
Platelet-Derived PCSK9 Is Associated with LDL Metabolism and Modulates Atherothrombotic Mechanisms in Coronary Artery Disease

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Platelet Count Is Associated with Plasma LDL Cholesterol
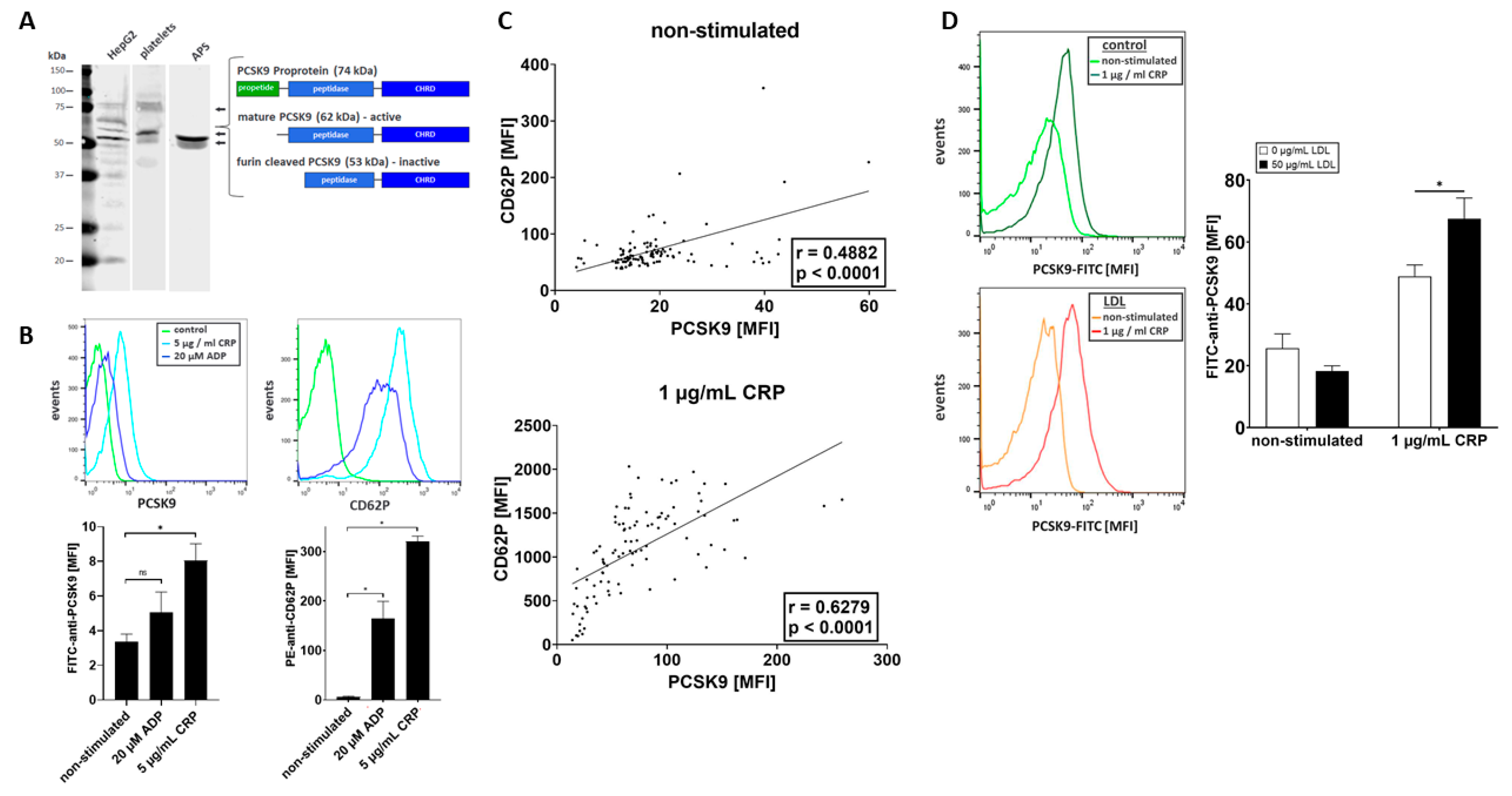
2.2. Platelets Store and Release PCSK9 upon Activation
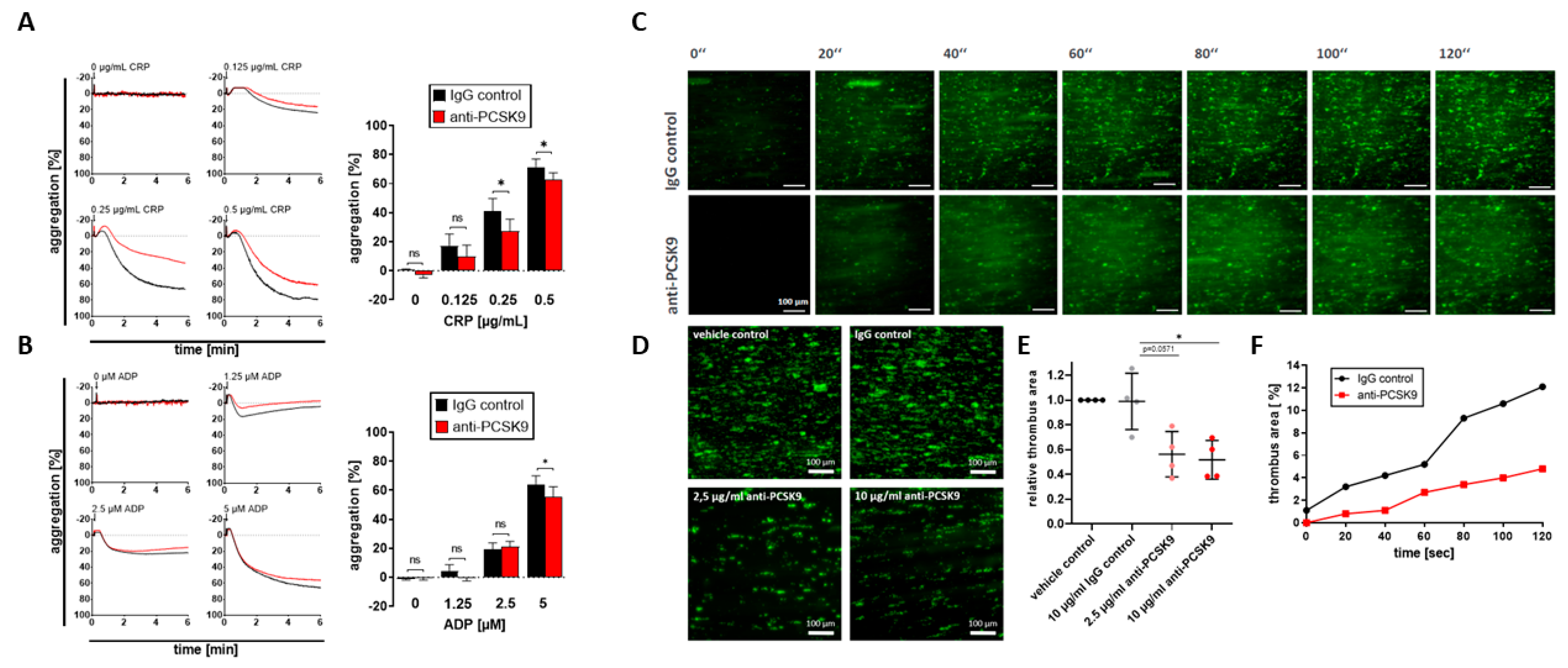
2.3. Platelet-Derived PCSK9 Promotes Platelet Aggregation and Thrombus Formation under Flow Dynamic Conditions
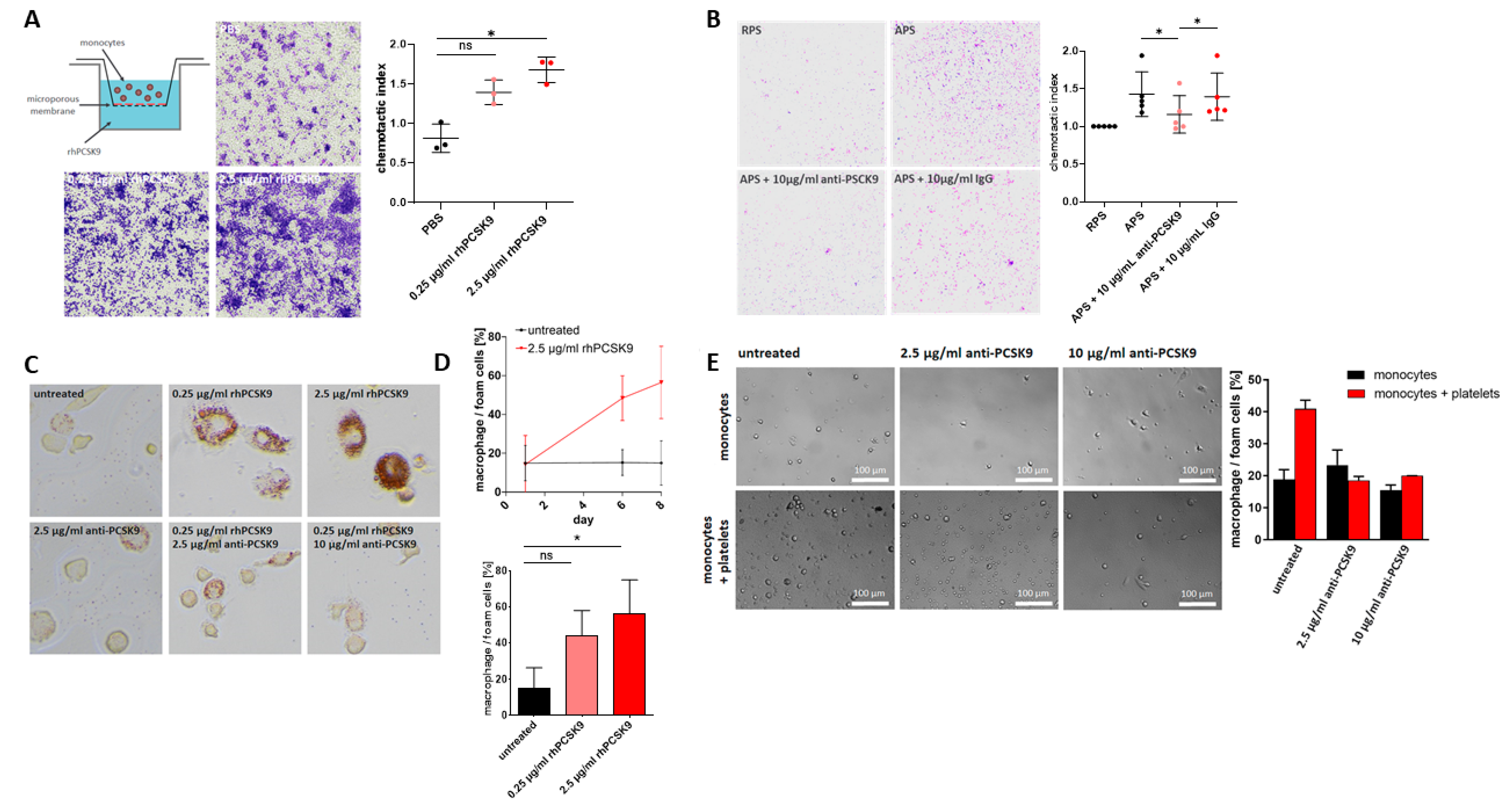
2.4. Platelet-Derived PCSK9 Stimulates Migration of Monocytes
2.5. Platelet-Derived PCSK9 Promotes Differentiation of Monocytes into Macrophages/Foam Cells
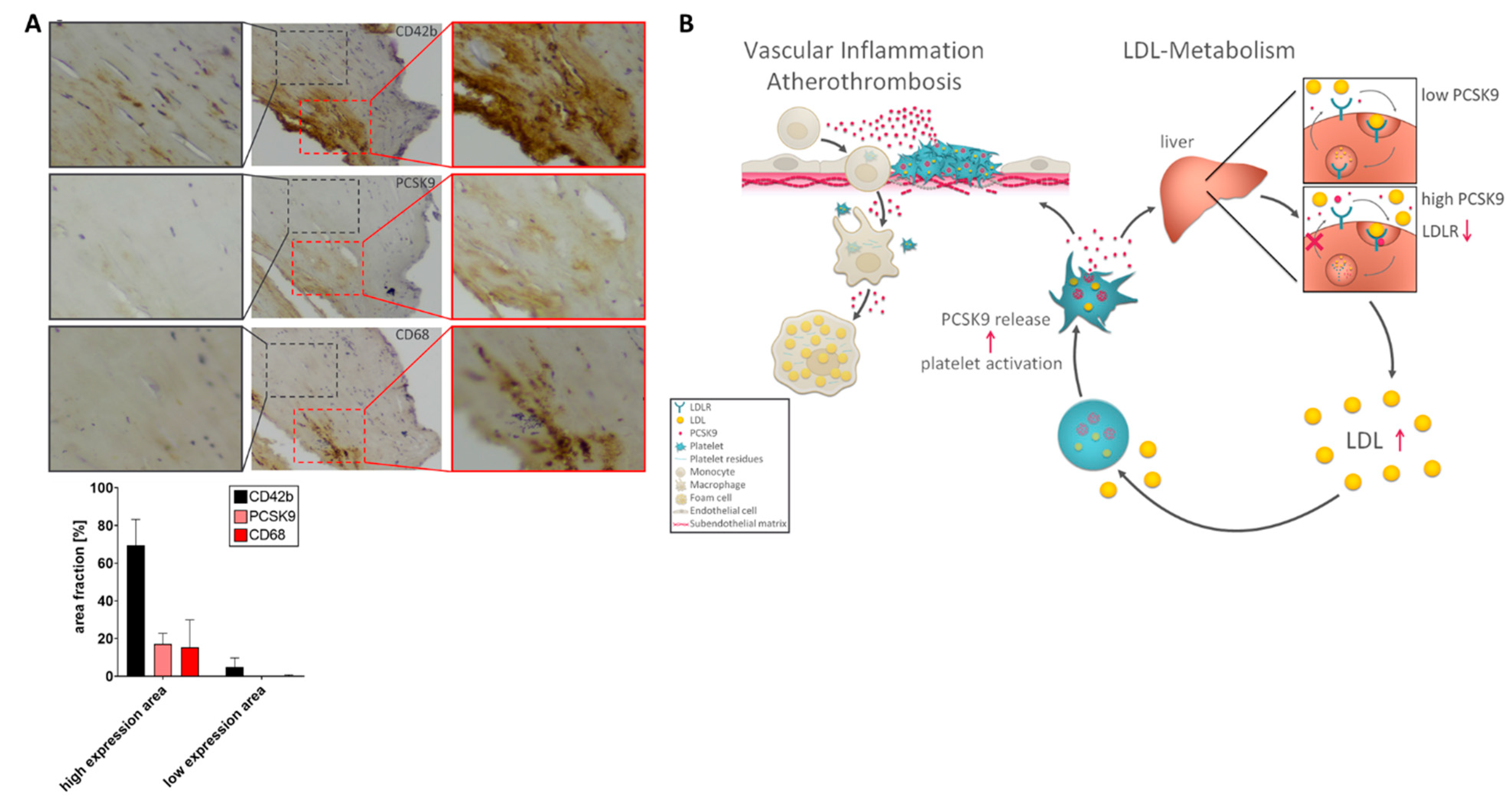
2.6. Expression of PCSK9 in Platelet/Macrophage-Rich Areas in Atherosclerotic Carotid Tissue
3. Discussion
4. Materials and Methods
4.1. Study Patients
4.2. Chemicals and Reagents
4.3. Isolation of Peripheral Blood Platelets and Monocytes and Platelet Flow Cytometry
4.4. Immunoblotting
4.5. In Vitro Monocyte Migration Assay
4.6. Platelet Aggregometry
4.7. Ex Vivo Thrombus Formation-Flow Chamber Assay
4.8. Monocyte Differentiation into Macrophages and Foam Cells
4.9. Immunostaining of Atherosclerotic Carotid Samples
4.10. Statistics
5. Limitations
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gawaz, M.; Langer, H.; May, A.E. Platelets in inflammation and atherogenesis. J. Clin. Investig. 2005, 115, 3378–3384. [Google Scholar] [CrossRef] [Green Version]
- Gawaz, M. Role of platelets in coronary thrombosis and reperfusion of ischemic myocardium. Cardiovasc. Res. 2004, 61, 498–511. [Google Scholar] [CrossRef] [PubMed]
- Massberg, S.; Brand, K.; Grüner, S.; Page, S.; Müller, E.; Müller, I.; Bergmeier, W.; Richter, T.; Lorenz, M.; Konrad, I.; et al. A Critical Role of Platelet Adhesion in the Initiation of Atherosclerotic Lesion Formation. J. Exp. Med. 2002, 196, 887–896. [Google Scholar] [CrossRef] [PubMed]
