
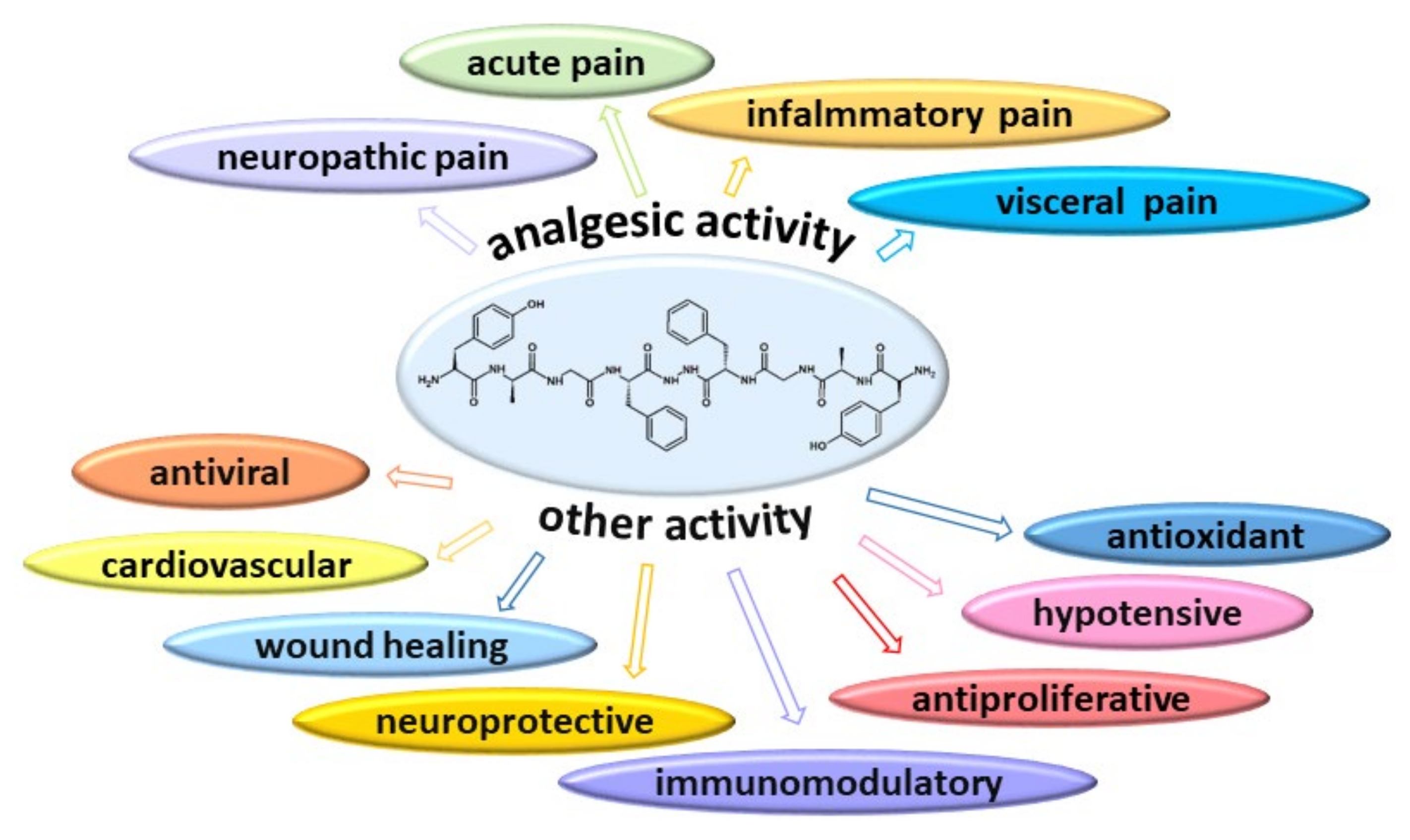
Biphalin—A Potent Opioid Agonist—As a Panacea for Opioid System-Dependent Pathophysiological Diseases? †



Abstract
:1. Introduction
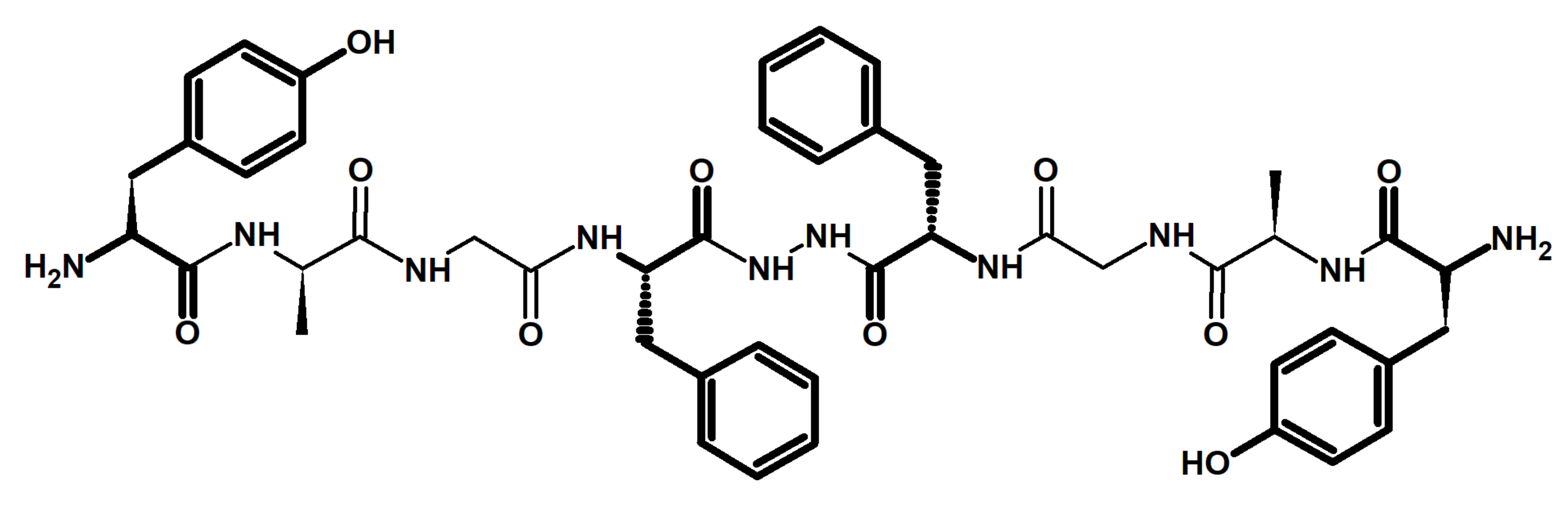
2. Chemistry and Pharmacology of Biphalin
3. Biphalin as an Analgesic Agent (In Vivo Studies)
4. Other Activity of Biphalin
4.1. Biphalin as an Antiviral and Antiproliferative Agent
4.2. Biphalin as an Immunomodulatory Agent
4.3. Biphalin as an Agent Improving Wound Healing
4.4. Biphalin as a Neuroprotective Agent
4.5. Cardiorespiratory Effect of Biphalin
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Hedner, T.; Cassuto, J. Opioids and opioid receptors. In The Essence of Analgesia and Analgesics; Cambridge University Press: Cambridge, UK, 2010; pp. 73–81. [Google Scholar] [CrossRef]
- Stein, S.; Zöllner, C. Opioids and Sensory Nerves. In Sensory Nerves; Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2009; Volume 194, pp. 139–183. ISBN 9783540790891. [Google Scholar]
- Al-Hasani, R.; Bruchas, M.R. Molecular Mechanisms of Opioid Receptor-Dependent Signalling and Behaviour. Anesthesiology 2011, 115, 1363–1381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodnar, R.J. Endogenous opiates and behavior: 2012. Peptides 2013, 50, 55–95. [Google Scholar] [CrossRef] [PubMed]
- Bodnar, R.J. Endogenous opioids and feeding behavior: A decade of further progress (2004–2014). A Festschrift to Dr. Abba Kastin. Peptides 2015, 72, 20–33. [Google Scholar] [CrossRef] [PubMed]
- Bodnar, R.J. Endogenous Opiates and Behavior: 2016. Peptides 2018, 101, 167–212. [Google Scholar] [CrossRef] [PubMed]
- Bodnar, R.J. Endogenous opiates and behavior: 2017. Peptides 2020, 124, 170223. [Google Scholar] [CrossRef] [PubMed]
- Bodnar, R.J. Endogenous Opiates and Behavior: 2018. Peptides 2020, 132, 170348. [Google Scholar] [CrossRef]
- Lipkowski, A.W.; Konecka, A.M.; Sroczyńska, I. Double-enkephalins—Synthesis, activity on guinea-pig ileum, and analgesic effect. Peptides 1982, 3, 697–700. [Google Scholar] [CrossRef]
- Cowell, S.M.; Sun Lee, Y. Biphalin: The Foundation of Bivalent Ligands. Curr. Med. Chem. 2016, 23, 3267–3284. [Google Scholar] [CrossRef]
- Feliciani, F.; Pinnen, F.; Stefanucci, A.; Costante, R.; Cacciatore, I.; Lucente, G.; Mollica, A. Structure-activity relationships of biphalin analogs and their biological evaluation on opioid receptors. Mini Rev. Med. Chem. 2013, 13, 11–33. [Google Scholar] [CrossRef]
- Tymecka, D.; Misicka, A. Solution Phase Peptide Synthesis: The Case of Biphalin. In Peptide Synthesis; Hussein, W., Skwarczynski, M., Toth, I., Eds.; Humana: New York, NY, USA, 2020; pp. 1–11. ISBN 978-1-0716-0226-3. [Google Scholar]
- Flippen-Anderson, J.L.; Deschamps, J.R.; George, C.; Hruby, V.J.; Misicka, A.; Lipkowski, A.W. Crystal structure of biphalin sulfate: A multireceptor opioid peptide. J. Pept. Res. 2002, 59, 123–133. [Google Scholar] [CrossRef]
- Urbańczyk-Lipkowska, Z.; Krajewski, J.W.; Gluziński, P.; Lipkowski, A.W.; Argay, G. The structure of N,N′-di-l-phenylalanine hydrazide determined by X-ray diffraction methods. J. Mol. Struct. 1986, 140, 151–157. [Google Scholar] [CrossRef]
- Konecka, A.M.; Sroczyńska, I.; Lipkowski, A.W. The effect of enkephalin dimers on body temperature in mice. Peptides 1987, 8, 431–435. [Google Scholar] [CrossRef]
- Lipkowski, A.W.; Konecka, A.M.; Sroczynska, I.; Przewlocki, R.; Stala, L.; Tam, S.W. Bivalent opioid peptide analogues with reduced distances between pharmacophores. Life Sci. 1987, 40, 2283–2288. [Google Scholar] [CrossRef]
- Stepinski, J.; Zajaczkowski, I.; Kazem-Bek, D.; Temeriusz, A.; Lipkowski, A.W.; Tam, W.S. Use of hydrophilic diamines for bridging of two opioid peptide pharmacophores. Int. J. Pept. Protein Res. 1991, 38, 588–592. [Google Scholar] [CrossRef]
- Misicka, A.; Lipkowski, A.W.; Horvath, R.; Davis, P.; Porreca, F.; Yamamura, H.I.; Hruby, V.J. Structure-activity relationship of biphalin. The synthesis and biological activities of new analogues with modifications in positions 3 and 4. Life Sci. 1997, 60, 1263–1269. [Google Scholar] [CrossRef]
- Slaninova, J.; Appleyard, S.M.; Misicka, A.; Lipkowski, A.W.; Knapp, R.J.; Weber, S.J.; Davis, T.P.; Yamamura, H.I.; Hruby, V.J. [125I-Tyr1]biphalin binding to opioid receptors of rat brain and NG108-15 cell membranes. Life Sci. 1998, 62, 199–204. [Google Scholar] [CrossRef]
- Li, G.; Haq, W.; Xiang, L.; Hughes, R.; De Leon, I.A.; Davis, P.; Gillespie, T.J.; Romanowski, M.; Zhu, X.; Misicka, A.; et al. Modification of the 4,4′-residues and SAR studies of biphalin, a highly potent opioid receptor active peptide. Bioorg. Med. Chem. Lett. 1998, 8, 555–560. [Google Scholar] [CrossRef]
