
Genome Study of a Novel Virulent Phage vB_SspS_KASIA and Mu-like Prophages of Shewanella sp. M16 Provides Insights into the Genetic Diversity of the Shewanella Virome


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
2.1. Identification and Characterization of Shewanella sp. M16
2.2. Identification and Characterization of the vB_SspS_KASIA Phage
2.3. Genomic Analysis of the vB_SspS_KASIA Phage
2.4. Analysis of the vB_SspS_KASIA Phage Introns
2.5. Identification and Characterization of Shewanella sp. M16 Prophages
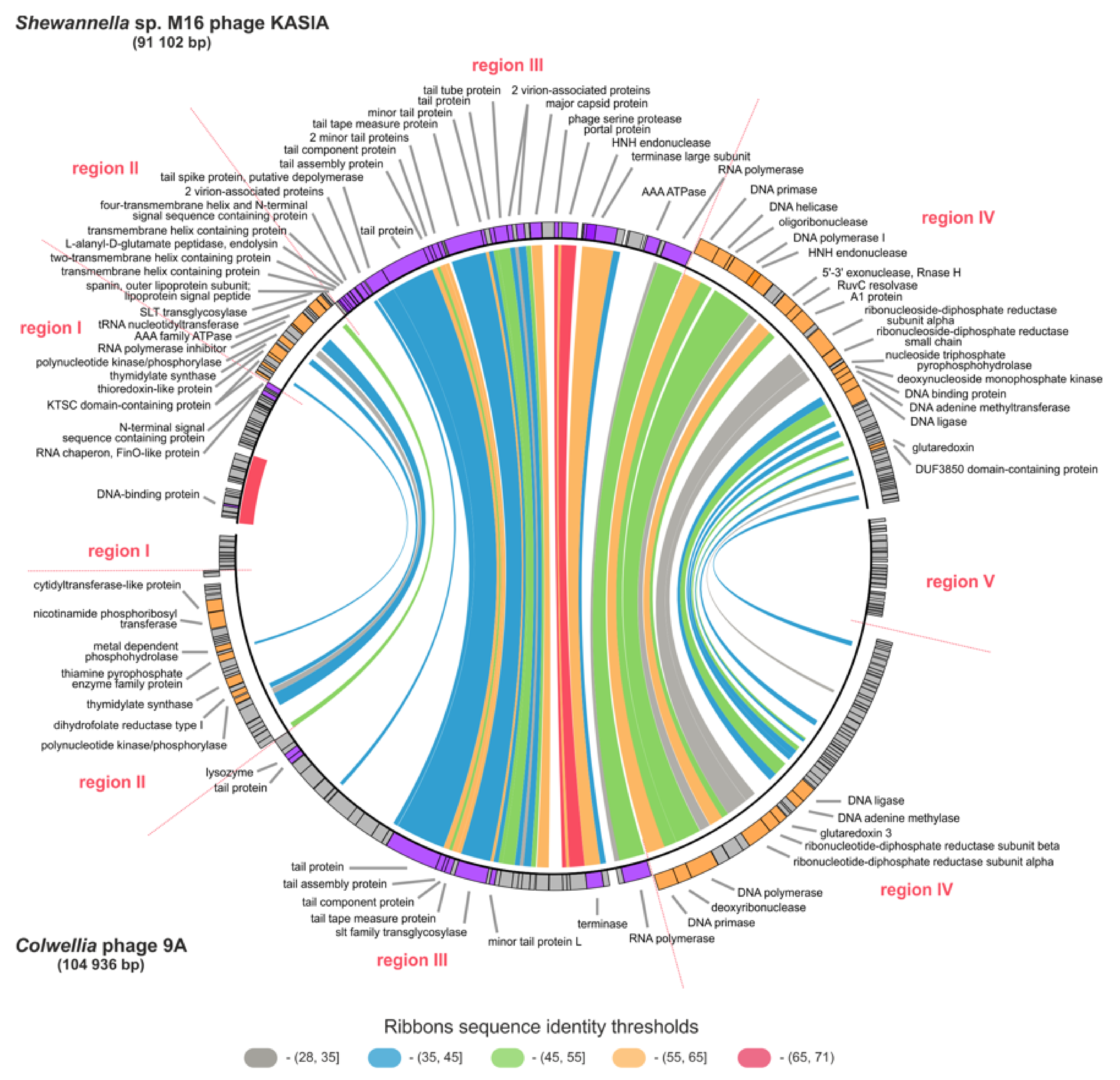
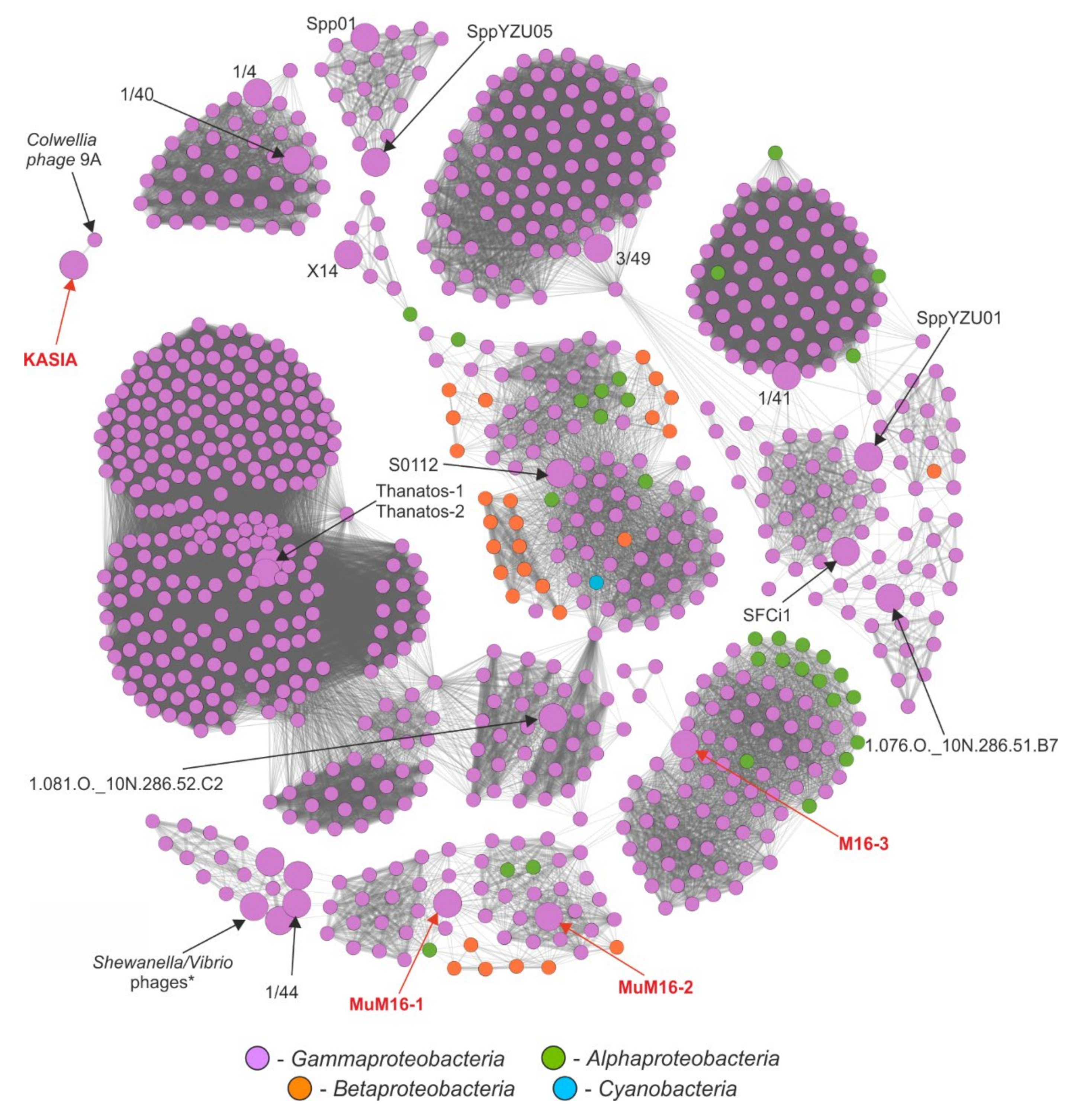
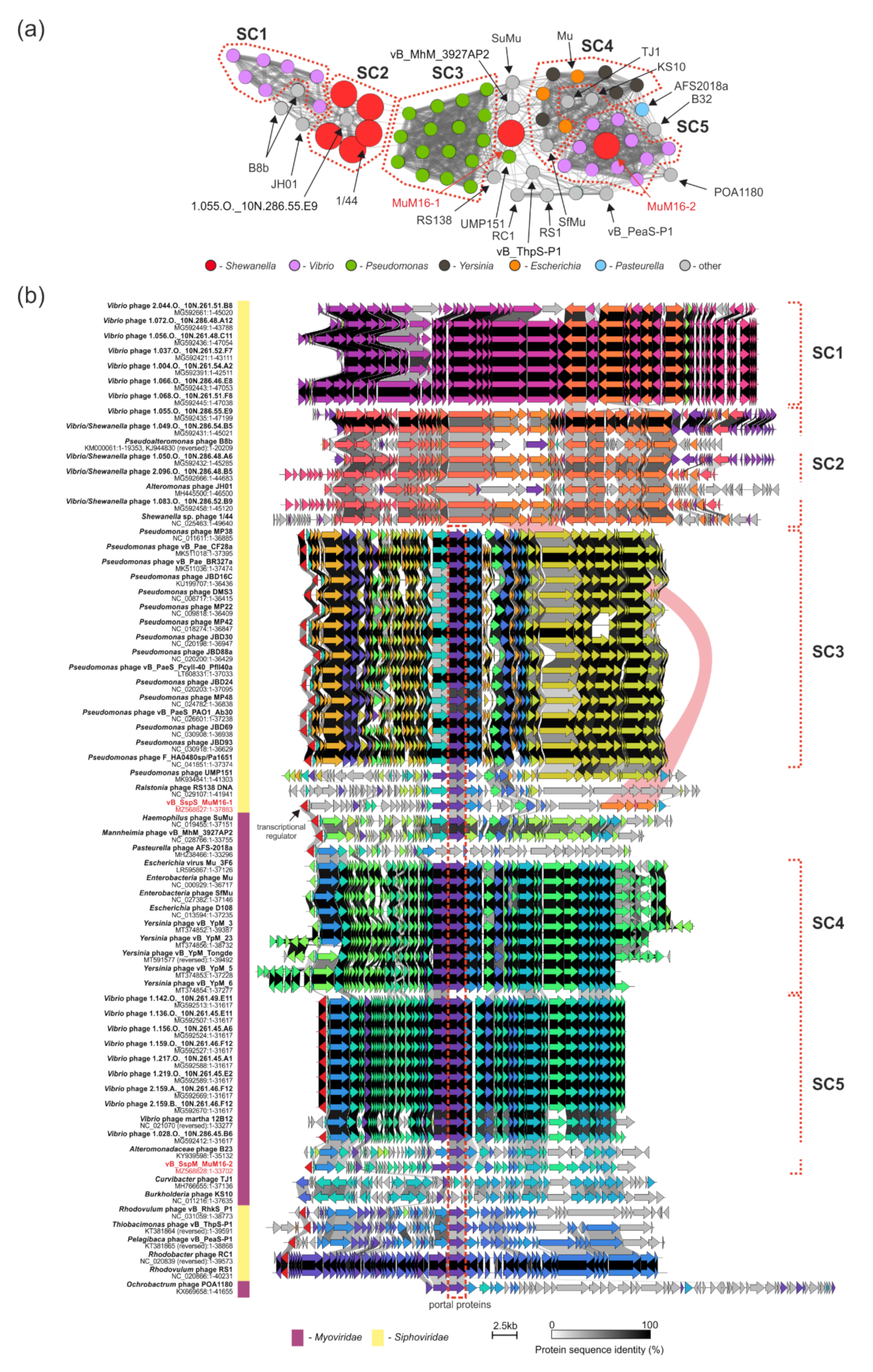
2.6. Comparative Genomic Analyses
2.7. Functional Characterization of vB_SspS_KASIA
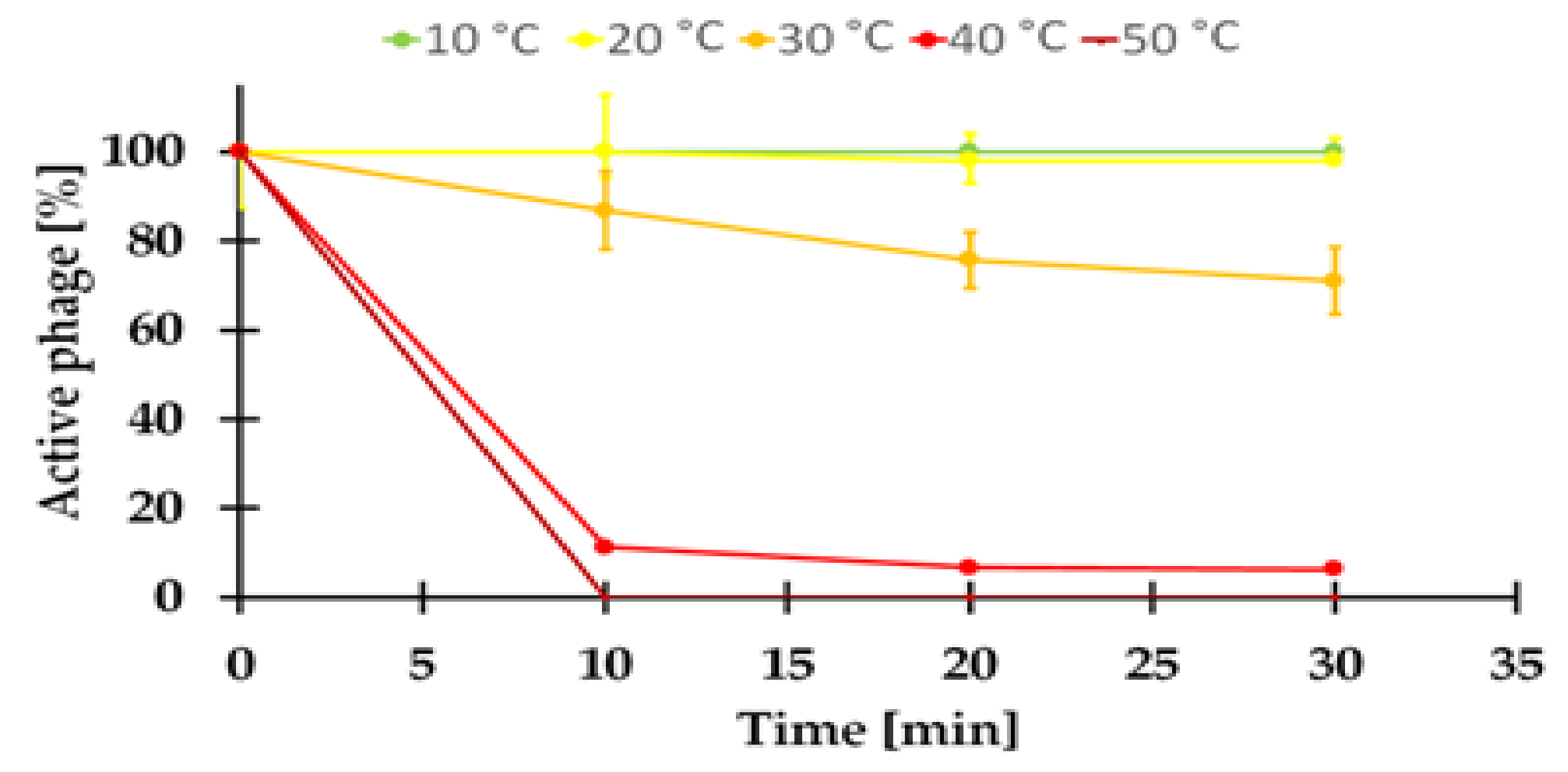
2.7.1. Sensitivity to Temperature of the KASIA Phage
2.7.2. The Influence of Various Conditions on the KASIA Phage Plaque Formation
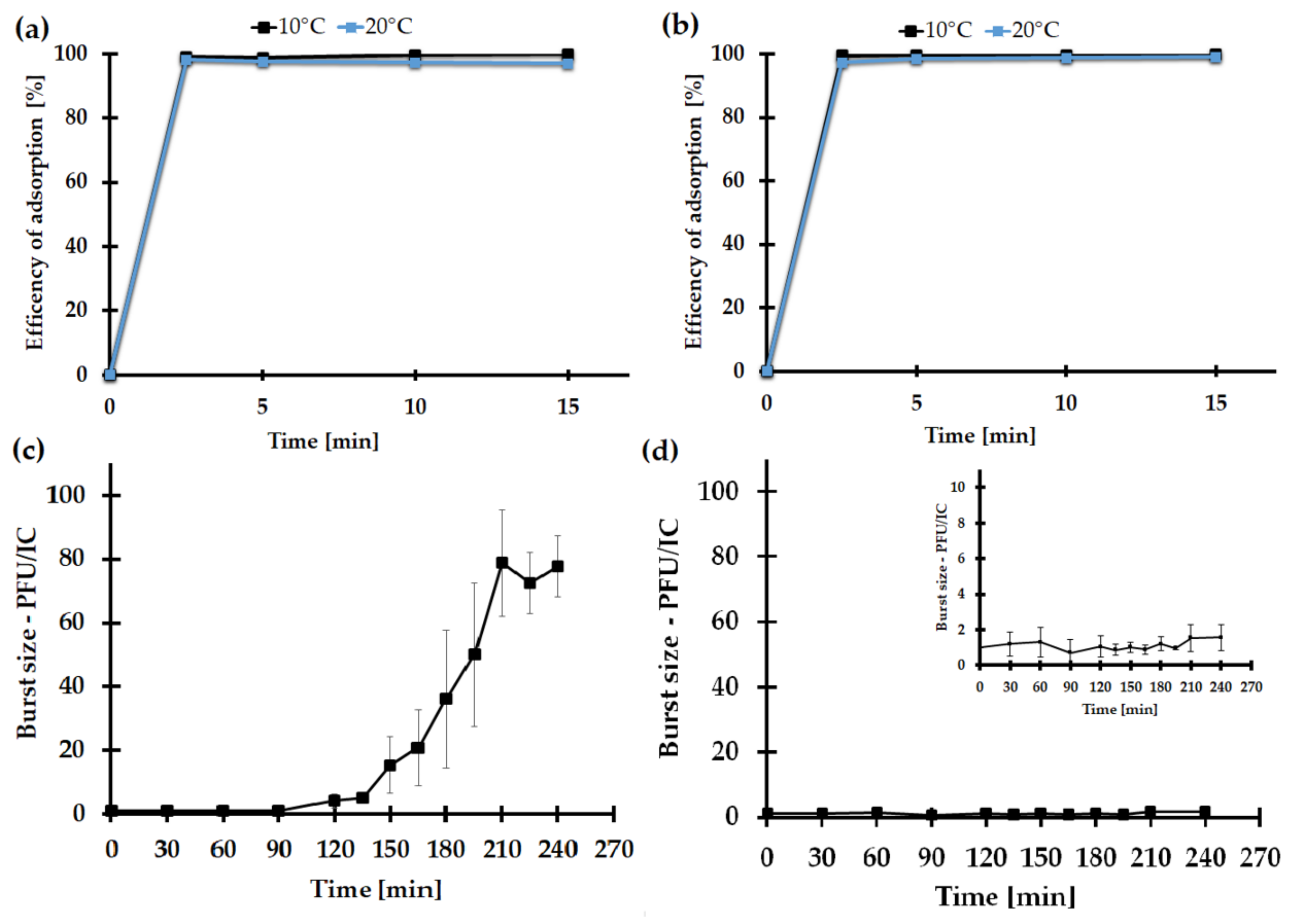
2.7.3. The Impact of the Medium Type on the KASIA Phage Adsorption Rate
2.7.4. One-Step Growth of the KASIA Phage
2.7.5. Host Range
3. Materials and Methods
3.1. Bacterial Strains, Plasmids, and Culture Conditions
3.2. Isolation of the KASIA Phage
3.3. Transmission Electron Microscopy (TEM)
3.4. DNA Isolation and Sequencing
3.5. Genome Annotation
3.6. Comparative Analysis
3.7. SDS-PAGE and Mass Spectrometry Protein Analysis
3.8. RNA Isolation and In Vivo Splicing Assay
3.9. Cloning, Overexpression, Purification, and Testing the Specificity of a Putative DNA MTase
3.10. Confirmation of the M16–3 Genome Assembly
3.11. Determination of the Phage Host Range by Spot Testing
3.12. Thermal Stability of the Phage
3.13. Adsorption Kinetics
3.14. One-Step Growth Curve
3.15. Nucleotide Sequence Accession Numbers
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gil, J.F.; Mesa, V.; Estrada-Ortiz, N.; Lopez-Obando, M.; Gómez, A.; Plácido, J. Viruses in Extreme Environments, Current Overview, and Biotechnological Potential. Viruses 2021, 13, 81. [Google Scholar] [CrossRef] [PubMed]
- Krupovic, M.; Dolja, V.V.K.; Eugene, V. Origin of viruses: Primordial replicators recruiting capsids from hosts. Nat. Rev. Microbiol. 2019, 17, 449–458. [Google Scholar] [CrossRef] [PubMed]
- Le Romancer, M.; Gaillard, M.; Geslin, C.; Prieur, D. Viruses in extreme environments. Rev. Environ. Sci. Biotechnol. 2006, 6, 17–31. [Google Scholar] [CrossRef] [Green Version]
- Dávila-Ramos, S.; Castelán-Sánchez, H.G.; Martínez-Ávila, L.; Sánchez-Carbente, M.d.R.; Peralta, R.; Hernández-Mendoza, A.; Dobson, A.D.W.; Gonzalez, R.A.; Pastor, N.; Batista-García, R.A. A Review on Viral Metagenomics in Extreme Environments. Front. Microbiol. 2019, 10, 403. [Google Scholar] [CrossRef] [Green Version]
- Wells, L.E.; Deming, J.W. Modelled and measured dynamics of viruses in Arctic winter sea-ice brines. Environ. Microbiol. 2006, 8, 1115–1121. [Google Scholar] [CrossRef] [PubMed]
- Wells, L.E.; Deming, J.W. Characterization of a cold-active bacteriophage on two psychrophilic marine hosts. Aquat. Microb. Ecol. 2006, 45, 15–29. [Google Scholar] [CrossRef]
- Tapia, P.; Flores, F.M.; Covarrubias, P.C.; Acuña, L.G.; Holmes, D.S.; Quatrini, R. Complete Genome Sequence of Temperate Bacteriophage phiML1 from the Extreme Acidophile Acidithiobacillus caldus ATCC 51756. J. Virol. 2012, 86, 12452–12453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atanasova, N.S.; Oksanen, H.M.; Bamford, D.H. Haloviruses of archaea, bacteria, and eukaryotes. Curr. Opin. Microbiol. 2015, 25, 40–48. [Google Scholar] [CrossRef]