- Geisler, T.; Schaeffeler, E.; Dippon, J.; Winter, S.; Buse, V.; Bischofs, C.; Zuern, C.; Moerike, K.; Gawaz, M.; Schwab, M. CYP2C19 and nongenetic factors predict poor responsiveness to clopidogrel loading dose after coronary stent implantation. Pharmacogenomics 2008, 9, 1251–1259. [Google Scholar] [CrossRef] [Green Version]
- DiMinno, G.; Silver, M.J.; Cerbone, A.M.; Rainone, A.; Postiglione, A.; Mancini, M. Increased fibrinogen binding to platelets from patients with familial hypercholesterolemia. Arter. Off. J. Am. Heart Assoc. Inc. 1986, 6, 203–211. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, M.; Rath, D.; Schlotterbeck, J.; Rheinlaender, J.; Walker-Allgaier, B.; Alnaggar, N.; Zdanyte, M.; Müller, I.; Borst, O.; Geisler, T.; et al. Regulation of oxidized platelet lipidome: Implications for coronary artery disease. Eur. Heart J. 2017, 38, 1993–2005. [Google Scholar] [CrossRef]
- Podrez, E.A.; Byzova, T.V.; Febbraio, M.; Salomon, R.G.; Ma, Y.; Valiyaveettil, M.; Poliakov, E.; Sun, M.; Finton, P.J.; Curtis, B.R.; et al. Platelet CD36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype. Nat. Med. 2007, 13, 1086–1095. [Google Scholar] [CrossRef] [Green Version]
- Magwenzi, S.; Woodward, C.; Wraith, K.S.; Aburima, A.; Raslan, Z.; Jones, H.; McNeil, C.; Wheatcroft, S.; Yuldasheva, N.Y.; Febbriao, M.; et al. Oxidized LDL activates blood platelets through CD36/NOX2–mediated inhibition of the cGMP/protein kinase G signaling cascade. Blood 2015, 125, 2693–2703. [Google Scholar] [CrossRef] [Green Version]
- Ragusa, R.; Basta, G.; Neglia, D.; De Caterina, R.; Del Turco, S.; Caselli, C. PCSK9 and atherosclerosis: Looking beyond LDL regulation. Eur. J. Clin. Investig. 2021, 51, e13459. [Google Scholar] [CrossRef] [PubMed]
- Glerup, S.; Schulz, R.; Laufs, U.; Schlüter, K.-D. Physiological and therapeutic regulation of PCSK9 activity in cardiovascular disease. Basic Res. Cardiol. 2017, 112, 1–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sabatine, M.S.; Giugliano, R.P.; Keech, A.C.; Honarpour, N.; Wiviott, S.D.; Murphy, S.A.; Kuder, J.F.; Wang, H.; Liu, T.; Wasserman, S.M.; et al. Evolocumab and Clinical Outcomes in Patients with Cardiovascular Disease. N. Engl. J. Med. 2017, 376, 1713–1722. [Google Scholar] [CrossRef]
