Oxidative Stress and Lipid Mediators Modulate Immune Cell Functions in Autoimmune Diseases



Abstract
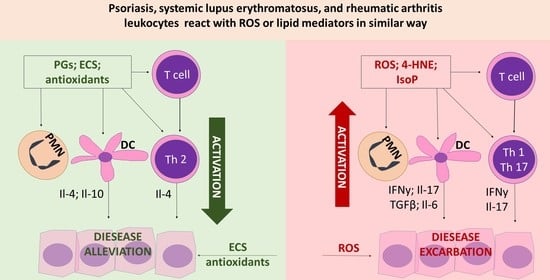
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Literature Review
1.1. Oxidative Stress
1.2. Lipid Mediators
1.2.1. Endocannabinoids
1.2.2. Eicosanoids
1.2.3. Non-Enzymatic Modifications
2. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| 2-AG | 2-Arachidonylglycerol |
| 4-HNE | 4-Hydroxynonenal |
| 4-ONE | 4-Oxynonenal |
| AEA | Anandamide |
| CB | Cannabinoid receptor |
| COX | Cyclooxygenase |
| DAGL | Diacylglycerol lipase |
| DP | Prostaglandin D receptor |
| EP | Prostaglandin E receptor |
| IFNγ | Interferon γ |
| IL | Interleukin |
| IMQ | Imiquimod |
| IPP | Prostaglandin I receptor |
| LOX | Lipoxygenases |
| LTB4 | Leukotriene B4 |
| MDA | Malondialdehyde |
| NAPE-PLD | N-acylphosphatidylethanolamine phospholipase D |
| NAT | N-acyltransferase |
| NFκB | Nuclear factor kappa-light-chain-enhancer of activated B cells |
| Nrf2 | Nuclear factor erythroid 2-like |
| OEA | Oleoylethanolamide |
| PGE | Prostaglandin D |
| PGE | Prostaglandin E |
| PGF | Prostaglandin F |
| PGI | Prostaglandin I |
| PGJ | Prostaglandin J |
| PLA2 | Phospholipase A2 |
| PLC | Phospholipase C |
| PUFAs | Polyunsaturated fatty acids |
| RA | Rheumatic arthritis |
| ROS | Reactive oxygen species |
| SLE | Systemic lupus erythematosus |
| Th | T helper lymphocytes |
| TLR | Toll like receptor |
| TNFα | Tumor necrosis factor α |
| TNFR | Tumor necrosis factor receptor |
| TRPV1 | The transient receptor potential cation channel subfamily V member 1 |
| TXA2 | Thromboxane A2 |
References
- Fortina, A.B.; Bardazzi, F.; Berti, S.; Carnevale, C.; Di Lernia, V.; El Hachem, M.; Neri, I.; Gelmetti, C.M.; Lora, V.; Mazzatenta, C.; et al. Treatment of severe psoriasis in children: Recommendations of an Italian expert group. Eur. J. Pediatr. 2017, 176, 1339–1354. [Google Scholar] [CrossRef] [PubMed]
- Jesus, D.; Matos, A.; Henriques, C.; Zen, M.; LaRosa, M.; Iaccarino, L.; Da Silva, J.A.P.; Doria, A.; Inês, L.S. Derivation and validation of the SLE Disease Activity Score (SLE-DAS): A new SLE continuous measure with high sensitivity for changes in disease activity. Ann. Rheum. Dis. 2019, 78, 365–371. [Google Scholar] [CrossRef] [PubMed]
- Gonçalves, R.S.G.; Pereira, G.A.; de Andrade Lima, E.; Martins, T.H.F.; Junior, J.O.A.; Carvalho, J.B.; Mariz, H.A.; Dantas, A.T.; Duarte, A.L.B.P. Validation of the Toronto Psoriatic Arthritis Screen II (ToPAS II) questionnaire in a Brazilian population. Clin. Rheumatol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Farid, E.; Mumtaz, M.; Hajji, F.; Ebrahim, R.A.; Abdulla, H.; Tabbara, K. T Regulatory Cells in Rheumatoid Arthritis with Reference to Anti-Citrullinated Peptide Antibody and TNF-alpha Inhibitor Therapy. Egypt J. Immunol. 2020, 27, 55–63. [Google Scholar] [PubMed]
- Dogra, S.; Mahajan, R. Psoriasis: Epidemiology, clinical features, co-morbidities, and clinical scoring. Indian Dermatol. Online J. 2016, 7, 471–480. [Google Scholar] [CrossRef]
- Capon, F. The Genetic Basis of Psoriasis. Int. J. Mol. Sci. 2017, 18, 2526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wasserman, A. Diagnosis and management of rheumatoid arthritis. Am. Fam. Physician 2011, 84, 1245–1252. [Google Scholar]
- Fava, A.; Petri, M. Systemic lupus erythematosus: Diagnosis and clinical management. J. Autoimmun. 2019, 96, 1–13. [Google Scholar] [CrossRef]
- Patente, T.A.; Pinho, M.P.; Oliveira, A.A.; Evangelista, G.C.M.; Bergami-Santos, P.C.; Barbuto, J.A.M. Human Dendritic Cells: Their Heterogeneity and Clinical Application Potential in Cancer Immunotherapy. Front. Immunol. 2019, 9, 3176. [Google Scholar] [CrossRef]
- Li, J.-G.; Du, Y.-M.; Yan, Z.-D.; Yan, J.; Zhuansun, Y.-X.; Chen, R.; Zhang, W.; Feng, S.-L.; Ran, P.-X. CD80 and CD86 knockdown in dendritic cells regulates Th1/Th2 cytokine production in asthmatic mice. Exp. Ther. Med. 2016, 11, 878–884. [Google Scholar] [CrossRef]