- Chlebicki, A.; Zielenkiewicz, U.; Wilczek, A.M. Fungi are not involved in biofilm formation on rock wall in subterranean arsenic mine in Poland. Nova Hedwig. 2014, 99, 255–269. [Google Scholar] [CrossRef]
- Przylibski, T.A. Radon and its daughter products behaviour in the air of an underground tourist route in the former arsenic and gold mine in Złoty Stok (Sudety Mountains, SW Poland). J. Environ. Radioact. 2001, 57, 87–103. [Google Scholar] [CrossRef]
- Drewniak, L.; Matlakowska, R.; Rewerski, B.; Sklodowska, A. Arsenic release from gold mine rocks mediated by the activity of indigenous bacteria. Hydrometallurgy 2010, 104, 437–442. [Google Scholar] [CrossRef]
- Drewniak, L.; Styczek, A.; Majder-Lopatka, M.; Sklodowska, A. Bacteria, hypertolerant to arsenic in the rocks of an ancient gold mine, and their potential role in dissemination of arsenic pollution. Environ. Pollut. 2008, 156, 1069–1074. [Google Scholar] [CrossRef]
- Drewniak, L.; Maryan, N.; Lewandowski, W.; Kaczanowski, S.; Sklodowska, A. The contribution of microbial mats to the arsenic geochemistry of an ancient gold mine. Environ. Pollut. 2012, 162, 190–201. [Google Scholar] [CrossRef]
- Uhrynowski, W.; Decewicz, P.; Dziewit, L.; Radlinska, M.; Krawczyk, P.S.; Lipinski, L.; Adamska, D.; Drewniak, L. Analysis of the Genome and Mobilome of a Dissimilatory Arsenate Reducing Aeromonas sp. O23A Reveals Multiple Mechanisms for Heavy Metal Resistance and Metabolism. Front. Microbiol. 2017, 8, 936. [Google Scholar] [CrossRef] [PubMed]
- Uhrynowski, W.; Radlinska, M.; Drewniak, L. Genomic Analysis of Shewanella sp. O23S—The Natural Host of the pSheB Plasmid Carrying Genes for Arsenic Resistance and Dissimilatory Reduction. Int. J. Mol. Sci. 2019, 20, 1018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bujak, K.; Decewicz, P.; Kaminski, J.; Radlinska, M. Identification, Characterization, and Genomic Analysis of Novel Serratia Temperate Phages from a Gold Mine. Int. J. Mol. Sci. 2020, 21, 6709. [Google Scholar] [CrossRef]
- Wang, F.; Wang, F.; Li, Q.; Xiao, X. A Novel Filamentous Phage from the Deep-Sea Bacterium Shewanella piezotolerans WP3 Is Induced at Low Temperature. J. Bacteriol. 2007, 189, 7151–7153. [Google Scholar] [CrossRef] [Green Version]
- Lemaire, O.N.; Méjean, V.; Iobbi-Nivol, C. The Shewanella genus: Ubiquitous organisms sustaining and preserving aquatic ecosystems. FEMS Microbiol. Rev. 2020, 44, 155–170. [Google Scholar] [CrossRef] [PubMed]
- Kauffman, K.M.; Brown, J.M.; Sharma, R.S.; VanInsberghe, D.; Elsherbini, J.; Polz, M.; Kelly, L. Viruses of the Nahant Collection, characterization of 251 marine Vibrionaceae viruses. Sci. Data 2018, 5, 180114. [Google Scholar] [CrossRef] [Green Version]
- Gödeke, J.; Paul, K.; Lassak, J.; Thormann, K.M. Phage-induced lysis enhances biofilm formation in Shewanella oneidensis MR-1. ISME J. 2011, 5, 613–626. [Google Scholar] [CrossRef] [Green Version]
- Zeng, Z.; Liu, X.; Yao, J.; Guo, Y.; Li, B.; Li, Y.; Jiao, N.; Wang, X. Cold adaptation regulated by cryptic prophage excision in Shewanella oneidensis. ISME J. 2016, 10, 2787–2800. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borriss, M.; Helmke, E.; Hanschke, R.; Schweder, T. Isolation and characterization of marine psychrophilic phage-host systems from Arctic sea ice. Extremophiles 2003, 7, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Drewniak, L.; Stasiuk, R.; Uhrynowski, W.; Sklodowska, A. Shewanella sp. O23S as a Driving Agent of a System Utilizing Dissimilatory Arsenate-Reducing Bacteria Responsible for Self-Cleaning of Water Contaminated with Arsenic. Int. J. Mol. Sci. 2015, 16, 14409–14427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knecht, L.E.; Veljkovic, M.; Fieseler, L. Diversity and Function of Phage Encoded Depolymerases. Front. Microbiol. 2020, 10, 2949. [Google Scholar] [CrossRef]