- Paciullo, F.; Momi, S.; Gresele, P. PCSK9 in Haemostasis and Thrombosis: Possible Pleiotropic Effects of PCSK9 Inhibitors in Cardiovascular Prevention. Thromb. Haemost. 2019, 119, 359–367. [Google Scholar] [CrossRef] [Green Version]
- Macchi, C.; Ferri, N.; Sirtori, C.R.; Corsini, A.; Banach, M.; Ruscica, M. Proprotein Convertase Subtilisin Kexin Type 9. Am. J. Pathol. 2021, 191, 1385–1397. [Google Scholar] [CrossRef]
- Urban, D.; Pöss, J.; Böhm, M.; Laufs, U. Targeting the Proprotein Convertase Subtilisin/Kexin Type 9 for the Treatment of Dyslipidemia and Atherosclerosis. J. Am. Coll. Cardiol. 2013, 62, 1401–1408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adorni, M.P.; Ruscica, M.; Ferri, N.; Bernini, F.; Zimetti, F. Proprotein Convertase Subtilisin/Kexin Type 9, Brain Cholesterol Homeostasis and Potential Implication for Alzheimer’s Disease. Front. Aging Neurosci. 2019, 11, 120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ricci, C.; Ruscica, M.; Camera, M.; Rossetti, L.; Macchi, C.; Colciago, A.; Zanotti, I.; Lupo, M.G.; Adorni, M.P.; Cicero, A.F.G.; et al. PCSK9 induces a pro-inflammatory response in macrophages. Sci. Rep. 2018, 8, 2267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camera, M.; Rossetti, L.; Barbieri, S.S.; Zanotti, I.; Canciani, B.; Trabattoni, D.; Ruscica, M.; Tremoli, E.; Ferri, N. PCSK9 as a Positive Modulator of Platelet Activation. J. Am. Coll. Cardiol. 2018, 71, 952–954. [Google Scholar] [CrossRef] [PubMed]
- Qi, Z.; Hu, L.; Zhang, J.; Yang, W.; Liu, X.; Jia, D.; Yao, Z.; Chang, L.; Pan, G.; Zhong, H.; et al. PCSK9 (Proprotein Convertase Subtilisin/Kexin 9) Enhances Platelet Activation, Thrombosis, and Myocardial Infarct Expansion by Binding to Platelet CD36. Circulation 2021, 143, 45–61. [Google Scholar] [CrossRef] [PubMed]