- Gilliet, M.; Lande, R. Antimicrobial peptides and self-DNA in autoimmune skin inflammation. Curr. Opin. Immunol. 2008, 20, 401–407. [Google Scholar] [CrossRef]
- Ueki, M.; Kimura-Kataoka, K.; Fujihara, J.; Iida, R.; Kawai, Y.; Kusaka, A.; Sasaki, T.; Takeshita, H.; Yasuda, T. Evaluation of the functional effects of genetic variants‒missense and nonsense SNPs, indels and copy number variations‒in the gene encoding human deoxyribonuclease I potentially implicated in autoimmunity. Sci. Rep. 2019, 9, 13660. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lund, J.M.; Alexopoulou, L.; Sato, A.; Karow, M.; Adams, N.C.; Gale, N.W.; Iwasaki, A.; Flavell, R.A. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc. Natl. Acad. Sci. USA 2004, 101, 5598–5603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Marken, J.; Chen, J.; Tran, V.B.; Li, Q.-Z.; Li, M.; Cerosaletti, K.; Elkon, K.B.; Zeng, X.; Giltiay, N.V. High TLR7 Expression Drives the Expansion of CD19+CD24hiCD38hi Transitional B Cells and Autoantibody Production in SLE Patients. Front. Immunol. 2019, 10, 1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García-Ortiz, H.; Velázquez-Cruz, R.; Espinosa-Rosales, F.; Jiménez-Morales, S.; Baca, V.; Orozco, L. Association of TLR7 copy number variation with susceptibility to childhood-onset systemic lupus erythematosus in Mexican population. Ann. Rheum. Dis. 2010, 69, 1861–1865. [Google Scholar] [CrossRef] [PubMed]
- Lai, C.-Y.; Su, Y.-W.; Lin, K.-I.; Hsu, L.-C.; Chuang, T.-H. Natural Modulators of Endosomal Toll-Like Receptor-Mediated Psoriatic Skin Inflammation. J. Immunol. Res. 2017, 2017, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Huang, Q.-Q.; Pope, R.M. The role of Toll-like receptors in rheumatoid arthritis. Curr. Rheumatol. Rep. 2009, 11, 357–364. [Google Scholar] [CrossRef] [Green Version]
- Chamberlain, N.D.; Kim, S.; Vila, O.M.; Volin, M.V.; Volkov, S.; Pope, R.M.; Arami, S.; Mandelln, A.M.; Shahrara, S. Ligation of TLR7 by rheumatoid arthritis synovial fluid single strand RNA induces transcription of TNFα in monocytes. Ann. Rheum. Dis. 2013, 72, 418–426. [Google Scholar] [CrossRef]
- Nie, F.; Ding, F.; Chen, B.; Huang, S.; Liu, Q.; Xu, C. Dendritic cells aggregate inflammation in experimental osteoarthritis through a toll-like receptor (TLR)-dependent machinery response to challenges. Life Sci. 2019, 238, 116920. [Google Scholar] [CrossRef]
- Bengtsson, A.A.; Sturfelt, G.; Truedsson, L.; Blomberg, J.; Alm, G.; Vallin, H.; Rönnblom, L. Activation of type I interferon system in systemic lupus erythematosus correlates with disease activity but not with antiretroviral antibodies. Lupus 2000, 9, 664–671. [Google Scholar] [CrossRef]
- Ohl, K.; Tenbrock, K. Inflammatory Cytokines in Systemic Lupus Erythematosus. J. Biomed. Biotechnol. 2011, 2011, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Prajeeth, C.K.; Kronisch, J.; Khorooshi, R.; Knier, B.; Toft-Hansen, H.; Gudi, V.; Floess, S.; Huehn, J.; Owens, T.; Korn, T.; et al. Effectors of Th1 and Th17 cells act on astrocytes and augment their neuroinflammatory properties. J. Neuroinflamm. 2017, 14, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Prinz, J.C.; Gross, B.; Vollmer, S.; Trommler, P.; Strobel, I.; Meurer, M.; Plewig, G. T cell clones from psoriasis skin lesions can promote keratinocyte proliferation in vitro via secreted products. Eur. J. Immunol. 1994, 24, 593–598. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, W.; Lakkis, F.G.; Chalasani, G. B Cells, Antibodies, and More. Clin. J. Am. Soc. Nephrol. 2016, 11, 137–154. [Google Scholar] [CrossRef]
- Dema, B.; Charles, N. Autoantibodies in SLE: Specificities, Isotypes and Receptors. Antibodies 2016, 5, 2. [Google Scholar] [CrossRef] [Green Version]
