelets alter tumor cell attributes to propel metastasis: Programming in transit. Cancer Cell 2011, 20, 553–554. [Google Scholar] [CrossRef] [Green Version]
- Takagi, S.; Takemoto, A.; Takami, M.; Oh-Hara, T.; Fujita, N. Platelets promote osteosarcoma cell growth through activation of the platelet-derived growth factor receptor-Akt signaling axis. Cancer Sci. 2014, 105, 983–988. [Google Scholar] [CrossRef] [Green Version]
- Schlesinger, M. Role of platelets and platelet receptors in cancer metastasis. J. Hematol. Oncol. 2018, 11, 125. [Google Scholar] [CrossRef]
- Lonsdorf, A.S.; Krämer, B.F.; Fahrleitner, M.; Schönberger, T.; Gnerlich, S.; Ring, S.; Gehring, S.; Schneider, S.W.; Kruhlak, M.J.; Meuth, S.G.; et al. Engagement of αIIbβ3 (GPIIb/IIIa) with ανβ3 integrin mediates interaction of melanoma cells with platelets: A connection to hematogenous metastasis. J. Biol. Chem. 2012, 287, 2168–2178. [Google Scholar] [CrossRef] [Green Version]
- Ekambaram, P.; Lambiv, W.; Cazzolli, R.; Ashton, A.W.; Honn, K.V. The thromboxane synthase and receptor signaling pathway in cancer: An emerging paradigm in cancer progression and metastasis. Cancer Metastasis Rev. 2011, 30, 397–408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiesner, T.; Bugl, S.; Mayer, F.; Hartmann, J.T.; Kopp, H.-G. Differential changes in platelet VEGF, Tsp, CXCL12, and CXCL4 in patients with metastatic cancer. Clin. Exp. Metastasis 2010, 27, 141–149. [Google Scholar] [CrossRef]
- Farooqi, A.A.; Siddik, Z.H. Platelet-derived growth factor (PDGF) signalling in cancer: Rapidly emerging signalling landscape. Cell Biochem. Funct. 2015, 33, 257–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McEver, R.P. Properties of GMP-140, an inducible granule membrane protein of platelets and endothelium. Blood Cells 1990, 16, 73–80; discussion 80–83. [Google Scholar]
- Schlossman, S.F.; Boumsell, L.; Gilks, W.; Harlan, J.M.; Kishimoto, T.; Morimoto, C.; Ritz, J.; Shaw, S.; Silverstein, R.L.; Springer, T.A. CD antigens 1993. Blood 1994, 83, 879–880. [Google Scholar] [CrossRef]
- Furie, B.; Celi, A.; Palabrica, T.M.; Larsen, E.; Wagner, D.D.; Furie, B.C. PADGEM, a leukocyte receptor on activated platelets. Biology and application to in vivo medical diagnostics. Curr. Stud. Hematol. Blood Transfus. 1991, 58, 32–36. [Google Scholar]
- Kim, Y.J.; Borsig, L.; Varki, N.M.; Varki, A. P-selectin deficiency attenuates tumor growth and metastasis. Proc. Natl. Acad. Sci. USA 1998, 95, 9325–9330. [Google Scholar] [CrossRef] [Green Version]
- Läubli, H.; Borsig, L. Selectins as mediators of lung metastasis. Cancer Microenviron. 2010, 3, 97–105. [Google Scholar] [CrossRef] [Green Version]
- Mannori, G.; Crottet, P.; Cecconi, O.; Hanasaki, K.; Aruffo, A.; Nelson, R.M.; Varki, A.; Bevilacqua, M.P. Differential colon cancer cell adhesion to E-, P-, and L-selectin: Role of mucin-type glycoproteins. Cancer Res. 1995, 55, 4425–4431. [Google Scholar]