- Cammisotto, V.; Baratta, F.; Castellani, V.; Bartimoccia, S.; Nocella, C.; D’Erasmo, L.; Cocomello, N.; Barale, C.; Scicali, R.; Di Pino, A.; et al. Proprotein Convertase Subtilisin Kexin Type 9 Inhibitors Reduce Platelet Activation Modulating ox-LDL Pathways. Int. J. Mol. Sci. 2021, 22, 7193. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, G.G.; Steg, P.G.; Szarek, M.; Bhatt, D.L.; Bittner, V.A.; Diaz, R.; Edelberg, J.M.; Goodman, S.G.; Hanotin, C.; Harrington, R.A.; et al. Alirocumab and Cardiovascular Outcomes after Acute Coronary Syndrome. N. Engl. J. Med. 2018, 379, 2097–2107. [Google Scholar] [CrossRef] [PubMed]
- Ray, K.; Wright, R.S.; Kallend, D.; Koenig, W.; Leiter, L.A.; Raal, F.J.; Bisch, J.A.; Richardson, T.; Jaros, M.; Wijngaard, P.L.; et al. Two Phase 3 Trials of Inclisiran in Patients with Elevated LDL Cholesterol. N. Engl. J. Med. 2020, 382, 1507–1519. [Google Scholar] [CrossRef] [PubMed]
- Pedreño, J.; de Castellarnau, C.; Cullaré, C.; Sánchez, J.; Gómez-Gerique, J.; Ordóñez-Llanos, J.; González-Sastre, F. LDL binding sites on platelets differ from the “classical” receptor of nucleated cells. Arter. Thromb. A J. Vasc. Biol. 1992, 12, 1353–1362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Witte, A.; Rohlfing, A.-K.; Dannenmann, B.; Dicenta, V.; Nasri, M.; Kolb, K.; Sudmann, J.; Castor, T.; Rath, D.; Borst, O.; et al. The chemokine CXCL14 mediates platelet function and migration via direct interaction with CXCR4. Cardiovasc. Res. 2021, 117, 903–917. [Google Scholar] [CrossRef] [PubMed]
- Malehmir, M.; Pfister, D.; Gallage, S.; Szydlowska, M.; Inverso, D.; Kotsiliti, E.; Leone, V.; Peiseler, M.; Surewaard, B.G.J.; Rath, D.; et al. Platelet GPIbα is a mediator and potential interventional target for NASH and subsequent liver cancer. Nat. Med. 2019, 25, 641–655. [Google Scholar] [CrossRef] [Green Version]