- Hatfull, G.F. Bacteriophage genomics. Curr. Opin. Microbiol. 2008, 11, 447–453. [Google Scholar] [CrossRef] [Green Version]
- Garneau, J.R.; Depardieu, F.; Fortier, L.-C.; Bikard, D.; Monot, M. PhageTerm: A tool for fast and accurate determination of phage termini and packaging mechanism using next-generation sequencing data. Sci. Rep. 2017, 7, 8292. [Google Scholar] [CrossRef]
- Jianbin, W.; Yan, J.; Myriam, V.; Yongqiao, S.; Hong, Y.; Jing, W.; Qiyu, B.; Huimin, K.; Songnian, H. Complete genome sequence of bacteriophage T5. Virology 2005, 332, 45–65. [Google Scholar] [CrossRef] [Green Version]
- Stewart, C.R.; Casjens Sherwood, R.; Cresawn Steven, G.; Houtz Jennifer, M.; Smith Alexis, L.; Ford Michael, E.; Peebles Craig, L.; Hatfull Graham, F.; Hendrix Roger, W.; Huang, W.M.; et al. The Genome of Bacillus subtilis Bacteriophage SPO1. J. Mol. Biol. 2009, 388, 48–70. [Google Scholar] [CrossRef] [Green Version]
- Casjens, S.R.; Gilcrease, E.B. Determining DNA Packaging Strategy by Analysis of the Termini of the Chromosomes in Tailed-Bacteriophage Virions. In Bacteriophages: Methods and Protocols; Volume 2 Molecular and Applied Aspects; Clokie, M.R.J., Kropinski, A.M., Eds.; Humana Press: Totowa, NJ, USA, 2009; pp. 91–111. [Google Scholar]
- Klumpp, J.; Lavigne, R.; Loessner, M.J.; Ackermann, H.W. The SPO1-related bacteriophages. Arch. Virol. 2010, 155, 1547–1561. [Google Scholar] [CrossRef]
- Methé, B.A.; Nelson, K.E.; Deming, J.W.; Momen, B.; Melamud, E.; Zhang, X.; Moult, J.; Madupu, R.; Nelson, W.C.; Dodson, R.J.; et al. The psychrophilic lifestyle as revealed by the genome sequence of Colwellia psychrerythraea 34H through genomic and proteomic analyses. Proc. Natl. Acad. Sci. USA 2005, 102, 10913–10918. [Google Scholar] [CrossRef] [Green Version]
- Colangelo-Lillis, J.R.; Deming, J.W. Genomic analysis of cold-active Colwelliaphage 9A and psychrophilic phage–host interactions. Extremophiles 2013, 17, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Olejniczak, M.; Storz, G. ProQ/FinO-domain proteins: Another ubiquitous family of RNA matchmakers? Mol. Microbiol. 2017, 104, 905–915. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, E.S.; Kutter, E.; Mosig, G.; Arisaka, F.; Kunisawa, T.; Rüger, W. Bacteriophage T4 Genome. Microbiol. Mol. Biol. Rev. 2003, 67, 86–156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathews, C.K.; Moen, L.K.; Yeong, W.; Sargent, R.G. Intracellular organization of DNA precursor biosynthetic enzymes. Trends Biochem. Sci. 1988, 13, 394–397. [Google Scholar] [CrossRef]
- Basta, T.; Boum, Y.; Briffotaux, J.; Becker, H.F.; Lamarre-Jouenne, I.; Lambry, J.-C.; Skouloubris, S.; Liebl, U.; Graille, M.; van Tilbeurgh, H.; et al. Mechanistic and structural basis for inhibition of thymidylate synthase ThyX. Open Biol. 2012, 2, 120120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, M.; Le, S.; Jin, X.; Li, G.; Tan, Y.; Li, M.; Zhao, X.; Shen, W.; Yang, Y.; Wang, J.; et al. Characterization and Comparative Genomic Analyses of Pseudomonas aeruginosa Phage PaoP5: New Members Assigned to PAK_P1-like Viruses. Sci. Rep. 2016, 6, 34067. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nechaev, S.; Severinov, K. Inhibition of Escherichia coli RNA Polymerase by Bacteriophage T7 Gene 2 Protein. J. Mol. Biol. 1999, 289, 815–826. [Google Scholar] [CrossRef] [PubMed]