- Tiwari, V.; Jandu, J.S.; Bergman, M.J. Rheumatoid Factor. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Singh, S.; Singh, U.; Singh, S. Prevalence of autoantibodies in patients of psoriasis. J. Clin. Lab. Anal. 2010, 24, 44–48. [Google Scholar] [CrossRef]
- Mishra, A.; Guo, Y.; Zhang, L.; More, S.; Weng, T.; Chintagari, N.R.; Huang, C.; Liang, Y.; Pushparaj, S.; Gou, D.; et al. A Critical Role for P2X7 Receptor-Induced VCAM-1 Shedding and Neutrophil Infiltration during Acute Lung Injury. J. Immunol. 2016, 197, 2828–2837. [Google Scholar] [CrossRef] [Green Version]
- Feng, D.; Nagy, J.A.; Pyne, K.; Dvorak, H.F.; Dvorak, A.M. Neutrophils Emigrate from Venules by a Transendothelial Cell Pathway in Response to FMLP. J. Exp. Med. 1998, 187, 903–915. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Qin, W.; Zhang, Y.; Zhang, H.; Sun, B. Endotoxin promotes neutrophil hierarchical chemotaxis via the p38-membrane receptor pathway. Oncotarget 2016, 7, 74247–74258. [Google Scholar] [CrossRef] [Green Version]
- Tang, D.; Kang, R.; Coyne, C.B.; Zeh, H.J.; Lotze, M.T. PAMPs and DAMPs: Signal 0s that spur autophagy and immunity. Immunol. Rev. 2012, 249, 158–175. [Google Scholar] [CrossRef]
- Brinkmann, V.; Zychlinsky, A. Neutrophil extracellular traps: Is immunity the second function of chromatin? J. Cell Biol. 2012, 198, 773–783. [Google Scholar] [CrossRef] [Green Version]
- Ambrożewicz, E.; Wójcik, P.; Wroński, A.; Łuczaj, W.; Jastrząb, A.; Žarković, N.; Skrzydlewska, E. Pathophysiological Alterations of Redox Signaling and Endocannabinoid System in Granulocytes and Plasma of Psoriatic Patients. Cells 2018, 7, 159. [Google Scholar] [CrossRef] [Green Version]
- Wójcik, P.; Biernacki, M.; Wroński, A.; Łuczaj, W.; Waeg, G.; Žarković, N.; Skrzydlewska, E. Altered Lipid Metabolism in Blood Mononuclear Cells of Psoriatic Patients Indicates Differential Changes in Psoriasis Vulgaris and Psoriatic Arthritis. Int. J. Mol. Sci. 2019, 20, 4249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asefi, M.; Vaisi-Raygani, A.; Bahrehmand, F.; Kiani, A.; Rahimi, Z.; Nomani, H.; Ebrahimi, A.; Tavilani, H.; Pourmotabbed, T. Paraoxonase 1 (PON1) 55 polymorphism, lipid profiles and psoriasis. Br. J. Dermatol. 2012, 167, 1279–1286. [Google Scholar] [CrossRef] [PubMed]
- Peluso, I.; Cavaliere, A.; Palmery, M. Plasma total antioxidant capacity and peroxidation biomarkers in psoriasis. J. Biomed. Sci. 2016, 23, 52. [Google Scholar] [CrossRef]
- Wang, W.; Yuhai; Wang, H.; Chasuna; Bagenna. Astilbin reduces ROS accumulation and VEGF expression through Nrf2 in psoriasis-like skin disease. Biol. Res. 2019, 52, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lightfoot, Y.L.; Blanco, L.P.; Kaplan, M.J. Metabolic abnormalities and oxidative stress in lupus. Curr. Opin. Rheumatol. 2017, 29, 442–449. [Google Scholar] [CrossRef]
- García-González, A.; Gaxiola-Robles, R.; Zenteno-Savín, T. Oxidative stress in patients with rheumatoid arthritis. Rev. Investig. Clin. 2015, 67, 46–53. [Google Scholar]
- Zhang, H.; Fu, R.; Guo, C.; Huang, Y.; Wang, H.; Wang, S.; Zhao, J.; Yang, N. Anti-dsDNA antibodies bind to TLR4 and activate NLRP3 inflammasome in lupus monocytes/macrophages. J. Transl. Med. 2016, 14, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Li, T.; Chen, S.; Gu, Y.; Ye, S. Neutrophil Extracellular Trap Mitochondrial DNA and Its Autoantibody in Systemic Lupus Erythematosus and a Proof-of-Concept Trial of Metformin. Arthritis Rheumatol. 2015, 67, 3190–3200. [Google Scholar] [CrossRef] [Green Version]
- Lai, Z.-W.; Hanczko, R.; Bonilla, E.; Caza, T.N.; Clair, B.; Bartos, A.; Miklossy, G.; Jimah, J.; Doherty, E.; Tily, H.; et al. N-acetylcysteine reduces disease activity by blocking mammalian target of rapamycin in T cells from systemic lupus erythematosus patients: A randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2012, 64, 2937–2946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, Y.; Terao, C. The Impact of Cigarette Smoking on Risk of Rheumatoid Arthritis: A Narrative Review. Cells 2020, 9, 475. [Google Scholar] [CrossRef] [Green Version]