- Miao, S.; Shu, D.; Zhu, Y.; Lu, M.; Zhang, Q.; Pei, Y.; He, A.-D.; Ma, R.; Zhang, B.; Ming, Z.-Y. Cancer cell-derived immunoglobulin G activates platelets by binding to platelet FcγRIIa. Cell Death Dis. 2019, 10, 87. [Google Scholar] [CrossRef] [PubMed]
- Ramsland, P.A.; Farrugia, W.; Bradford, T.M.; Sardjono, C.T.; Esparon, S.; Trist, H.M.; Powell, M.S.; Tan, P.S.; Cendron, A.C.; Wines, B.D.; et al. Structural basis for Fc gammaRIIa recognition of human IgG and formation of inflammatory signaling complexes. J. Immunol. 2011, 187, 3208–3217. [Google Scholar] [CrossRef] [PubMed]
- Sondermann, P.; Huber, R.; Oosthuizen, V.; Jacob, U. The 3.2-A crystal structure of the human IgG1 Fc fragment-Fc gammaRIII complex. Nature 2000, 406, 267–273. [Google Scholar] [CrossRef]
- Bruhns, P.; Iannascoli, B.; England, P.; Mancardi, D.A.; Fernandez, N.; Jorieux, S.; Daëron, M. Specificity and affinity of human Fcgamma receptors and their polymorphic variants for human IgG subclasses. Blood 2009, 113, 3716–3725. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Ravetch, J.V. Divergent immunoglobulin g subclass activity through selective Fc receptor binding. Science 2005, 310, 1510–1512. [Google Scholar] [CrossRef] [Green Version]
- Pincetic, A.; Bournazos, S.; DiLillo, D.J.; Maamary, J.; Wang, T.T.; Dahan, R.; Fiebiger, B.-M.; Ravetch, J.V. Type I and type II Fc receptors regulate innate and adaptive immunity. Nat. Immunol. 2014, 15, 707–716. [Google Scholar] [CrossRef] [PubMed]
- Sondermann, P.; Pincetic, A.; Maamary, J.; Lammens, K.; Ravetch, J.V. General mechanism for modulating immunoglobulin effector function. Proc. Natl. Acad. Sci. USA 2013, 110, 9868–9872. [Google Scholar] [CrossRef] [Green Version]
- Anthony, R.M.; Wermeling, F.; Karlsson, M.C.I.; Ravetch, J.V. Identification of a receptor required for the anti-inflammatory activity of IVIG. Proc. Natl. Acad. Sci. USA 2008, 105, 19571–19578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.T.; Maamary, J.; Tan, G.S.; Bournazos, S.; Davis, C.W.; Krammer, F.; Schlesinger, S.J.; Palese, P.; Ahmed, R.; Ravetch, J.V. Anti-HA Glycoforms Drive B Cell Affinity Selection and Determine Influenza Vaccine Efficacy. Cell 2015, 162, 160–169. [Google Scholar] [CrossRef] [Green Version]
- Anthony, R.M.; Kobayashi, T.; Wermeling, F.; Ravetch, J.V. Intravenous gammaglobulin suppresses inflammation through a novel T(H)2 pathway. Nature 2011, 475, 110–113. [Google Scholar] [CrossRef]
- Kanakaraj, P.; Duckworth, B.; Azzoni, L.; Kamoun, M.; Cantley, L.C.; Perussia, B. Phosphatidylinositol-3 kinase activation induced upon Fc gamma RIIIA-ligand interaction. J. Exp. Med. 1994, 179, 551–558. [Google Scholar] [CrossRef]
- Sánchez-Mejorada, G.; Rosales, C. Fcgamma receptor-mediated mitogen-activated protein kinase activation in monocytes is independent of Ras. J. Biol. Chem. 1998, 273, 27610–27619. [Google Scholar] [CrossRef] [Green Version]
- Odin, J.A.; Edberg, J.C.; Painter, C.J.; Kimberly, R.P.; Unkeless, J.C. Regulation of phagocytosis and Ca2+i flux by distinct regions of an Fc receptor. Science 1991, 254, 1785–1788. [Google Scholar] [CrossRef]