- Evseev, P.V.; Lukianova, A.A.; Shneider, M.M.; Korzhenkov, A.A.; Bugaeva, E.N.; Kabanova, A.P.; Miroshnikov, K.K.; Kulikov, E.E.; Toshchakov, S.V.; Ignatov, A.N.; et al. Origin and Evolution of Studiervirinae Bacteriophages Infecting Pectobacterium: Horizontal Transfer Assists Adaptation to New Niches. Microorganisms 2020, 8, 1707. [Google Scholar] [CrossRef]
- Lu, S.; Wang, J.; Chitsaz, F.; Derbyshire, M.K.; Geer, R.C.; Gonzales, N.R.; Gwadz, M.; Hurwitz, D.I.; Marchler, G.H.; Song, J.S.; et al. CDD/SPARCLE: The conserved domain database in 2020. Nucleic Acids Res. 2019, 48, D265–D268. [Google Scholar] [CrossRef] [Green Version]
- Lavigne, R.; Seto, D.; Mahadevan, P.; Ackermann, H.-W.; Kropinski, A.M. Unifying classical and molecular taxonomic classification: Analysis of the Podoviridae using BLASTP-based tools. Res. Microbiol. 2008, 159, 406–414. [Google Scholar] [CrossRef]
- McAllister, W.T. Structure and function of the bacteriophage T7 RNA polymerase (or, the virtues of simplicity). Cell. Mol. Biol. Res. 1993, 39, 385–391. [Google Scholar]
- Ceyssens, P.-J.; Lavigne, R.; Mattheus, W.; Chibeu, A.; Hertveldt, K.; Mast, J.; Robben, J.; Volckaert, G. Genomic Analysis of Pseudomonas aeruginosa Phages LKD16 and LKA1: Establishment of the phiKMV Subgroup within the T7 Supergroup. J. Bacteriol. 2006, 188, 6924–6931. [Google Scholar] [CrossRef] [Green Version]
- Yuzenkova, J.; Nechaev, S.; Berlin, J.; Rogulja, D.; Kuznedelov, K.; Inman, R.; Mushegian, A.; Severinov, K. Genome of Xanthomonas oryzae Bacteriophage Xp10: An Odd T-Odd Phage. J. Mol. Biol. 2003, 330, 735–748. [Google Scholar] [CrossRef]
- Lee, C.-N.; Hu, R.-M.; Chow, T.-Y.; Lin, J.-W.; Chen, H.-Y.; Tseng, Y.-H.; Weng, S.-F. Comparison of Genomes of Three Xanthomonas oryzae Bacteriophages. BMC Genom. 2007, 8, 442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoue, Y.; Matsuura, T.; Ohara, T.; Azegami, K. Bacteriophage OP1, lytic for Xanthomonas oryzae pv. oryzae, changes its host range by duplication and deletion of the small domain in the deduced tail fiber gene. J. Gen. Plant Pathol. 2006, 72, 111–118. [Google Scholar] [CrossRef]
- Kalatzis, P.G.; Rørbo, N.I.; Castillo, D.; Mauritzen, J.J.; Jørgensen, J.; Kokkari, C.; Zhang, F.; Katharios, P.; Middelboe, M. Stumbling across the Same Phage: Comparative Genomics of Widespread Temperate Phages Infecting the Fish Pathogen Vibrio anguillarum. Viruses 2017, 9, 122. [Google Scholar] [CrossRef] [PubMed]
- Piya, D.; Yicheng, X.; Hernandez Morales, A.C.; Everett, G.F.K. Complete Genome Sequence of Salmonella enterica Serovar Typhimurium Siphophage Shivani. Genome Announc. 2015, 3, e01443–14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikoulinskaia, G.V.; Odinokova, I.V.; Zimin, A.A.; Lysanskaya, V.Y.; Feofanov, S.A.; Stepnaya, O.A. Identification and characterization of the metal ion-dependent 1,72alanoyl-d-glutamate peptidase encoded by bacteriophage T5. FEBS J. 2009, 276, 7329–7342. [Google Scholar] [CrossRef] [PubMed]
- Dik, D.A.; Marous, D.R.; Fisher, J.F.; Mobashery, S. Lytic transglycosylases: Concinnity in concision of the bacterial cell wall. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 503–542. [Google Scholar] [CrossRef] [PubMed]
- Dwivedi, B.; Xue, B.; Lundin, D.; Edwards, R.A.; Breitbart, M. A bioinformatic analysis of ribonucleotide reductase genes in phage genomes and metagenomes. BMC Evol. Biol. 2013, 13, 33. [Google Scholar] [CrossRef] [Green Version]