- Lesniak, A.; Bochynska-Czyz, M.; Sacharczuk, M.; Benhye, S.; Misicka, A.; Bujalska-Zadrozny, M.; Lipkowski, A.W. Biphalin preferentially recruits peripheral opioid receptors to facilitate analgesia in a mouse model of cancer pain—A comparison with morphine. Eur. J. Pharm. Sci. 2016, 89, 39–49. [Google Scholar] [CrossRef] [PubMed]
- Stefanucci, A.; Carotenuto, A.; Macedonio, G.; Novellino, E.; Pieretti, S.; Marzoli, F.; Szücs, E.; Erdei, A.I.; Zádor, F.; Benyhe, S.; et al. Cyclic Biphalin Analogues Incorporating a Xylene Bridge: Synthesis, Characterization, and Biological Profile. ACS Med. Chem. Lett. 2017, 8, 858–863. [Google Scholar] [CrossRef] [PubMed]
- Mollica, A.; Carotenuto, A.; Novellino, E.; Limatola, A.; Costante, R.; Pinnen, F.; Stefanucci, A.; Pieretti, S.; Borsodi, A.; Samavati, R.; et al. Novel cyclic biphalin analogue with improved antinociceptive properties. ACS Med. Chem. Lett. 2014, 5, 1032–1036. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quock, R.M.; Burkey, T.H.; Varga, E.; Hosohata, Y.; Hosohata, K.; Cowell, S.M.; Slate, C.A.; Ehlert, F.J.; Roeske, W.R.; Yamamura, H.I. The δ-opioid receptor: Molecular pharmacology, signal transduction, and the determination of drug efficacy. Pharmacol. Rev. 1999, 51, 503–532. [Google Scholar] [PubMed]
- Lipkowski, A.W.; Misicka, A.; Davis, P.; Stropova, D.; Janders, J.; Lachwa, M.; Porreca, F.; Yamamura, H.I.; Hruby, V.J. Biological activity of fragments and analogues of the potent dimeric opioid peptide, biphalin. Bioorg. Med. Chem. Lett. 1999, 9, 2763–2766. [Google Scholar] [CrossRef]
- Dyniewicz, J.; Lipiński, P.F.J.; Kosson, P.; Leśniak, A.; Bochyńska-Czyż, M.; Muchowska, A.; Tourwe, D.; Ballet, S.; Misicka, A.; Lipkowski, A.W. Hydrazone Linker as a Useful Tool for Preparing Chimeric Peptide/Nonpeptide Bifunctional Compounds. ACS Med. Chem. Lett. 2017, 8, 73–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dyniewicz, J.; Lipinski, P.F.J.; Kosson, P.; Bochynska-Czyz, M.; Matalinska, J.; Misicka, A. Antinociceptive and cytotoxic activity of opioid peptides with hydrazone and hydrazide moieties at the C-terminus. Molecules 2020, 25, 3429. [Google Scholar] [CrossRef] [PubMed]
- Nair, P.; Yamamoto, T.; Largent-Milnes, T.M.; Cowell, S.; Kulkarni, V.; Moye, S.; Navratilova, E.; Davis, P.; Ma, S.W.; Vanderah, T.W.; et al. Truncation of the peptide sequence in bifunctional ligands with mu and delta opioid receptor agonist and neurokinin 1 receptor antagonist activities. Bioorg. Med. Chem. Lett. 2013, 23, 4975–4978. [Google Scholar] [CrossRef] [Green Version]
- Bonney Maszczynska, I.; Foran, S.E.; Marchand, J.E.; Lipkowski, A.W.; Carr, D.B. Spinal antinociceptive effects of AA501, a novel chimeric peptide with opioid receptor agonist and tachykinin receptor antagonist moieties. Eur. J. Pharmacol. 2004, 488, 91–99. [Google Scholar] [CrossRef]
- Yamamoto, T.; Nair, P.; Ma, S.-w.; Davis, P.; Yamamura, H.I.; Vanderah, T.W.; Porreca, F.; Lai, J.; Hruby, V.J. The biological activity and metabolic stability of peptidic bifunctional compounds that are opioid receptor agonists and neurokinin-1 receptor antagonists with a cystine moiety. Bioorg. Med. Chem. 2009, 17, 7337–7343. [Google Scholar] [CrossRef] [Green Version]
- Hruby, V.J.; Agnes, R.S.; Davis, P.; Ma, S.W.; Lee, Y.S.; Vanderah, T.W.; Lai, J.; Porreca, F. Design of novel peptide ligands which have opioid agonist activity and CCK antagonist activity for the treatment of pain. Life Sci. 2003, 73, 699–704. [Google Scholar] [CrossRef] [Green Version]
- Nair, P.; Yamamoto, T.; Cowell, S.; Kulkarni, V.; Moye, S.; Navratilova, E.; Davis, P.; Ma, S.-W.; Vanderah, T.W.; Lai, J.; et al. Discovery of tripeptide-derived multifunctional ligands possessing delta/mu opioid receptor agonist and neurokinin 1 receptor antagonist activities. Bioorg. Med. Chem. Lett. 2015, 25, 3716–3720. [Google Scholar] [CrossRef] [Green Version]
- Starnowska-Sokół, J.; Piotrowska, A.; Bogacka, J.; Makuch, W.; Mika, J.; Witkowska, E.; Godlewska, M.; Osiejuk, J.; Gątarz, S.; Misicka, A.; et al. Novel hybrid compounds, opioid agonist+melanocortin 4 receptor antagonist, as efficient analgesics in mouse chronic constriction injury model of neuropathic pain. Neuropharmacology 2020, 178, 108232. [Google Scholar] [CrossRef]
- Mollica, A.; Davis, P.; Ma, S.-W.; Porreca, F.; Lai, J.; Hruby, V.J. Synthesis and biological activity of the first cyclic biphalin analogues. Bioorg. Med. Chem. Lett. 2006, 16, 367–372. [Google Scholar] [CrossRef]