- Costenbader, K.H.; Feskanich, D.; Mandl, L.A.; Karlson, E.W. Smoking Intensity, Duration, and Cessation, and the Risk of Rheumatoid Arthritis in Women. Am. J. Med. 2006, 119, 503.e1–503.e9. [Google Scholar] [CrossRef] [PubMed]
- Di Giuseppe, D.; Discacciati, A.; Orsini, N.; Wolk, A. Cigarette smoking and risk of rheumatoid arthritis: A dose-response meta-analysis. Arthritis Res. Ther. 2014, 16, R61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Datta, S.; Kundu, S.; Ghosh, P.; De, S.; Ghosh, A.; Chatterjee, M. Correlation of oxidant status with oxidative tissue damage in patients with rheumatoid arthritis. Clin. Rheumatol. 2014, 33, 1557–1564. [Google Scholar] [CrossRef]
- Toukap, A.N.; Delporte, C.; Noyon, C.; Franck, T.; Rousseau, A.; Serteyn, D.; Raes, M.; Vanhaeverbeek, M.; Moguilevsky, N.; Nève, J.; et al. Myeloperoxidase and its products in synovial fluid of patients with treated or untreated rheumatoid arthritis. Free Radic. Res. 2014, 48, 461–465. [Google Scholar] [CrossRef] [Green Version]
- Jaganjac, M.; Cipak, A.; Schaur, R.J.; Zarkovic, N. Pathophysiology of neutrophil-mediated extracellular redox reactions. Front. Biosci. 2016, 21, 839–855. [Google Scholar] [CrossRef] [Green Version]
- Wójcik, P.; Žarković, N.; Gęgotek, A.; Skrzydlewska, E. Involvement of Metabolic Lipid Mediators in the Regulation of Apoptosis. Biomolecules 2020, 10, 402. [Google Scholar] [CrossRef] [Green Version]
- Sokolova, O.; Naumann, M. NF-κB Signaling in Gastric Cancer. Toxins 2017, 9, 119. [Google Scholar] [CrossRef]
- Miraghazadeh, B.; Cook, M.C. Nuclear Factor-kappaB in Autoimmunity: Man and Mouse. Front. Immunol. 2018, 9, 613. [Google Scholar] [CrossRef] [Green Version]
- Farrugia, M.; Baron, B. The role of TNF-α in rheumatoid arthritis: A focus on regulatory T cells. J. Clin. Transl. Res. 2016, 2, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Perdriger, A. Infliximab in the treatment of rheumatoid arthritis. Biologics 2009, 3, 183–191. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Sarabia, F.; Ariza-Ariza, R.; Hernández-Cruz, B.; Villanueva, I. Adalimumab for treating rheumatoid arthritis. J. Rheumatol. 2006, 33, 1075–1081. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Mysler, E.; Moots, R.J. Etanercept for the treatment of rheumatoid arthritis. Immunotherapy 2018, 10, 433–445. [Google Scholar] [CrossRef]
- Nguyen, T.U.; Koo, J. Etanercept in the treatment of plaque psoriasis. Clin. Cosmet. Investig. Dermatol. 2009, 2, 77–84. [Google Scholar] [CrossRef] [Green Version]
- Buendia, I.; Michalska, P.; Navarro, E.; Gameiro, I.; Egea, J.; León, R. Nrf2-ARE pathway: An emerging target against oxidative stress and neuroinflammation in neurodegenerative diseases. Pharmacol. Ther. 2016, 157, 84–104. [Google Scholar] [CrossRef]
- Yang, L.; Fan, X.; Cui, T.; Dang, E.; Wang, G. Nrf2 Promotes Keratinocyte Proliferation in Psoriasis through Up-Regulation of Keratin 6, Keratin 16, and Keratin 17. J. Investig. Dermatol. 2017, 137, 2168–2176. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Stein, T.D.; Johnson, J.A. Genetic dissection of systemic autoimmune disease in Nrf2-deficient mice. Physiol. Genom. 2004, 18, 261–272. [Google Scholar] [CrossRef]
- Ebihara, S.; Tajima, H.; Ono, M. Nuclear factor erythroid 2-related factor 2 is a critical target for the treatment of glucocorticoid-resistant lupus nephritis. Arthritis Res. Ther. 2016, 18, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Ferrándiz, M.L.; Nacher-Juan, J.; Alcaraz, M.J. Nrf2 as a therapeutic target for rheumatic diseases. Biochem. Pharmacol. 2018, 152, 338–346. [Google Scholar] [CrossRef]