- Tseng, M.J.; He, P.; Hilfinger, J.M.; Greenberg, G.R. Bacteriophage T4 nrdA and nrdB genes, encoding ribonucleotide reductase, are expressed both separately and coordinately: Characterization of the nrdB promoter. J. Bacteriol. 1990, 172, 6323–6332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiu, C.S.; Cook, K.S.; Greenberg, G.R. Characteristics of a bacteriophage T4-induced complex synthesizing deoxyribonucleotides. J. Biol. Chem. 1982, 257, 15087–15097. [Google Scholar] [CrossRef]
- Chiu, C.S.; Tomich, P.K.; Greenberg, G.R. Simultaneous initiation of synthesis of bacteriophage T4 DNA and of deoxyribonucleotides. Proc. Natl. Acad. Sci. USA 1976, 73, 757–761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavysh, D.; Sokolova, M.; Minakhin, L.; Yakunina, M.; Artamonova, T.; Kozyavkin, S.; Makarova, K.S.; Koonin, E.V.; Severinov, K. The genome of AR9, a giant transducing Bacillus phage encoding two multisubunit RNA polymerases. Virology 2016, 495, 185–196. [Google Scholar] [CrossRef]
- Niyogi, S.K.; Datta, A.K. A novel oligoribonuclease of Escherichia coli. I. Isolation and properties. J. Biol. Chem. 1975, 250, 7307–7312. [Google Scholar] [CrossRef]
- Mikoulinskaia, G.V.; Gubanov, S.I.; Zimin, A.A.; Kolesnikov, I.V.; Feofanov, S.A.; Miroshnikov, A.I. Purification and characterization of the deoxynucleoside monophosphate kinase of bacteriophage T5. Protein Expr. Purif. 2003, 27, 195–201. [Google Scholar] [CrossRef]
- Sváb, D.; Falgenhauer, L.; Rohde, M.; Szabó, J.; Chakraborty, T.; Tóth, I. Identification and Characterization of T5-Like Bacteriophages Representing Two Novel Subgroups from Food Products. Front. Microbiol. 2018, 9, 202. [Google Scholar] [CrossRef]
- Davison, J. Pre-early functions of bacteriophage T5 and its relatives. Bacteriophage 2015, 5, e1086500. [Google Scholar] [CrossRef] [Green Version]
- McCorquodale, D.J.; Chen, C.W.; Joseph, M.K.; Woychik, R. Modification of RNA polymerase from Escherichia coli by pre-early gene products of bacteriophage T5. J. Virol. 1981, 40, 958–962. [Google Scholar] [CrossRef] [Green Version]
- Gill, J.J.; Berry, J.D.; Russell, W.K.; Lessor, L.; Escobar-Garcia, D.A.; Hernandez, D.; Kane, A.; Keene, J.; Maddox, M.; Martin, R.; et al. The Caulobacter crescentus phage phiCbK: Genomics of a canonical phage. BMC Genom. 2012, 13, 542. [Google Scholar] [CrossRef] [Green Version]
- Adhikari, S.; Curtis, P.D. DNA methyltransferases and epigenetic regulation in bacteria. FEMS Microbiol. Rev. 2016, 40, 575–591. [Google Scholar] [CrossRef]
- Hattman, S.; Wilkinson, J.; Swinton, D.; Schlagman, S.; Macdonald, P.M.; Mosig, G. Common evolutionary origin of the phage T4 dam and host Escherichia coli dam DNA-adenine methyltransferase genes. J. Bacteriol. 1985, 164, 932–937. [Google Scholar] [CrossRef] [Green Version]
- Horton, J.R.; Liebert, K.; Bekes, M.; Jeltsch, A.; Cheng, X. Structure and Substrate Recognition of the Escherichia coli DNA Adenine Methyltransferase. J. Mol. Biol. 2006, 358, 559–570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elsawy, H.; Podobinschi, S.; Chahar, S.; Jeltsch, A. Transition from EcoDam to T4Dam DNA Recognition Mechanism without Loss of Activity and Specificity. ChemBioChem 2009, 10, 2488–2493. [Google Scholar] [CrossRef] [PubMed]
- Hattman, S.; Malygin, E.G. Bacteriophage T2Dam and T4Dam DNA-[N6-adenine]-methyltransferases1. In Progress in Nucleic Acid Research and Molecular Biology; Academic Press: Cambridge, MA, USA, 2004; Volume 77, pp. 67–126. [Google Scholar]
- Deschavanne, P.; Radman, M. Counterselection of GATC sequences in enterobacteriophages by the components of the methyl-directed mismatch repair system. J. Mol. Evol. 1991, 33, 125–132. [Google Scholar] [CrossRef]