- Yamamoto, T.; Nair, P.; Vagner, J.; Largent-Milnes, T.; Davis, P.; Ma, S.; Navratilova, E.; Moye, S.; Tumati, S.; Lai, J.; et al. A Structure–Activity Relationship Study and Combinatorial Synthetic Approach of C-Terminal Modified Bifunctional Peptides That Are δ/μ Opioid Receptor Agonists and Neurokinin 1 Receptor Antagonists. J. Med. Chem. 2008, 51, 1369–1376. [Google Scholar] [CrossRef] [Green Version]
- Kramer, T.H.; Davis, P.; Hruby, V.J.; Burks, T.F.; Porreca, F. In vitro potency, affinity and agonist efficacy of highly selective delta opioid receptor ligands. J. Pharmacol. Exp. Ther. 1993, 266, 577–584. [Google Scholar]
- Kamei, J.; Kasuya, Y. Effects of Double-enkephalin (Biphalin), an Enkephalin Analogue, on Respiration and the Cough Reflex in Rats. J. Pharmacobiodyn. 1988, 11, 645–650. [Google Scholar] [CrossRef] [Green Version]
- Weber, S.J.; Abbruscato, T.J.; Brownson, E.A.; Lipkowski, A.W.; Polt, R.; Misicka, A.; Haaseth, R.C.; Bartosz, H.; Hruby, V.J.; Davis, T.P. Assessment of an in vitro blood-brain barrier model using several [Met5]enkephalin opioid analogs. J. Pharmacol. Exp. Ther. 1993, 266, 1649–1655. [Google Scholar]
- Abbruscato, T.J.; Williams, S.A.; Misicka, A.; Lipkowski, A.W.; Hruby, V.J.; Davis, T.P. Blood-to-central nervous system entry and stability of biphalin, a unique double-enkephalin analog, and its halogenated derivatives. J. Pharmacol. Exp. Ther. 1996, 276, 1049–1057. [Google Scholar] [PubMed]
- Abbruscato, T.J.; Thomas, S.A.; Hruby, V.J.; Davis, T.P. Brain and spinal cord distribution of biphalin: Correlation with opioid receptor density and mechanism of CNS entry. J. Neurochem. 1997, 69, 1236–1245. [Google Scholar] [CrossRef]
- Egleton, R.D.; Abbruscato, T.J.; Thomas, S.A.; Davis, T.P. Transport of Opioid Peptides into the Central Nervous System. J. Pharm. Sci. 1998, 87, 1433–1439. [Google Scholar] [CrossRef] [PubMed]
- Albekairi, T.H.; Vaidya, B.; Patel, R.; Nozohouri, S.; Villalba, H.; Zhang, Y.; Lee, Y.S.; Al-Ahmad, A.; Abbruscato, T.J. Brain delivery of a potent opioid receptor agonist, biphalin during ischemic stroke: Role of organic anion transporting polypeptide (OATP). Pharmaceutics 2019, 11, 467. [Google Scholar] [CrossRef] [Green Version]
- Lipkowski, A.W.; Carr, D.B.; Bonney, I.; Misicka, A. Biphalin: A multireceptor opioid ligand. In The Delta Receptor; Chang, K.-J., Porreca, F., Woods, J.H., Eds.; CRC Press: New York, NY, USA, 2003. [Google Scholar]
- Romanowski, M.; Zhu, X.; Kim, K.; Hruby, V.J.; O’Brien, D.F. Interaction of enkephalin peptides with anionic model membranes. Biochim. Biophys. Acta Biomembr. 2002, 1558, 45–53. [Google Scholar] [CrossRef] [Green Version]
- Romanowski, M.; Zhu, X.; Ramaswami, V.; Misicka, A.; Lipkowski, A.W.; Hruby, V.J.; O’Brien, D.F. Interaction of a highly potent dimeric enkephalin analog, biphalin, with model membranes. Biochim. Biophys. Acta Biomembr. 1997, 1329, 245–258. [Google Scholar] [CrossRef] [Green Version]
- Shen, K.F.; Crain, S.M. Biphalin, an enkephalin analog with unexpectedly high antinociceptive potency and low dependence liability in vivo, selectively antagonizes excitatory opioid receptor functions of sensory neurons in culture. Brain Res. 1995, 701, 158–166. [Google Scholar] [CrossRef]
- Horan, P.J.; Mattia, A.; Bilsky, E.J.; Weber, S.; Davis, T.P.; Yamamura, H.I.; Malatynska, E.; Appleyard, S.M.; Slaninova, J.; Misicka, A. Antinociceptive profile of biphalin, a dimeric enkephalin analog. J. Pharmacol. Exp. Ther. 1993, 265, 1446–1454. [Google Scholar] [PubMed]
- Silbert, B.S.; Cepeda, M.S.; Szyfelbein, S.K.; Osgood, P.E.; Carr, D.B. Analgesic activity of a novel bivalent opioid peptide compared to morphine via different routes of administration. Comp. Study 1991, 33, 382–387. [Google Scholar] [CrossRef] [PubMed]
- Silbert, B.S.; Lipkowski, A.W.; Soledad Cepeda, M.S.; Szyfelbein, S.K.; Osgood, P.F.; Carr, D.B. Enhanced potency of receptor-selective opioids after acute burn injury. Anesth. Analg. 1991, 73, 427–433. [Google Scholar] [CrossRef]
- Lipkowski, A.W.; Carr, D.B.; Langlade, A.; Osgood, P.F.; Szyfelbein, S.K. Morphine-3-glucoronide: Silent regulator of morphine actions. Life Sci. 1994, 55, 149–154. [Google Scholar] [CrossRef]
- Misterek, K.; Maszczynska, I.; Dorociak, A.; Gumulka, S.W.; Carr, D.B.; Szyfelbein, S.K.; Lipkowski, A.W. Spinal co-administration of peptide substance P antagonist increases antinociceptive effect of the opioid peptide biphalin. Life Sci. 1994, 54, 939–944. [Google Scholar] [CrossRef]
- Huber, J.D.; Campos, C.R.; Egleton, R.D.; Witt, K.; Guo, L.; Roberts, M.J.; Bentley, M.D.; Davis, T.P. Conjugation of low molecular weight poly(ethylene glycol) to biphalin enhances antinociceptive profile. J. Pharm. Sci. 2003, 92, 1377–1385. [Google Scholar] [CrossRef]