- Latour, S.; Fridman, W.H.; Daëron, M. Identification, molecular cloning, biologic properties, and tissue distribution of a novel isoform of murine low-affinity IgG receptor homologous to human Fc gamma RIIB1. J. Immunol. 1996, 157, 189–197. [Google Scholar]
- Amigorena, S.; Salamero, J.; Davoust, J.; Fridman, W.H.; Bonnerot, C. Tyrosine-containing motif that transduces cell activation signals also determines internalization and antigen presentation via type III receptors for IgG. Nature 1992, 358, 337–341. [Google Scholar] [CrossRef]
- Bonnerot, C.; Briken, V.; Brachet, V.; Lankar, D.; Cassard, S.; Jabri, B.; Amigorena, S. syk protein tyrosine kinase regulates Fc receptor gamma-chain-mediated transport to lysosomes. EMBO J. 1998, 17, 4606–4616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergtold, A.; Desai, D.D.; Gavhane, A.; Clynes, R. Cell surface recycling of internalized antigen permits dendritic cell priming of B cells. Immunity 2005, 23, 503–514. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swanson, J.A.; Hoppe, A.D. The coordination of signaling during Fc receptor-mediated phagocytosis. J. Leukoc. Biol. 2004, 76, 1093–1103. [Google Scholar] [CrossRef] [Green Version]
- Desai, D.D.; Harbers, S.O.; Flores, M.; Colonna, L.; Downie, M.P.; Bergtold, A.; Jung, S.; Clynes, R. Fc gamma receptor IIB on dendritic cells enforces peripheral tolerance by inhibiting effector T cell responses. J. Immunol. 2007, 178, 6217–6226. [Google Scholar] [CrossRef] [Green Version]
- Dhodapkar, K.M.; Kaufman, J.L.; Ehlers, M.; Banerjee, D.K.; Bonvini, E.; Koenig, S.; Steinman, R.M.; Ravetch, J.V.; Dhodapkar, M.V. Selective blockade of inhibitory Fcgamma receptor enables human dendritic cell maturation with IL-12p70 production and immunity to antibody-coated tumor cells. Proc. Natl. Acad. Sci. USA 2005, 102, 2910–2915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clynes, R.A.; Towers, T.L.; Presta, L.G.; Ravetch, J.V. Inhibitory Fc receptors modulate in vivo cytotoxicity against tumor targets. Nat. Med. 2000, 6, 443–446. [Google Scholar] [CrossRef]
- Kalergis, A.M.; Ravetch, J.V. Inducing tumor immunity through the selective engagement of activating Fcgamma receptors on dendritic cells. J. Exp. Med. 2002, 195, 1653–1659. [Google Scholar] [CrossRef]
- Kaneko, Y.; Nimmerjahn, F.; Ravetch, J.V. Anti-inflammatory activity of immunoglobulin G resulting from Fc sialylation. Science 2006, 313, 670–673. [Google Scholar] [CrossRef] [Green Version]
- Nimmerjahn, F.; Ravetch, J.V. The antiinflammatory activity of IgG: The intravenous IgG paradox. J. Exp. Med. 2007, 204, 11–15. [Google Scholar] [CrossRef]
- Padet, L.; Bazin, R. IVIg prevents the in vitro activation of T cells by neutralizing the T cell activators. Immunol. Lett. 2013, 150, 54–60. [Google Scholar] [CrossRef]
- Schwab, I.; Nimmerjahn, F. Intravenous immunoglobulin therapy: How does IgG modulate the immune system? Nat. Rev. Immunol. 2013, 13, 176–189. [Google Scholar] [CrossRef] [PubMed]
- Galeotti, C.; Kaveri, S.V.; Bayry, J. IVIG-mediated effector functions in autoimmune and inflammatory diseases. Int. Immunol. 2017, 29, 491–498. [Google Scholar] [CrossRef] [PubMed]