- Sandegren, L.; Sjöberg, B.-M. Self-Splicing of the Bacteriophage T4 Group I Introns Requires Efficient Translation of the Pre-mRNA In Vivo and Correlates with the Growth State of the Infected Bacterium. J. Bacteriol. 2007, 189, 980–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luhtanen, A.-M.; Eronen-Rasimus, E.; Kaartokallio, H.; Rintala, J.-M.; Autio, R.; Roine, E. Isolation and characterization of phage—host systems from the Baltic Sea ice. Extremophiles 2014, 18, 121–130. [Google Scholar] [CrossRef]
- Foley, S.; Bruttin, A.; Brüssow, H. Widespread Distribution of a Group I Intron and Its Three Deletion Derivatives in the Lysin Gene of Streptococcus thermophilus Bacteriophages. J. Virol. 2000, 74, 611–618. [Google Scholar] [CrossRef] [Green Version]
- Bechhofer, D.H.; Hue, K.K.; Shub, D.A. An intron in the thymidylate synthase gene of Bacillus bacteriophage beta 22: Evidence for independent evolution of a gene, its group I intron, and the intron open reading frame. Proc. Natl. Acad. Sci. USA 1994, 91, 11669–11673. [Google Scholar] [CrossRef] [Green Version]
- Goodrich-Blair, H.; David, A.S. The DNA polymerase genes of several HMU-bacteriophages have similar group I introns with highly divergent open reading frames. Nucleic Acids Res. 1994, 22, 3715–3721. [Google Scholar] [CrossRef] [Green Version]
- Landthaler, M.; Begley, U.; Lau, N.C.; Shub, D.A. Two self-splicing group I introns in the ribonucleotide reductase large subunit gene of Staphylococcus aureus phage Twort. Nucleic Acids Res. 2002, 30, 1935–1943. [Google Scholar] [CrossRef] [PubMed]
- Geuskens, V.; Vogel, J.L.; Grimaud, R.; Desmet, L.; Higgins, N.P.; Toussaint, A. Frameshift mutations in the bacteriophage Mu repressor gene can confer a trans-dominant virulent phenotype to the phage. J. Bacteriol. 1991, 173, 6578–6585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howe, M.M. Transduction by bacteriophage MU-1. Virology 1973, 55, 103–117. [Google Scholar] [CrossRef]
- Fogg, P.C.M.; Hynes, A.P.; Digby, E.; Lang, A.S.; Beatty, J.T. Characterization of a newly discovered Mu-like bacteriophage, RcapMu, in Rhodobacter capsulatus strain SB1003. Virology 2011, 421, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Q.; Zhang, R.; Xu, Y.; Iii, R.A.W.; Wang, Y.; Luo, T.; Jiao, N. A marine inducible prophage vB_CibM-P1 isolated from the aerobic anoxygenic phototrophic bacterium Citromicrobium bathyomarinum JL354. Sci. Rep. 2014, 4, 7118. [Google Scholar] [CrossRef] [Green Version]
- Lin, W.; Chen, Q.; Liu, Y.; Jiao, N.; Zheng, Q. Characteristics of two myoviruses induced from the coastal photoheterotrophic bacterium Porphyrobacter sp. YT40. FEMS Microbiol. Lett. 2019, 366, 9. [Google Scholar] [CrossRef]
- Lin, D.; Tang, K.; Han, Y.; Li, C.; Chen, X. Genome sequence of an inducible phage in Rhodovulum sp. P5 isolated from the shallow-sea hydrothermal system. Mar. Genom. 2016, 30, 93–95. [Google Scholar] [CrossRef]
- Tang, K.; Lin, D.; Zheng, Q.; Liu, K.; Yang, Y.; Han, Y.; Jiao, N. Genomic, proteomic and bioinformatic analysis of two temperate phages in Roseobacter clade bacteria isolated from the deep-sea water. BMC Genom. 2017, 18, 485. [Google Scholar] [CrossRef]
- Laanto, E.; Ravantti, J.J.; Sundberg, L.-R. Prophages and Past Prophage-Host Interactions Revealed by CRISPR Spacer Content in a Fish Pathogen. Microorganisms 2020, 8, 1919. [Google Scholar] [CrossRef]
- Little, J.W. Autodigestion of lexA and phage lambda repressors. Proc. Natl. Acad. Sci. USA 1984, 81, 1375–1379. [Google Scholar] [CrossRef] [Green Version]