- Kosson, D.; Bonney, I.; Carr, D.B.; Mayzner-Zawadzka, E.; Lipkowski, A.W. Antinociception after intrathecal biphalin application in rats: A reevaluation and novel, rapid method to confirm correct catheter tip position. Pharmacol. Rep. 2005, 57, 545–549. [Google Scholar]
- Kosson, D.; Klinowiecka, A.; Kosson, P.; Bonney, I.; Carr, D.B.; Mayzner-Zawadzka, E.; Lipkowski, A.W. Intrathecal antinociceptive interaction between the NMDA antagonist ketamine and the opioids, morphine and biphalin. Eur. J. Pain 2008, 12, 611–616. [Google Scholar] [CrossRef] [PubMed]
- Leone, S.; Chiavaroli, A.; Orlando, G.; Mollica, A.; Di Nisio, C.; Brunetti, L.; Vacca, M. The analgesic activity of biphalin and its analog {AM} 94 in rats. Eur. J. Pharmacol. 2012, 685, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Popiolek-Barczyk, K.; Piotrowska, A.; Makuch, W.; Mika, J. Biphalin, a Dimeric Enkephalin, Alleviates LPS-Induced Activation in Rat Primary Microglial Cultures in Opioid Receptor-Dependent and Receptor-Independent Manners. Neural Plast. 2017, 2017, 3829472. [Google Scholar] [CrossRef] [PubMed]
- Sobczak, M.; Pilarczyk, A.; Jonakowski, M.; Jarmuz, A.; Sałaga, M.; Lipkowski, A.W.; Fichna, J. Anti-inflammatory and antinociceptive action of the dimeric enkephalin peptide biphalin in the mouse model of colitis: New potential treatment of abdominal pain associated with inflammatory bowel diseases. Peptides 2014, 60, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, M.; Suzuki, T.; Narita, M.; Lipkowski, A.W. The opioid peptide analogue biphalin induces less physical dependence than morphine. Life Sci. 2001, 69, 1023–1028. [Google Scholar] [CrossRef]
- Tang, J.L.; Lipkowski, A.W.; Specter, S. Inhibitory effect of biphalin and azt on murine Friend leukemia virus infection in vitro. Int. J. Immunopharmacol. 1998, 20, 457–466. [Google Scholar] [CrossRef]
- Tang, J.L.; Lipkowski, A.W.; Specter, S. Molecular assessment of the potential combination therapy of cytokines with biphalin and AZT for friend leukemia virus infection in vitro. Pharmacol. Rep. 2008, 60, 190–198. [Google Scholar]
- Lazarczyk, M.; Matyja, E.; Lipkowski, A.W. A comparative study of morphine stimulation and biphalin inhibition of human glioblastoma T98G cell proliferation in vitro. Peptides 2010, 31, 1606–1612. [Google Scholar] [CrossRef]
- Mehrotra, S.; Prajapati, R.K.; Haq, W.; Singh, V.K. Immunomodulation by biphalin, dimeric synthetic opioid peptide, and its analog. Immunopharmacol. Immunotoxicol. 2002, 24, 83–96. [Google Scholar] [CrossRef]
- Zielińska, M.; Jarmuz, A.; Sałaga, M.; Lipkowski, A.W.; Fichna, J. Mixed MOP/DOP agonist biphalin elicits anti-transit effect in mouse models mimicking diarrhea-predominant irritable bowel syndrome symptoms. Pharmacol. Rep. 2016, 68, 32–36. [Google Scholar] [CrossRef]
- Yıldız, E.; Totuk, Ö.M.G.; Mollica, A.; Kabadayı, K.; Şahin, A. Effects of Biphalin on Corneal Epithelial Wound Healing. Proceedings 2018, 2, 1552. [Google Scholar] [CrossRef] [Green Version]
- Muchowska, A.; Redkiewicz, P.; Różycki, K.; Matalińska, J.; Lipiński, P.F.J.; Czuwara, J.; Kosson, P. The analgesic hybrid of dermorphin/substance P and analog of enkephalin improve wound healing in streptozotocin-induced diabetic rats. Wound Repair Regen. 2020, 28, 177–184. [Google Scholar] [CrossRef]
- Kawalec, M.; Kowalczyk, J.E.; Beresewicz, M.; Lipkowski, A.W.; Zablocka, B. Neuroprotective potential of biphalin, multireceptor opioid peptide, against excitotoxic injury in hippocampal organotypic culture. Neurochem. Res. 2011, 36, 2091–2095. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Wang, H.; Shah, K.; Karamyan, V.T.; Abbruscato, T.J. Opioid receptor agonists reduce brain edema in stroke. Brain Res. 2011, 1383, 307–316. [Google Scholar] [CrossRef]
- Yang, L.; Shah, K.; Wang, H.; Karamyan, V.T.; Abbruscato, T.J. Characterization of Neuroprotective Effects of Biphalin, an Opioid Receptor Agonist, in a Model of Focal Brain Ischemia. J. Pharmacol. Exp. Ther. 2011, 339, 499–508. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Islam, M.R.; Karamyan, V.T.; Abbruscato, T.J. In vitro and in vivo efficacy of a potent opioid receptor agonist, biphalin, compared to subtype-selective opioid receptor agonists for stroke treatment. Brain Res. 2015, 1609, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Lesniak, A.; Pick, C.G.; Misicka, A.; Lipkowski, A.W.; Sacharczuk, M. Biphalin protects against cognitive deficits in a mouse model of mild traumatic brain injury (mTBI). Neuropharmacology 2016, 101, 506–518. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.J.; Li, Y.; Chen, S.; Yang, X.F.; Min, J.W. Biphalin, a dimeric opioid peptide, reduces neonatal hypoxia-ischemia brain injury in mice by the activation of PI3K/Akt signaling pathway. J. Chem. Neuroanat. 2021, 115, 101967. [Google Scholar] [CrossRef] [PubMed]