- Rath, D.; Chatterjee, M.; Borst, O.; Müller, K.; Stellos, K.; Mack, A.F.; Bongartz, A.; Bigalke, B.; Langer, H.; Schwab, M.; et al. Expression of stromal cell-derived factor-1 receptors CXCR4 and CXCR7 on circulating platelets of patients with acute coronary syndrome and association with left ventricular functional recovery. Eur. Heart J. 2014, 35, 386–394. [Google Scholar] [CrossRef] [Green Version]
- Daub, K.; Langer, H.; Seizer, P.; Stellos, K.; May, A.E.; Goyal, P.; Bigalke, B.; Schönberger, T.; Geisler, T.; Siegel-Axel, D.; et al. Platelets induce differentiation of human CD34 + progenitor cells into foam cells and endothelial cells. FASEB J. 2006, 20, 2559–2561. [Google Scholar] [CrossRef] [Green Version]
- Daub, K.; Siegel-Axel, D.; Schönberger, T.; Leder, C.; Seizer, P.; Müller, K.; Schaller, M.; Penz, S.; Menzel, D.; Büchele, B.; et al. Inhibition of foam cell formation using a soluble CD68-Fc fusion protein. J. Mol. Med. 2010, 88, 909–920. [Google Scholar] [CrossRef]
- Müller, I.; Schönberger, T.; Schneider, M.; Borst, O.; Ziegler, M.; Seizer, P.; Leder, C.; Müller, K.; Lang, M.; Appenzeller, F.; et al. Gremlin-1 Is an Inhibitor of Macrophage Migration Inhibitory Factor and Attenuates Atherosclerotic Plaque Growth in ApoE−/− Mice. J. Biol. Chem. 2013, 288, 31635–31645. [Google Scholar] [CrossRef] [Green Version]
- Chatterjee, M.; I Von Ungern-Sternberg, S.N.; Seizer, P.; Schlegel, F.; Büttcher, M.; Sindhu, N.A.; Muller, S.; Mack, A.F.; Gawaz, M. Platelet-derived CXCL12 regulates monocyte function, survival, differentiation into macrophages and foam cells through differential involvement of CXCR4–CXCR7. Cell Death Dis. 2015, 6, e1989. [Google Scholar] [CrossRef] [Green Version]
- Seizer, P.; Schönberger, T.; Schött, M.; Lang, M.R.; Langer, H.F.; Bigalke, B.; Krämer, B.F.; Borst, O.; Daub, K.; Heidenreich, O.; et al. EMMPRIN and its ligand cyclophilin A regulate MT1-MMP, MMP-9 and M-CSF during foam cell formation. Atherosclerosis 2010, 209, 51–57. [Google Scholar] [CrossRef]