- Pazmandi, K.; Magyarics, Z.; Boldogh, I.; Csillag, A.; Rajnavolgyi, E.; Bacsi, A. Modulatory effects of low-dose hydrogen peroxide on the function of human plasmacytoid dendritic cells. Free Radic. Biol. Med. 2012, 52, 635–645. [Google Scholar] [CrossRef] [Green Version]
- Akhter, N.; Madhoun, A.; Arefanian, H.; Wilson, A.; Kochumon, S.; Thomas, R.; Shenouda, S.; Al-Mulla, F.; Ahmad, R.; Sindhu, S. Oxidative Stress Induces Expression of the Toll-Like Receptors (TLRs) 2 and 4 in the Human Peripheral Blood Mononuclear Cells: Implications for Metabolic Inflammation. Cell Physiol. Biochem. 2019, 53, 1–18. [Google Scholar]
- Tan, P.H.; Sagoo, P.; Chan, C.; Yates, J.B.; Campbell, J.; Beutelspacher, S.C.; Foxwell, B.M.J.; Lombardi, G.; George, A.J.T. Inhibition of NF-κB and Oxidative Pathways in Human Dendritic Cells by Antioxidative Vitamins Generates Regulatory T Cells. J. Immunol. Res. 2005, 174, 7633–7644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koga, Y.; Tsurumaki, H.; Aoki-Saito, H.; Sato, M.; Yatomi, M.; Takehara, K.; Hisada, T. Roles of Cyclic AMP Response Element Binding Activation in the ERK1/2 and p38 MAPK Signalling Pathway in Central Nervous System, Cardiovascular System, Osteoclast Differentiation and Mucin and Cytokine Production. Int. J. Mol. Sci. 2019, 20, 1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuadrado, A.; Nebreda, A.R. Mechanisms and functions of p38 MAPK signalling. Biochem. J. 2010, 429, 403–417. [Google Scholar] [CrossRef] [Green Version]
- Qin, T.; Yin, Y.; Yu, Q.; Yang, Q. Bursopentin (BP5) Protects Dendritic Cells from Lipopolysaccharide-Induced Oxidative Stress for Immunosuppression. PLoS ONE 2015, 10, e0117477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rutault, K.; Alderman, C.; Chain, B.M.; Katz, D.R. Reactive oxygen species activate human peripheral blood dendritic cells. Free Radic. Biol. Med. 1999, 26, 232–238. [Google Scholar] [CrossRef]
- Yang, J.; Yang, X.; Zou, H.; Li, M. Oxidative Stress and Treg and Th17 Dysfunction in Systemic Lupus Erythematosus. Oxidative Med. Cell. Longev. 2016, 2016, 2526174. [Google Scholar] [CrossRef]
- Delgoffe, G.M.; Pollizzi, K.N.; Waickman, A.T.; Heikamp, E.; Meyers, D.J.; Horton, M.R.; Xiao, B.; Worley, P.F.; Powell, J.D. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat. Immunol. 2011, 12, 295–303. [Google Scholar] [CrossRef] [Green Version]
- Park, D.; Jeong, H.; Lee, M.N.; Koh, A.; Kwon, O.; Yang, Y.R.; Noh, J.; Suh, P.-G.; Park, H.; Ryu, S.H. Resveratrol induces autophagy by directly inhibiting mTOR through ATP competition. Sci. Rep. 2016, 6, 21772. [Google Scholar] [CrossRef] [Green Version]
- Sizzano, F.; Collino, S.; Cominetti, O.; Monti, D.; Garagnani, P.; Ostan, R.; Pirazzini, C.; Bacalini, M.G.; Mari, D.; Passarino, G.; et al. Evaluation of Lymphocyte Response to the Induced Oxidative Stress in a Cohort of Ageing Subjects, including Semisupercentenarians and Their Offspring. Mediat. Inflamm. 2018, 2018, 7109312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zabłocka-Słowińska, K.; Płaczkowska, S.; Skórska, K.; Prescha, A.; Pawełczyk, K.; Porębska, I.; Kosacka, M.; Grajeta, H. Oxidative stress in lung cancer patients is associated with altered serum markers of lipid metabolism. PLoS ONE 2019, 14, e0215246. [Google Scholar] [CrossRef]
- Hu, L.; Tian, K.; Zhang, T.; Fan, C.-H.; Zhou, P.; Zeng, D.; Zhao, S.; Li, L.-S.; Smith, H.S.; Li, J.; et al. Cyanate Induces Oxidative Stress Injury and Abnormal Lipid Metabolism in Liver through Nrf2/HO-1. Molecules 2019, 24, 3231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liaras, K.; Fesatidou, M.; Geronikaki, A. Thiazoles and Thiazolidinones as COX/LOX Inhibitors. Molecules 2018, 23, 685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hao, M.; Jiang, L.; Fang, N.; Pu, J.; Hu, L.; Shen, L.-H.; Song, W.; He, B. The cannabinoid WIN55,212-2 protects against oxidized LDL-induced inflammatory response in murine macrophages. J. Lipid Res. 2010, 51, 2181–2190. [Google Scholar] [CrossRef] [Green Version]