- Wojciechowski, P.; Szereda-Przestaszewska, M.; Lipkowski, A.W. Respiratory and cardiovascular effects of biphalin in anaesthetized rats. Eur. J. Pharmacol. 2009, 602, 50–53. [Google Scholar] [CrossRef] [PubMed]
- Wojciechowski, P.; Szereda-Przestaszewska, M.; Lipkowski, A.W. Delta opioid receptors contribute to the cardiorespiratory effects of biphalin in anesthetized rats. Pharmacol. Rep. 2011, 63, 1235–1242. [Google Scholar] [CrossRef]
- Bądzyńska, B.; Lipkowski, A.W.; Sadowski, J. An antihypertensive opioid: Biphalin, a synthetic non-addictive enkephalin analog decreases blood pressure in spontaneously hypertensive rats. Pharmacol. Rep. 2016, 68, 51–55. [Google Scholar] [CrossRef]
- Bądzyńska, B.; Lipkowski, A.W.; Olszyński, K.H.; Sadowski, J. Different blood pressure responses to opioids in three rat hypertension models: Role of the baseline status of sympathetic and renin-angiotensin systems. Can. J. Physiol. Pharmacol. 2016, 94, 1159–1169. [Google Scholar] [CrossRef]
- Stein, C.; Küchler, S. Targeting inflammation and wound healing by opioids. Trends Pharmacol. Sci. 2013, 34, 303–312. [Google Scholar] [CrossRef]
- Marotti, T.; Burek, B.; Rabatic, S.; Balog, T.; Hrsak, I. Modulation of lipopolysaccharide-induced production of cytokines by methionine-enkephalin. Immunol. Lett. 1994, 40, 43–47. [Google Scholar] [CrossRef]
- Marotti, T.; Balog, T.; Maeuran, R.; Ro, B. The role of cytokines in MET-enkephalin-modulated nitric oxide release. Neuropeptides 1998, 32, 57–62. [Google Scholar] [CrossRef]
- Liang, Y.; Chu, H.; Jiang, Y.; Yuan, L. Morphine enhances IL-1β release through toll-like receptor 4-mediated endocytic pathway in microglia. Purinergic Signal. 2016, 12, 637–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, N.; Li, H.; Wei, D.; LeSage, G.; Chen, L.; Wang, S.; Zhang, Y.; Chi, L.; Ferslew, K.; He, L.; et al. Glycogen synthase kinase-3 and p38 MAPK are required for opioid-induced microglia apoptosis. Neuropharmacology 2010, 59, 444–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merighi, S.; Gessi, S.; Varani, K.; Fazzi, D.; Stefanelli, A.; Borea, P.A. Morphine mediates a proinflammatory phenotype via m-opioid receptor-PKCε-Akt-ERK1/2 signaling pathway in activated microglial cells. Biochem. Pharmacol. 2013, 86, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Gessi, S.; Borea, P.A.; Bencivenni, S.; Fazzi, D.; Varani, K.; Merighi, S. The activation of μ-opioid receptor potentiates LPS-induced NF-kB promoting an inflammatory phenotype in microglia. FEBS Lett. 2016, 590, 2813–2826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horvath, R.J.; DeLeo, J.A. Morphine enhances microglial migration through modulation of P2X 4 receptor signaling. J. Neurosci. 2009, 29, 998–1005. [Google Scholar] [CrossRef] [PubMed]
- Corder, G.; Tawfik, V.L.; Wang, D.; Sypek, E.I.; Low, S.A.; Dickinson, J.R.; Sotoudeh, C.; Clark, J.D.; Barres, A.; Bohlen, C.J.; et al. Loss of µ-opioid receptor signaling in nociceptors, and not spinal microglia, abrogates morphine tolerance without disrupting analgesic efficacy. Nat. Med. 2017, 23, 164–173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lam, C.-F.; Chang, P.-J.; Huang, Y.-S.; Sung, Y.-H.; Huang, C.-C.; Lin, M.-W.; Liu, Y.-C.; Tsai, Y.-C. Prolonged Use of High-Dose Morphine Impairs Angiogenesis and Mobilization of Endothelial Progenitor Cells in Mice. Anesth. Analg. 2008, 107, 686–692. [Google Scholar] [CrossRef]
- Gupta, M.; Poonawala, T.; Farooqui, M.; Ericson, M.E.; Gupta, K. Topical fentanyl stimulates healing of ischemic wounds in diabetic rats. J. Diabetes 2015, 7, 573–583. [Google Scholar] [CrossRef] [Green Version]
- Rook, J.M.; Hasan, W.; McCarson, K.E. Temporal Effects of Topical Morphine Application on Cutaneous Wound Healing. Anesthesiology 2008, 109, 130–136. [Google Scholar] [CrossRef] [Green Version]
- Garbuz, O.; Gulea, A.; Dyniewicz, J.; Zablocka, B.; Lipkowski, A.W. The non-opioid receptor, antioxidant properties of morphine and the opioid peptide analog biphalin. Peptides 2015, 63, 1–3. [Google Scholar] [CrossRef]
- Stefanucci, A.; Dimmito, M.P.; Macedonio, G.; Ciarlo, L.; Pieretti, S.; Novellino, E.; Lei, W.; Barlow, D.; Houseknecht, K.L.; Streicher, J.M.; et al. Potent, Efficacious, and Stable Cyclic Opioid Peptides with Long Lasting Antinociceptive Effect after Peripheral Administration. J. Med. Chem. 2020, 63, 2673–2687. [Google Scholar] [CrossRef]
- Stefanucci, A.; Dimmito, M.P.; Molnar, G.; Streicher, J.M.; Novellino, E.; Zengin, G.; Mollica, A. Developing Cyclic Opioid Analogues: Fluorescently Labeled Bioconjugates of Biphalin. ACS Med. Chem. Lett. 2020, 11, 720–726. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Source of the Opioid Receptors | Ki (nM) ± S.E.M. * | IC50 (nM) ± S.E.M. * | Ref. | ||||