- Büll, C.; Heise, T.; Adema, G.J.; Boltje, T.J. Sialic Acid Mimetics to Target the Sialic Acid-Siglec Axis. Trends Biochem. Sci. 2016, 41, 519–531. [Google Scholar] [CrossRef] [PubMed]
- Crocker, P.R.; Paulson, J.C.; Varki, A. Siglecs and their roles in the immune system. Nat. Rev. Immunol. 2007, 7, 255–266. [Google Scholar] [CrossRef]
- Lee, C.Y.; Chen, K.W.; Sheu, F.S.; Tsang, A.; Chao, K.C.; Ng, H.T. Studies of a tumor-associated antigen, COX-1, recognized by a monoclonal antibody. Cancer Immunol. Immunother. 1992, 35, 19–26. [Google Scholar] [CrossRef]
- Lee, G.; Cheung, A.P.; Ge, B.; Zhu, M.; Giolma, B.; Li, B.; Wong, E.; Li, Y.; Wang, Y.; Chen, Z.; et al. CA215 and GnRH receptor as targets for cancer therapy. Cancer Immunol. Immunother. 2012, 61, 1805–1817. [Google Scholar] [CrossRef]
- Lee, G.; Laflamme, E.; Chien, C.-H.; Ting, H.H. Molecular identity of a pan cancer marker, CA215. Cancer Biol. Ther. 2008, 7, 2007–2014. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Zhang, J.; Liu, Y.; Liao, Q.; Huang, J.; Geng, Z.; Xu, W.; Sheng, Z.; Lee, G.; Zhang, Y.; et al. Lung squamous cell carcinoma cells express non-canonically glycosylated IgG that activates integrin-FAK signaling. Cancer Lett. 2018, 430, 148–159. [Google Scholar] [CrossRef] [PubMed]
- Fisher, M.L.; Kerr, C.; Adhikary, G.; Grun, D.; Xu, W.; Keillor, J.W.; Eckert, R.L. Transglutaminase Interaction with α6/β4-Integrin Stimulates YAP1-Dependent ΔNp63α Stabilization and Leads to Enhanced Cancer Stem Cell Survival and Tumor Formation. Cancer Res. 2016, 76, 7265–7276. [Google Scholar] [CrossRef] [Green Version]
- Soung, Y.H.; Gil, H.J.; Clifford, J.L.; Chung, J. Role of α6β4 integrin in cell motility, invasion and metastasis of mammary tumors. Curr. Protein Pept. Sci. 2011, 12, 23–29. [Google Scholar] [CrossRef] [PubMed]
- Vieira, A.F.; Ribeiro, A.S.; Dionísio, M.R.; Sousa, B.; Nobre, A.R.; Albergaria, A.; Santiago-Gómez, A.; Mendes, N.; Gerhard, R.; Schmitt, F.; et al. P-cadherin signals through the laminin receptor α6β4 integrin to induce stem cell and invasive properties in basal-like breast cancer cells. Oncotarget 2014, 5, 679–692. [Google Scholar] [CrossRef] [Green Version]
- Yoshioka, T.; Otero, J.; Chen, Y.; Kim, Y.-M.; Koutcher, J.A.; Satagopan, J.; Reuter, V.; Carver, B.; de Stanchina, E.; Enomoto, K.; et al. β4 Integrin signaling induces expansion of prostate tumor progenitors. J. Clin. Investig. 2013, 123, 682–699. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; de La Cruz, C.C.; Sayles, L.C.; Alleyne-Chin, C.; Vaka, D.; Knaak, T.D.; Bigos, M.; Xu, Y.; Hoang, C.D.; Shrager, J.B.; et al. A rare population of CD24(+)ITGB4(+)Notch(hi) cells drives tumor propagation in NSCLC and requires Notch3 for self-renewal. Cancer Cell 2013, 24, 59–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, L. Metastatic Inefficiency. Adv. Cancer Res. 1990, 54, 159–211. [Google Scholar] [CrossRef]