- Rajesh, M.; Mukhopadhyay, P.; Haskó, G.; Liaudet, L.; Mackie, K.; Pacher, P. Cannabinoid-1 receptor activation induces reactive oxygen species-dependent and -independent mitogen-activated protein kinase activation and cell death in human coronary artery endothelial cells. Br. J. Pharmacol. 2010, 160, 688–700. [Google Scholar] [CrossRef] [Green Version]
- Honda, T.; Segi-Nishida, E.; Miyachi, Y.; Narumiya, S. Prostacyclin-IP signaling and prostaglandin E2-EP2/EP4 signaling both mediate joint inflammation in mouse collagen-induced arthritis. J. Exp. Med. 2006, 203, 325–335. [Google Scholar] [CrossRef]
- Tsuboi, K.; Uyama, T.; Okamoto, Y.; Ueda, N. Endocannabinoids and related N-acylethanolamines: Biological activities and metabolism. Inflamm. Regen. 2018, 38, 28. [Google Scholar] [CrossRef]
- Matthews, A.T.; Lee, J.H.; Borazjani, A.; Mangum, L.C.; Hou, X.; Ross, M.K. Oxyradical stress increases the biosynthesis of 2-arachidonoylglycerol: Involvement of NADPH oxidase. Am. J. Physiol. Cell Physiol. 2016, 311, C960–C974. [Google Scholar] [CrossRef] [Green Version]
- Turner, S.E.; Williams, C.M.; Iversen, L.; Whalley, B.J. Molecular Pharmacology of Phytocannabinoids. Prog. Chem. Org. Nat. Prod. 2017, 103, 61–101. [Google Scholar] [CrossRef]
- Atalay, S.; Jarocka-Karpowicz, I.; Skrzydlewska, E. Antioxidative and Anti-Inflammatory Properties of Cannabidiol. Antioxidants 2019, 9, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atalay, S.; Dobrzyńska, I.; Gęgotek, A.; Skrzydlewska, E. Cannabidiol protects keratinocyte cell membranes following exposure to UVB and hydrogen peroxide. Redox Biol. 2020, 36, 101613. [Google Scholar] [CrossRef] [PubMed]
- Turcotte, C.; Blanchet, M.-R.; LaViolette, M.; Flamand, N. The CB2 receptor and its role as a regulator of inflammation. Cell. Mol. Life Sci. 2016, 73, 4449–4470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gouda, H.M.; Kamel, N.M. Cannabinoid CB2 receptor gene (CNR2) polymorphism is associated with chronic childhood immune thrombocytopenia in Egypt. Blood Coagul. Fibrinolysis 2013, 24, 247–251. [Google Scholar] [CrossRef]
- Rossi, F.; Bellini, G.; Tolone, C.; Luongo, L.; Mancusi, S.; Papparella, A.; Sturgeon, C.; Fasano, A.; Nobili, B.; Perrone, L.; et al. The Cannabinoid Receptor type 2 Q63R variant increases the risk of celiac disease: Implication for a novel molecular biomarker and future therapeutic intervention. Pharmacol. Res. 2012, 66, 88–94. [Google Scholar] [CrossRef]
- Hohmann, U.; Pelzer, M.; Kleine, J.; Hohmann, T.; Ghadban, C.; Dehghani, F. Opposite Effects of Neuroprotective Cannabinoids, Palmitoylethanolamide, and 2-Arachidonoylglycerol on Function and Morphology of Microglia. Front. Neurosci. 2019, 13, 1180. [Google Scholar] [CrossRef]
- Souza, M.C.; Rosas, E.C. Cannabinoid Receptors as Regulators of Neutrophil Activity in Inflammatory Diseases. In Neutrophils; IntechOpen: London, UK, 2018. [Google Scholar] [CrossRef] [Green Version]
- Kurihara, R.; Tohyama, Y.; Matsusaka, S.; Naruse, H.; Kinoshita, E.; Tsujioka, T.; Katsumata, Y.; Yamamura, H. Effects of Peripheral Cannabinoid Receptor Ligands on Motility and Polarization in Neutrophil-like HL60 Cells and Human Neutrophils. J. Biol. Chem. 2006, 281, 12908–12918. [Google Scholar] [CrossRef] [Green Version]
- Balenga, N.A.B.; Aflaki, E.; Kargl, J.; Platzer, W.; Schröder, R.; Blättermann, S.; Kostenis, E.; Brown, A.J.; Heinemann, A.; Waldhoer, M. GPR55 regulates cannabinoid 2 receptor-mediated responses in human neutrophils. Cell Res. 2011, 21, 1452–1469. [Google Scholar] [CrossRef] [Green Version]
- Chiurchiù, V.; Cencioni, M.T.; Bisicchia, E.; Bardi, M.D.; Gasperini, C.; Borsellino, G.; Centonze, D.; Battistini, L.; Maccarrone, M. Distinct modulation of human myeloid and plasmacytoid dendritic cells by anandamide in multiple sclerosis. Ann. Neurol. 2013, 73, 626–636. [Google Scholar] [CrossRef]