|---|---|---|---|---|---|---|---|
| MOR | DOR | KOR | MOR | DOR | KOR | ||
| Guinea pig brain membrane | 12 ± 2 a | 4.6 ± 0.2 b | 270 ± 15 c | [16,17] | |||
| Rat brain membrane | 1.4 ± 0.4 d | 2.6 ± 0.3 e | [18] | ||||
| Rat brain membrane | 0.74 ± 0.26 d | 2.96 ± 0.22 e | 35.1 ± 2.0 f | [19] | |||
| Guinea pig brain homogenate | 2.8 ± 0.4 d | 5.2 ± 0.3 g | [20] | ||||
| Rat brain homogenate | 0.19 (0.12-0.29) h (95% CI) * | 1.04 (0.69-1.55) i (95% CI) * | [21] | ||||
| Rat brain membrane | 0.79 h | 3.5 j | [22] | ||||
| Rat brain membrane | 2.6 ± 0.7 h | 15 ± 2.3 k | 283.1 ± 18 f | [23] | |||
| CHO cell transfected with cloned human δ-opioid receptor membrane | 46.5 ± 1.5 l | [24] | |||||
| Compound | DOR | MOR | KOR | Ref. | |||
|---|---|---|---|---|---|---|---|
| Emax ± S.E.M * (%) | EC50 ± S.E.M * (nM) | Emax ± S.E.M * (%) | EC50 ± S.E.M * (nM) | Emax ± S.E.M * (%) | EC50 ± S.E.M * (nM) | ||
| Biphalin | 176.9 ± 4.9 | 75.4 | [22] | ||||
| 219.6 ± 5.7 | 90.5 ± 25 | 178.2 ± 3.6 | 12 ± 4.6 | 108.9 ± 4.1 | Amb # | [23] | |
| 98 ± 10 | 34.0 ± 13.1 | [24] | |||||
| 83 ± 4 | 1.1 | [35] | |||||
| 27 ± 3.5 | 2.5 ± 0.5 | 25 ± 4.7 | 6 ± 0.2 | [34] | |||
| 238 ± 4.9 | pEC50 ± S.E.M * −7.0 ± 0.08 | [21] | |||||
| Deltrophin II | 96 ± 2 | 9.3 ± 4.2 | [24] | ||||
| Compound | Bioassay IC50 (nM) ± S.E.M * | Ref. | |
|---|---|---|---|
| GPI | MVD | ||
| Biphalin | 1.94 ± 0.29 | [9] | |
| 8.8 | 27 | [25] | |
| 8.8 ± 0.3 | 27 ± 1.5 | [35,36] | |
| D-Ala2-Met-enkephalinamide | 22.8 ± 3.4 | [9] | |
| Pain Model/Animal/Test | Route of Administration/ Dose | Effect | Ref. |
|---|---|---|---|
| Mouse, hot-plate assay | i.p./ 5, 10, 20 mg/kg | The dose of 20 mg/kg increased the response latency 60 min after the injection by 185.8% compared to the pre-injection control. | [9] |
| Rat, hot plate test | i.p./ 10, 20 mg/kg | The dose of 20 mg/kg increased the latency of the response 60 min after administration by 177.4% of the pre-injection control value. | [37] |
| Rat, tail-flick test Rat, tail pinch test | s.c./2.5, 5, 10, 40, 80 μmol/kg i.v./10, 20, 40 μmol/kg i.t./0.5, 10, 20 nmol | s.c.—the ED50 (95% Cl) was 7.88 nmol/kg (6.33–9.81) for tail flick and 5.58 nmol/kg (4.80–6.48) for tail pinch. i.v.—the ED50 (95% Cl) were 17.87µmol/kg (15.06–21.19) for tail flick and 18.9 µmol/kg (15.0–23.8) for tail pinch test. i.t.—ED50 (95% Cl) were 2.88 nmol (1.09–7.54) for tail flick and greater than 250 nmol for tail pinch. | [48] |
| Non burned (NB) rat, Burned (B) rat, tail-flick test. | i.v./ NB group: 5, 10, 15 μmol/kg B group: 5, 7.5, 10 μmol/kg | ED50 (95% Cl) are 7.34 (6.37–8.30) and 10.69 (8.66–12.73) μmol/kg in the B and NB groups, respectively. | [49] |
| Rat, tail flick test | i.v./5 μmol/kg | ~45% MPE at 5 min after administration. | [50] |
| Rat, tail-flick test | i.t./0.75, 2.5, 5.0 μg | ~40% MPE at a dose of 0.75 μg at 15 min after administration. ~50% MPE at a dose of 2.5 μg at 15 min after administration. ~80% MPE at a dose of 5 μg at 30 min after administration. | [51] |
| ICR mice tail flick test | i.c.v./1, 3, 10, 30, 100 pmol/mouse i.t./8.8, 880, 8800 pmol/mouse i.p./2.6, 4, 5.3, 8.8 μmol/kg | i.c.v.—A50 (95% Cl) of 4.9 (1.6–15.3) pmol/mouse and a time to peak effect of 20 min. i.t.—~60% antinociceptive response up to a dose of 8.8 nmol/mouse. i.p.—A50 (95% Cl) of 5.7 (3.7–8.7) μmol/kg; the peak effect after 20 min. | [47] |
| ICR mice, tail-flick test | i.c.v./0.4 nmol/kg i.v./685 nmol/kg | i.c.v.—68% MPE at 20 min then quickly dropped to <10% MPE by 45 min. i.v.—83% MPE at 30–60 min. | [52] |
| Rat, tail-flick test | i.v./150, 300, 600, 1200 nmol/kg i.m./4300, 8600, 17200 nmol/kg s.c./4300, 8600, 17200 nmol/kg | i.v.—A50 ± S.E.M 523 ± 9 nmol/kg. i.m.—A50 ± S.E.M 236 ± 42 nmol/kg. s.c.—A50 ± S.E.M 9276 ± 1290 nmol/kg. | |
| Rat with encephalomyelitis (EAE) | i.v. | 83% MPE at 15 min after administration. The analgesic potency correlated well with the progression of EAE | [43] |
| Rat, tail-flick test | i.t./0.001, 0.005, 0.0125, 0.025, 0.5, 2, 20 nmol | 60–70% MPE at a dose of 0.005 nmol at 15 min after administration. 100% MPE at doses of 0.5 and 2.0 nmol at 15–30 min after administration. At a dose of 20 nmol, long-lasting analgesia, body rigidity. | [53] |
| Rat, tail-flick test | i.t./0.005 μmol | 60–70% MPE at 15 min after administration. | [54] |
| Mouse, hot plate test | i.c.v./0.1 nmol/mouse i.v./1500 nmol/kg | i.c.v.—~90% of MPE at 30 min after administration. i.v.—~40% of MPE at 30 min after administration. | [23] |