- Rosenson, R.S.; Hegele, R.A.; Fazio, S.; Cannon, C.P. The Evolving Future of PCSK9 Inhibitors. J. Am. Coll. Cardiol. 2018, 72, 314–329. [Google Scholar] [CrossRef]
- Cesaro, A.; Gragnano, F.; Fimiani, F.; Moscarella, E.; Diana, V.; Pariggiano, I.; Concilio, C.; Natale, F.; Limongelli, G.; Bossone, E.; et al. Impact of PCSK9 inhibitors on the quality of life of patients at high cardiovascular risk. Eur. J. Prev. Cardiol. 2020, 27, 556–558. [Google Scholar] [CrossRef]
- Lloyd-Jones, D.; Morris, P.B.; Ballantyne, C.M.; Birtcher, K.K.; Daly, D.D.; DePalma, S.; Minissian, M.B.; Orringer, C.E.; Smith, S.C. 2017 Focused Update of the 2016 ACC Expert Consensus Decision Pathway on the Role of Non-Statin Therapies for LDL-Cholesterol Lowering in the Management of Atherosclerotic Cardiovascular Disease Risk. J. Am. Coll. Cardiol. 2017, 70, 1785–1822. [Google Scholar] [CrossRef]
- Li, S.; Zhu, C.-G.; Guo, Y.-L.; Xu, R.-X.; Zhang, Y.; Sun, J.; Li, J.-J. The Relationship between the Plasma PCSK9 Levels and Platelet Indices in Patients with Stable Coronary Artery Disease. J. Atheroscler. Thromb. 2015, 22, 76–84. [Google Scholar] [CrossRef] [Green Version]
- Werner, C.; Hoffmann, M.M.; Winkler, K.; Böhm, M.; Laufs, U. Risk prediction with proprotein convertase subtilisin/kexin type 9 (PCSK9) in patients with stable coronary disease on statin treatment. Vasc. Pharmacol. 2014, 62, 94–102. [Google Scholar] [CrossRef]
- Zhu, Y.M.; Anderson, T.J.; Sikdar, K.; Fung, M.; McQueen, M.J.; Lonn, E.M.; Verma, S. Association of Proprotein Convertase Subtilisin/Kexin Type 9 (PCSK9) With Cardiovascular Risk in Primary Prevention. Arter. Thromb. Vasc. Biol. 2015, 35, 2254–2259. [Google Scholar] [CrossRef] [Green Version]
- Ridker, P.M.; Rifai, N.; Bradwin, G.; Rose, L.M. Plasma proprotein convertase subtilisin/kexin type 9 levels and the risk of first cardiovascular events. Eur. Heart J. 2016, 37, 554–560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gencer, B.; Montecucco, F.; Nanchen, D.; Carbone, F.; Klingenberg, R.; Vuilleumier, N.; Aghlmandi, S.; Heg, D.; Räber, L.; Auer, R.; et