- Chiurchiù, V.; Battistini, L.; Maccarrone, M. Endocannabinoid signalling in innate and adaptive immunity. Immunology 2015, 144, 352–364. [Google Scholar] [CrossRef]
- Smith, S.R.; Terminelli, C.; Denhardt, G. Effects of cannabinoid receptor agonist and antagonist ligands on production of inflammatory cytokines and anti-inflammatory interleukin-10 in endotoxemic mice. J. Pharmacol. Exp. Ther. 2000, 293, 136–150. [Google Scholar] [PubMed]
- Jarocka-Karpowicz, I.; Biernacki, M.; Wroński, A.; Gęgotek, A.; Skrzydlewska, E. Cannabidiol Effects on Phospholipid Metabolism in Keratinocytes from Patients with Psoriasis Vulgaris. Biomolecules 2020, 10, 367. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wójcik, P.; Garley, M.; Wroński, A.; Jabłońska, E.; Skrzydlewska, E. Cannabidiol Modifies the Formation of NETs in Neutrophils of Psoriatic Patients. Int. J. Mol. Sci. 2020, 21, 6795. [Google Scholar] [CrossRef] [PubMed]
- Palmieri, B.; Laurino, C.; Vadalà, M. A therapeutic effect of cbd-enriched ointment in inflammatory skin diseases and cutaneous scars. Clin. Ter. 2019, 170, e93–e99. [Google Scholar] [CrossRef]
- Richardson, D.; Pearson, R.G.; Kurian, N.; Latif, M.L.; Garle, M.J.; Barrett, D.A.; Kendall, D.A.; Scammell, B.E.; Reeve, A.J.; Chapman, V. Characterisation of the cannabinoid receptor system in synovial tissue and fluid in patients with osteoarthritis and rheumatoid arthritis. Arthritis Res. Ther. 2008, 10, R43. [Google Scholar] [CrossRef] [Green Version]
- Kinsey, S.G.; Mahadevan, A.; Zhao, B.; Sun, H.; Naidu, P.S.; Razdan, R.K.; Selley, D.E.; Imad Damaj, M.; Lichtman, A.H. The CB2 cannabinoid receptor-selective agonist O-3223 reduces pain and inflammation without apparent cannabinoid behavioral effects. Neuropharmacology 2011, 60, 244–251. [Google Scholar] [CrossRef] [Green Version]
- Fukuda, S.; Kohsaka, H.; Takayasu, A.; Yokoyama, W.; Miyabe, C.; Miyabe, Y.; Harigai, M.; Miyasaka, N.; Nanki, T. Cannabinoid receptor 2 as a potential therapeutic target in rheumatoid arthritis. BMC Musculoskelet. Disord. 2014, 15, 275. [Google Scholar] [CrossRef] [Green Version]
- Al-Kofahi, M.; Omura, S.; Tsunoda, I.; Sato, F.; Becker, F.; Gavins, F.N.E.; Woolard, M.D.; Pattillo, C.; Zawieja, D.; Muthuchamy, M.; et al. IL-1β reduces cardiac lymphatic muscle contraction via COX-2 and PGE2 induction: Potential role in myocarditis. Biomed. Pharmacother. 2018, 107, 1591–1600. [Google Scholar] [CrossRef]
- McAdam, B.F.; Mardini, I.A.; Habib, A.; Burke, A.; Lawson, J.A.; Kapoor, S.; Fitzgerald, G.A. Effect of regulated expression of human cyclooxygenase isoforms on eicosanoid and isoeicosanoid production in inflammation. J. Clin. Investig. 2000, 105, 1473–1482. [Google Scholar] [CrossRef] [Green Version]
- Gabbs, M.; Leng, S.; Devassy, J.G.; Monirujjaman, M.; Aukema, H.M. Advances in Our Understanding of Oxylipins Derived from Dietary PUFAs. Adv. Nutr. 2015, 6, 513–540. [Google Scholar] [CrossRef] [Green Version]
- Marone, G.; Galdiero, M.R.; Pecoraro, A.; Pucino, V.; Criscuolo, G.; Triassi, M.; Varricchi, G. Prostaglandin D2receptor antagonists in allergic disorders: Safety, efficacy, and future perspectives. Expert Opin. Investig. Drugs 2019, 28, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Ghandour, R.A.; Giroud, M.; Vegiopoulos, A.; Herzig, S.; Ailhaud, G.; Amri, E.-Z.; Pisani, D.F. IP-receptor and PPARs trigger the conversion of human white to brite adipocyte induced by carbaprostacyclin. Biochim. Biophys. Acta 2016, 1861, 285–293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, H.; Aladelokun, O.; Ideta, T.; Giardina, C.; Ellis, L.M.; Rosenberg, D.W. Inhibition of PGE 2/EP4 receptor signaling enhances oxaliplatin efficacy in resistant colon cancer cells through modulation of oxidative stress. Sci. Rep. 2019, 9, 4954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sales, K.; Boddy, S.; Jabbour, H. F-prostanoid receptor alters adhesion, morphology and migration of endometrial adenocarcinoma cells. Oncogene 2008, 27, 2466–2477. [Google Scholar] [CrossRef] [Green Version]