| Mouse, tail-flick test | i.c.v./0.1 nmol/mouse i.v./1500 nmol/kg | i.c.v.—~85% of MPE at 15–45 min after administration. i.v.—~40% of MPE at 30 min after administration. | |
| Rat, hot-plate test | i.c.v./1 nmol/kg i.v./1200 nmol/kg | i.c.v.—71% MPE at 30 min after administration. i.v.—68.32% MPE at 45 min after administration. | [55] |
| Mouse, tail-flick test | i.t./0.01 nmol/animal i.c.v/0.01 nmol/animal | ~80% of MPE were obtained at 15 min after injection in both tests i.t. and i.c.v. | [22] |
| Pain Model/Animal/Test | Route of Administration/Dose | Effect | Ref. |
|---|---|---|---|
| Cancer pain/Mouse, paw withdrawal test, tail-flick test | i.v./5, 10, 15, 20 μmol/kg | Dose-dependent increase in the total analgesic effect, higher doses caused motor impairments and muscle rigidity. The complete alleviation of thermally-induced pain was observed for a dose of 20 μmol/kg, %MPE reached 100% in most mice. At a dose of 20 μmol/kg, a strong peak analgesic effect in the tumor-bearing paw, %MPE: 55.5 ± 4.5. ED50 (μmol/kg, 95% CI) for: tumor-bearing paw: 19.10 (18.0–20.2); intact paw: 17.6 (15.6–18.7); tail-flick: 11.8 (10.9–12.6). | [21] |
| Acute and inflammatory pain/Mouse, formalin test | s.c./0.1 nmol/animal | Reduced formalin-induced pain behavior both in the early (acute pain) and in the late phase (inflammatory pain) of the test. | [22] |
| Neuropathic pain/Rat, mechanical and thermal hypersensitivity as measured by von Frey and cold plate tests. | i.t./20, 200, 1000 µM | Attenuated the development of tactile hypersensitivity as measured by von Frey test 30 min after drug injection, as compared to the vehicle-treated CCI (chronic constriction injury)-exposed rats (12.78 g ±0.55 versus 19.88 g ± 0.63, 25.58 g ± 0.32, and 25.91 g ± 0.09). Attenuated the development of thermal hypersensitivity as measured by cold plate test 35 min after drug administration, as compared to the vehicle-treated CCI-exposed animals (6.93 s ± 2.97 versus 20.11 s ± 2.81, 26.27 s ± 1.67, and 29.90 s ± 0.11, respectively, for administered doses). | [56] |
| Visceral pain/Mouse with acute colitis, colonic inflammation, mustard oil-induced pain responses and hot plate test | i.p./5 mg/kg i.c./5 mg/kg | Produced a strong analgesic effect in inflamed mice (mustard oil-induced pain) after i.p. injection (10 ± 1 vs. 51 ± 8 number of pain responses for vehicle-treated mice) and after i.c. injection. Delay in rearing (91 ± 12 vs. 48 ± 4 for biphalin vs. vehicle-treated mice) and jumping (183 ± 17 vs. 83 ± 13, respectively) times in the hot plate test. | [57] |
| Activity | Cell Line/Animal Model | Route of Administration/Dose | Effect | Ref. |
|---|---|---|---|---|
| Antiviral | Mus Dunni cells infected with FLV/ in vitro | 10−6–10−8 M | Inhibition of FLV RT activity. | [59,60] |
| 15–30 µg mixed with 0.5 ng/mL of AZT | Inhibition of FLV replication by 50%. | |||
| 100 μg/mL mixed with 10−6 of splenocytes | Inhibition of FLV replication by 58%. | |||
| 100 pg/mL mixed with 1 ng/mL of AZT and 10−6 of splenocytes | Inhibition of FLV replication by 68%. | |||
| 50 μg/mL mixed with 250 ng of INF-γ | Inhibition of FLV RT activity by 94%. | |||
| Antiproliferative (anticancer) | Human glioblastoma T98G/in vitro | 50 nM–40 μM | Inhibition of tumor cell growth and decrease in proliferation rate. Decline of cell ability to form colonies. Modulation of Ki69 proliferation index. | [61] |
| Immunomodulatory | Lymphocyte T, NK cells, suspension of human PMBCs and mouse macrophage RAW 264.7/in vitro | 10−8 or 10−10 M | Increase in cytotoxicity of NK cells. Stimulation of lymphocyte T proliferation. Increase in IL-2 production. Increase in chemotactic activity of monocytes. Marginal decrease in TNF-α and NO production by LPS. | [62] |
| Microglia cell culture LPS-stimulated/in vitro | 10 µM | Decrease in NO production, expression of Iba1, iNOS, IL-1β, IL-18, IL-6, IL-10, TNFα, pSTAT3, pERK1/2, p-NF-κB, p-IκB, p-p38MAPK, TRIF, and upregulation of SOCS3, TLR4, MyD88. | [56] | |
| CCI, chronic construction injury model of neuropathic pain in Wistar rats/in vivo | i.t./20, 200, 1000 μM | Diminished symptoms of neuropathy in von Frey test and cold plate test. | ||
| Semi-chronic colitis model in Balb/C mice/in vivo | i.c./5 mg/kg | Decrease in macroscopic and ulcer scores. | [57] | |
| i.p./5 mg/kg | No noticeable effect on colitis. | |||
| Ileum and distal colon from Balb/C mice/in vitro | 10−10–10−6 M into organs baths | Inhibition of colonic and ileal smooth muscle contractions. | [63] | |
| Balb/C mice/in vivo | i.p./5 mg/kg | Inhibition of colon motility. Prolongation of GI-transit and inhibition of colonic bead expulsion. Reversed hypermotility and exertion of anti-diarrheal effect. | ||