- Summer, E.J.; Gonzalez, C.F.; Carlisle, T.; Mebane, L.M.; Cass, A.M.; Savva, C.G.; LiPuma, J.; Young, R. Burkholderia cenocepacia Phage BcepMu and a Family of Mu-like Phages Encoding Potential Pathogenesis Factors. J. Mol. Biol. 2004, 340, 49–65. [Google Scholar] [CrossRef]
- Jakhetia, R.; Verma, N.K. Identification and Molecular Characterisation of a Novel Mu-Like Bacteriophage, SfMu, of Shigella flexneri. PLoS ONE 2015, 10, e0124053. [Google Scholar] [CrossRef]
- Mestre, M.R.; González-Delgado, A.; Gutiérrez-Rus, L.I.; Martínez-Abarca, F.; Toro, N. Systematic prediction of genes functionally associated with bacterial retrons and classification of the encoded tripartite systems. Nucleic Acids Res. 2020, 48, 12632–12647. [Google Scholar] [CrossRef] [PubMed]
- Millman, A.; Bernheim, A.; Stokar-Avihail, A.; Fedorenko, T.; Voichek, M.; Leavitt, A.; Oppenheimer-Shaanan, Y.; Sorek, R. Bacterial Retrons Function in Anti-Phage Defense. Cell 2020, 183, 1551–1561.e1512. [Google Scholar] [CrossRef] [PubMed]
- Eddy, S.R. Accelerated Profile HMM Searches. PLOS Comput. Biol. 2011, 7, e1002195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bin Jang, H.; Bolduc, B.; Zablocki, O.; Kuhn, J.H.; Roux, S.; Adriaenssens, E.M.; Brister, J.R.; Kropinski, A.M.; Krupovic, M.; Lavigne, R.; et al. Taxonomic assignment of uncultivated prokaryotic virus genomes is enabled by gene-sharing networks. Nat. Biotechnol. 2019, 37, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Cantu, V.A.; Salamon, P.; Seguritan, V.; Redfield, J.; Salamon, D.; Edwards, R.A.; Segall, A.M. PhANNs, a fast and accurate tool and web server to classify phage structural proteins. PLOS Comput. Biol. 2020, 16, e1007845. [Google Scholar] [CrossRef]
- Gilchrist, C.L.M.; Chooi, Y.-H. Clinker & clustermap.js: Automatic generation of gene cluster comparison figures. Bioinformatics 2021, 37, 2473–2475. [Google Scholar] [CrossRef]
- Burroughs, A.; Iyer, L.; Aravind, L. Comparative Genomics and Evolutionary Trajectories of Viral ATP Dependent DNA-Packaging Systems. Genome Dyn. 2007, 3, 48–65. [Google Scholar] [CrossRef] [PubMed]
- Iyer, L.M.; Tahiliani, M.; Rao, A.; Aravind, L. Prediction of novel families of enzymes involved in oxidative and other complex modifications of bases in nucleic acids. Cell Cycle 2009, 8, 1698–1710. [Google Scholar] [CrossRef] [Green Version]
- Ramesh, N.; Archana, L.; Madurantakam Royam, M.; Manohar, P.; Eniyan, K. Effect of various bacteriological media on the plaque morphology of. Access Microbiol. 2019, 1, e000036. [Google Scholar] [CrossRef]
- Hadas, H.; Einav, M.; Fishov, I.; Zaritsky, A. Bacteriophage T4 development depends on the physiology of its host Escherichia coli. Microbiology 1997, 143 Pt 1, 179–185. [Google Scholar] [CrossRef] [Green Version]
- Siwek, W.; Czapinska, H.; Bochtler, M.; Bujnicki, J.M.; Skowronek, K. Crystal structure and mechanism of action of the N6-methyladenine-dependent type IIM restriction endonuclease R.DpnI. Nucleic Acids Res. 2012, 40, 7563–7572. [Google Scholar] [CrossRef] [Green Version]
- Sambrook, J.; Russell, D.W. Molecular Cloning: A Laboratory Manual, 3rd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001. [Google Scholar]
- Wingett, S.W.; Andrews, S. FastQ Screen: A tool for multi-genome mapping and quality control. Research 2018, 7, 1338. [Google Scholar] [CrossRef]