- Luzzi, K.J.; MacDonald, I.C.; Schmidt, E.E.; Kerkvliet, N.; Morris, V.L.; Chambers, A.F.; Groom, A.C. Multistep Nature of Metastatic Inefficiency. Am. J. Pathol. 1998, 153, 865–873. [Google Scholar] [CrossRef]
- Talmadge, J.E.; Gabrilovich, D.I. History of myeloid-derived suppressor cells. Nat. Rev. Cancer 2013, 13, 739–752. [Google Scholar] [CrossRef]
- Ugel, S.; de Sanctis, F.; Mandruzzato, S.; Bronte, V. Tumor-induced myeloid deviation: When myeloid-derived suppressor cells meet tumor-associated macrophages. J. Clin. Investig. 2015, 125, 3365–3376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Veglia, F.; Perego, M.; Gabrilovich, D. Myeloid-derived suppressor cells coming of age. Nat. Immunol. 2018, 19, 108–119. [Google Scholar] [CrossRef] [PubMed]
- Alkasalias, T.; Moyano-Galceran, L.; Arsenian-Henriksson, M.; Lehti, K. Fibroblasts in the Tumor Microenvironment: Shield or Spear? Int. J. Mol. Sci. 2018, 19, 1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cancer Entity | Ig Isotype | Localization | Reference(s) |
|---|---|---|---|
| colon | IgG | tissue, cell line, serum | [12,14,17,32] |
| breast | IgG, IgA | tissue, cell line, serum | [12,15,32,33] |
| liver | IgG | tissue, cell line, serum | [12,32] |
| lung | IgG | tissue, cell line, serum | [12,32] |
| ovarian | IgG | tissue, cell line, serum | [12,32,34] |
| cervix | IgG, IgA, Igκ | tissue, cell line, serum | [12,32,35] |
| kidney | IgG | serum | [32] |
| lymphoma | IgG | serum | [32] |
| esophageal | IgG, Igκ, Igλ | tissue, cell line, serum | [36] |
| stomach | IgG | serum | [32] |
| pancreatic | IgG | tissue, cell line, serum | [32,37,38] |
| prostate | IgG, Igκ | tissue, cell line, serum | [32,35,39] |
| gastric | IgG | tissue, cell line | [40,41] |
| bladder | IgG | tissue, cell line | [13,42] |
| renal | IgG | tissue, cell line | [43,44] |
| nasopharyngeal | IgA, Igκ | cell line | [45,46] |
| laryngeal | IgM | tissue, cell line | [47,48] |
| oral | IgG, IgA | tissue, cell line | [49,50] |
| papillary thyroid | IgG | tissue | [18] |
| fibrous histiocytoma | IgG (serum only) | serum | [32] |
| malignant fibrous histiocytoma | IgG (serum only) | serum | [32] |
| fibroma | IgG (serum only) | serum | [32] |
| fibrosarcoma | IgG (serum only) | serum | [32] |
| leiomyoma | IgG (serum only) | serum | [32] |
| leiomyosarcoma | IgG (serum only) | serum | [32] |
| rhabdomyosarcoma | IgG (serum only) | serum | [32] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kdimati, S.; Mullins, C.S.; Linnebacher, M. Cancer-Cell-Derived IgG and Its Potential Role in Tumor Development. Int. J. Mol. Sci. 2021, 22, 11597. https://doi.org/10.3390/ijms222111597
Kdimati S, Mullins CS, Linnebacher M. Cancer-Cell-Derived IgG and Its Potential Role in Tumor Development. International Journal of Molecular Sciences. 2021; 22(21):11597. https://doi.org/10.3390/ijms222111597
Chicago/Turabian StyleKdimati, Said, Christina Susanne Mullins, and Michael Linnebacher. 2021. "Cancer-Cell-Derived IgG and Its Potential Role in Tumor Development" International Journal of Molecular Sciences 22, no. 21: 11597. https://doi.org/10.3390/ijms222111597