| Wound healing | Corneal epithelial cell culture (HCEC)/in vitro | 1 μM, 10 μM | Increased wound closure in in vitro wound healing model and increase in cell migration in transwell migration assay. | [64] |
| Streptozotocin-induced diabetic Wistar rats/in vivo | 1 mM | Reduction in wound size by 77% after 14 days of healing. Increase in the number of macrophages on day 4. Increase in the thickness of the epidermis on day 21. | [65] | |
| Neuroprotective | Hippocampal organotypic culture/in vitro | 0.025–0.1 μM | Reduction in NMDA-induced neuronal damage. Reduction in the number of dead cells. | [66] |
| Mouse primary cortical neurons exposed to OGD/in vitro | 0.001 nM–1 nM | Decrease in cell volume after OGD treatment. | [67] | |
| Hippocampal slices exposed to OGD/in situ | 0.01 µM–10 µM | Decrease in water content compared with selective agonists. | ||
| pMCAO model of CD-1 male mice/in vivo | i.p./5.7 µmol/kg | Decrease in edema (53%) and infarct ratios (48%) and neuronal recovery from stroke. | ||
| Mouse primary cortical neurons OGD treatment/in vitro | 0.01 nM | Decrease in cell volume after OGD treatment. | [68] | |
| tMCAO and pMCAO model of CD-1 male mice/in vivo | i.p./5.7 μmol/kg | Decrease in edema ratios by 66.6% tMCAO and by 58.3% pMCAO; decreased infarct ratios by 52.2% tMCAO and by 56.4% pMCAO. Improvement of neurological scores and locomotor activity. Decrease in penumbral expression of Na+, K+, 2Cl− cotransporter and translocation of isoforms of protein kinase C. | ||
| Mouse primary cortical neurons challenged with glutamate and hypoxic/aglycemic (H/A)/in vitro | 10 nM | Decrease in neuronal death; decrease in ROS production. | [69] | |
| tMCAO with reperfusion/in vivo | i.p./5 mg/kg | Reduction in the edema ratios by 76.4% and reduction in the infarct ratio by 77.3%. | ||
| Mouse model of mild traumatic brain injury (mTBI)/in vivo | i.v./10 mg/kg | Improvement of recognition memory in mTBI mice. Only a partial reversion of depressive-like immobility in mTBI mice; failed to reverse spatial memory deficits in mTBI mice. Immediate, delayed, or chronic biphalin administration improved spatial memory in mTBI mice. | [70] | |
| Mouse neonatal HI model/in vitro | i.p./5, 10, 20 mg/kg immediately after HI | Reduction in the infarct volume, brain edema, and brain atrophy. Improvement of neurobehavioral outcomes in neonatal HI in mice. Reduction in the infarct volume and brain edema required the activation of the opioid receptor/PI3K/Akt signaling pathway. Inhibition of HI-induced brain atrophy after long-term survival. Regulation in the expression level of phosphorylated Akt and apoptotic proteins after HI. | [71] | |
| Cardiorespiratory | Wistar rats/in vivo | i.v./0.3 mg/kg | Evocation of apnoea with a mean duration of 13.5 ± 1.25 s. Evocation of significant increases in Vt from immediate post-apnoeic phase to later time points. Reduction in respiratory rate. Decrease in breathing rate and increase in tidal volume, hypotension, and bradycardia. | [72,73] |
| Blood pressure and renal flows | Spontaneously hypertensive rats (SHR) Normotensive Wistar-Kyoto rats (WKY)/in vitro | i.v./150 μg/kg/h | Decrease in mean arterial blood pressure (MAP). Modest increase in renal blood flow and decrease in renal and hind limb vascular resistance. | [74] |
| Normotensive S-D rats Spontaneously hypertensive rats (SHR) Male S-D rats on a high-salt diet (HS/UNX) Male S-D rats with f angiotensin-induced hypertension (Ang-iH)/in vivo | i.v./300 µg/kg/h | Decrease in blood pressure in SHR but not in the HS/UNX and Ang-iH or normotensive WKY and S-D rats. In anesthetized and conscious SHR, decrease in MAP by ~10 and ~20 mmHg, respectively. In anesthetized HS/UNX and normotensive rats, increase in MAP by~6–7 mmHg. No changes in the MAP of Ang-iH rats. | [75] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Redkiewicz, P.; Dyniewicz, J.; Misicka, A. Biphalin—A Potent Opioid Agonist—As a Panacea for Opioid System-Dependent Pathophysiological Diseases? . Int. J. Mol. Sci. 2021, 22, 11347. https://doi.org/10.3390/ijms222111347
Redkiewicz P, Dyniewicz J, Misicka A. Biphalin—A Potent Opioid Agonist—As a Panacea for Opioid System-Dependent Pathophysiological Diseases? . International Journal of Molecular Sciences. 2021; 22(21):11347. https://doi.org/10.3390/ijms222111347
Chicago/Turabian StyleRedkiewicz, Patrycja, Jolanta Dyniewicz, and Aleksandra Misicka. 2021. "Biphalin—A Potent Opioid Agonist—As a Panacea for Opioid System-Dependent Pathophysiological Diseases? " International Journal of Molecular Sciences 22, no. 21: 11347. https://doi.org/10.3390/ijms222111347