al. Prognostic value of PCSK9 levels in patients with acute coronary syndromes. Eur. Heart J. 2016, 37, 546–553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Macchi, C.; Ferri, N.; Favero, C.; Cantone, L.; Vigna, L.; Pesatori, A.C.; Lupo, M.G.; Sirtori, C.R.; Corsini, A.; Bollati, V.; et al. Long-term exposure to air pollution raises circulating levels of proprotein convertase subtilisin/kexin type 9 in obese individuals. Eur. J. Prev. Cardiol. 2019, 26, 578–588. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, S.J.; Puri, R.; Anderson, T.; Ballantyne, C.M.; Cho, L.; Kastelein, J.J.P.; Koenig, W.; Somaratne, R.; Kassahun, H.; Yang, J.; et al. Effect of Evolocumab on Progression of Coronary Disease in Statin-Treated Patients. JAMA 2016, 316, 2373–2384. [Google Scholar] [CrossRef] [PubMed]
- Cheng, J.M.; Oemrawsingh, R.M.; Garcia-Garcia, H.M.; Boersma, E.; van Geuns, R.-J.; Serruys, P.W.; Kardys, I.; Akkerhuis, K.M. PCSK9 in relation to coronary plaque inflammation: Results of the ATHEROREMO-IVUS study. Atherosclerosis 2016, 248, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Ferri, N.; Tibolla, G.; Pirillo, A.; Cipollone, F.; Mezzetti, A.; Pacia, S.; Corsini, A.; Catapano, A.L. Proprotein convertase subtilisin kexin type 9 (PCSK9) secreted by cultured smooth muscle cells reduces macrophages LDLR levels. Atherosclerosis 2012, 220, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Cesaro, A.; Bianconi, V.; Gragnano, F.; Moscarella, E.; Fimiani, F.; Monda, E.; Scudiero, O.; Limongelli, G.; Pirro, M.; Calabrò, P. Beyond cholesterol metabolism: The pleiotropic effects of proprotein convertase subtilisin/kexin type 9 (PCSK9). Genetics, mutations, expression, and perspective for long-term inhibition. BioFactors 2020, 46, 367–380. [Google Scholar] [CrossRef] [PubMed]