- Chen, S.; Zhou, Y.; Chen, Y.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, i884–i890. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; Subgroup, G.P.D.P. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, J.T.; Thorvaldsdóttir, H.; Winckler, W.; Guttman, M.; Lander, E.S.; Getz, G.; Mesirov, J.P. Integrative genomics viewer. Nat. Biotechnol. 2011, 29, 24–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carver, T.; Berriman, M.; Tivey, A.; Patel, C.; Böhme, U.; Barrell, B.G.; Parkhill, J.; Rajandream, M.-A. Artemis and ACT: Viewing, annotating and comparing sequences stored in a relational database. Bioinformatics 2008, 24, 2672–2676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brettin, T.; Davis, J.J.; Disz, T.; Edwards, R.A.; Gerdes, S.; Olsen, G.J.; Olson, R.; Overbeek, R.; Parrello, B.; Pusch, G.D.; et al. RASTtk: A modular and extensible implementation of the RAST algorithm for building custom annotation pipelines and annotating batches of genomes. Sci. Rep. 2015, 5, 8365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wattam, A.R.; Davis, J.J.; Assaf, R.; Boisvert, S.; Brettin, T.; Bun, C.; Conrad, N.; Dietrich, E.M.; Disz, T.; Gabbard, J.L.; et al. Improvements to PATRIC, the all-bacterial Bioinformatics Database and Analysis Resource Center. Nucleic Acids Res. 2016, 45, D535–D542. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, A.; Remmert, M.; Biegert, A.; Söding, J. Fast and accurate automatic structure prediction with HHpred. Proteins Struct. Funct. Bioinform. 2009, 77, 128–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schattner, P.; Brooks, A.N.; Lowe, T.M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005, 33, W686–W689. [Google Scholar] [CrossRef]
- Laslett, D.; Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef]
- Lopes, A.; Tavares, P.; Petit, M.-A.; Guérois, R.; Zinn-Justin, S. Automated classification of tailed bacteriophages according to their neck organization. BMC Genom. 2014, 15, 1027. [Google Scholar] [CrossRef] [Green Version]
- Darzentas, N. Circoletto: Visualizing sequence similarity with Circos. Bioinformatics 2010, 26, 2620–2621. [Google Scholar] [CrossRef]
- Jacomy, M.; Venturini, T.; Heymann, S.; Bastian, M. ForceAtlas2, a Continuous Graph Layout Algorithm for Handy Network Visualization Designed for the Gephi Software. PLoS ONE 2014, 9, e98679. [Google Scholar] [CrossRef]
- Fruchterman, T.M.J.; Reingold, E.M. Graph drawing by force-directed placement. Softw. Pract. Exp. 1991, 21, 1129–1164. [Google Scholar] [CrossRef]
- Drozdz, M.; Piekarowicz, A.; Bujnicki, J.M.; Radlinska, M. Novel non-specific DNA adenine methyltransferases. Nucleic Acids Res. 2011, 40, 2119–2130. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bujak, K.; Decewicz, P.; Rosinska, J.M.; Radlinska, M. Genome Study of a Novel Virulent Phage vB_SspS_KASIA and Mu-like Prophages of Shewanella sp. M16 Provides Insights into the Genetic Diversity of the Shewanella Virome. Int. J. Mol. Sci. 2021, 22, 11070. https://doi.org/10.3390/ijms222011070
Bujak K, Decewicz P, Rosinska JM, Radlinska M. Genome Study of a Novel Virulent Phage vB_SspS_KASIA and Mu-like Prophages of Shewanella sp. M16 Provides Insights into the Genetic Diversity of the Shewanella Virome. International Journal of Molecular Sciences. 2021; 22(20):11070. https://doi.org/10.3390/ijms222011070
Chicago/Turabian StyleBujak, Katarzyna, Przemyslaw Decewicz, Joanna M. Rosinska, and Monika Radlinska. 2021. "Genome Study of a Novel Virulent Phage vB_SspS_KASIA and Mu-like Prophages of Shewanella sp. M16 Provides Insights into the Genetic Diversity of the Shewanella Virome" International Journal of Molecular Sciences 22, no. 20: 11070. https://doi.org/10.3390/ijms222011070