- Geue, S.; Aurbach, K.; Manke, M.-C.; Manukjan, G.; Münzer, P.; Stegner, D.; Brähler, C.; Walker-Allgaier, B.; Märklin, M.; Borst, C.E.; et al. Pivotal role of PDK1 in megakaryocyte cytoskeletal dynamics and polarization during platelet biogenesis. Blood 2019, 134, 1847–1858. [Google Scholar] [CrossRef] [PubMed]
- Münzer, P.; Walker-Allgaier, B.; Geue, S.; Geuss, E.; Hron, G.; Rath, D.; Eißler, D.; Winter, S.; Schaeffeler, E.; Meinert, M.; et al. PDK1 Determines Collagen-Dependent Platelet Ca 2+ Signaling and Is Critical to Development of Ischemic Stroke In Vivo. Arter. Thromb. Vasc. Biol. 2016, 36, 1507–1516. [Google Scholar] [CrossRef] [Green Version]
- Borst, O.; Münzer, P.; Alnaggar, N.; Geue, S.; Tegtmeyer, R.; Rath, D.; Droppa, M.; Seizer, P.; Heitmeier, S.; Heemskerk, J.W.M.; et al. Inhibitory mechanisms of very low–dose rivaroxaban in non–ST-elevation myocardial infarction. Blood Adv. 2018, 2, 715–730. [Google Scholar] [CrossRef] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Petersen-Uribe, Á.; Kremser, M.; Rohlfing, A.-K.; Castor, T.; Kolb, K.; Dicenta, V.; Emschermann, F.; Li, B.; Borst, O.; Rath, D.; et al. Platelet-Derived PCSK9 Is Associated with LDL Metabolism and Modulates Atherothrombotic Mechanisms in Coronary Artery Disease. Int. J. Mol. Sci. 2021, 22, 11179. https://doi.org/10.3390/ijms222011179
Petersen-Uribe Á, Kremser M, Rohlfing A-K, Castor T, Kolb K, Dicenta V, Emschermann F, Li B, Borst O, Rath D, et al. Platelet-Derived PCSK9 Is Associated with LDL Metabolism and Modulates Atherothrombotic Mechanisms in Coronary Artery Disease. International Journal of Molecular Sciences. 2021; 22(20):11179. https://doi.org/10.3390/ijms222011179
Chicago/Turabian StylePetersen-Uribe, Álvaro, Marcel Kremser, Anne-Katrin Rohlfing, Tatsiana Castor, Kyra Kolb, Valerie Dicenta, Frederic Emschermann, Bo Li, Oliver Borst, Dominik Rath, and et al. 2021. "Platelet-Derived PCSK9 Is Associated with LDL Metabolism and Modulates Atherothrombotic Mechanisms in Coronary Artery Disease" International Journal of Molecular Sciences 22, no. 20: 11179. https://doi.org/10.3390/ijms222011179