- Elhassanny, A.E.M.; Ladin, D.A.; Soliman, E.; Albassam, H.; Morris, A.; Kobet, R.; Thayne, K.; Burns, C.; Danell, A.S.; Van Dross, R. Prostaglandin D2-ethanolamide induces skin cancer apoptosis by suppressing the activity of cellular antioxidants. Prostaglandins Other Lipid Mediat. 2019, 142, 9–23. [Google Scholar] [CrossRef]
- Kaliński, P.; Hilkens, C.M.; Snijders, A.; Snijdewint, F.G.; Kapsenberg, M.L. IL-12-deficient dendritic cells, generated in the presence of prostaglandin E2, promote type 2 cytokine production in maturing human naive T helper cells. J. Immunol. 1997, 159, 28–35. [Google Scholar]
- Muthuswamy, R.; Mueller-Berghaus, J.; Haberkorn, U.; Reinhart, T.A.; Schadendorf, D.; Kalinski, P. PGE(2) transiently enhances DC expression of CCR7 but inhibits the ability of DCs to produce CCL19 and attract naive T cells. Blood 2010, 116, 1454–1459. [Google Scholar] [CrossRef]
- Joo, M.; Sadikot, R.T. PGD Synthase and PGD2 in Immune Resposne. Mediat. Inflamm. 2012, 2012, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Moalli, F.; Cupovic, J.; Thelen, F.; Halbherr, P.; Fukui, Y.; Narumiya, S.; Ludewig, B.; Stein, J.V. Thromboxane A2 acts as tonic immunoregulator by preferential disruption of low-avidity CD4+ T cell-dendritic cell interactions. J. Exp. Med. 2014, 211, 2507–2517. [Google Scholar] [CrossRef] [Green Version]
- Nagamachi, M.; Sakata, D.; Kabashima, K.; Furuyashiki, T.; Murata, T.; Segi-Nishida, E.; Soontrapa, K.; Matsuoka, T.; Miyachi, Y.; Narumiya, S. Facilitation of Th1-mediated immune response by prostaglandin E receptor EP1. J. Exp. Med. 2007, 204, 2865–2874. [Google Scholar] [CrossRef]
- Boniface, K.; Bak-Jensen, K.S.; Li, Y.; Blumenschein, W.M.; McGeachy, M.J.; McClanahan, T.K.; McKenzie, B.S.; Kastelein, R.A.; Cua, D.J.; de Waal Malefyt, R. Prostaglandin E2 regulates Th17 cell differentiation and function through cyclic AMP and EP2/EP4 receptor signaling. J. Exp. Med. 2009, 206, 535–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razali, N.; Hohjoh, H.; Inazumi, T.; Maharjan, B.D.; Nakagawa, K.; Konishi, M.; Sugimoto, Y.; Hasegawa, H. Induced Prostanoid Synthesis Regulates the Balance between Th1- and Th2-Producing Inflammatory Cytokines in the Thymus of Diet-Restricted Mice. Biol. Pharm. Bull. 2020, 43, 649–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernard, M.P.; Phipps, R.P. Inhibition of cyclooxygenase-2 impairs the expression of essential plasma cell transcription factors and human B-lymphocyte differentiation. Immunology 2010, 129, 87–96. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Zhang, L.; Kang, D.; Yang, D.; Tang, Y. Activation of PGE2/EP2 and PGE2/EP4 signaling pathways positively regulate the level of PD-1 in infiltrating CD8+ T cells in patients with lung cancer. Oncol. Lett. 2018, 15, 552–558. [Google Scholar] [CrossRef] [Green Version]
- Epp, N.; Frstenberger, G.; Mller, K.; De Juanes, S.; Leitges, M.; Hausser, I.; Thieme, F.; Liebisch, G.; Schmitz, G.; Krieg, P. 12R-lipoxygenase deficiency disrupts epidermal barrier function. J. Cell Biol. 2007, 177, 173–182. [Google Scholar] [CrossRef]
- Mayatepek, E.; Hoffmann, G.F. Leukotrienes: Biosynthesis, Metabolism, and Pathophysiologic Significance. Pediatr. Res. 1995, 37, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Bacon, K.B.; Camp, R.D.; Cunningham, F.M.; Woollard, P.M. Contrasting in vitro lymphocyte chemotactic activity of the hydroxyl enantiomers of 12-hydroxy-5,8,10,14-eicosatetraenoic acid. Br. J. Pharmacol. 1988, 95, 966–974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Y.; Kumar, R.K.; Zhou, J.; Foster, P.S.; Webb, D.C. Ym1/2 Promotes Th2 Cytokine Expression by Inhibiting 12/15(S)-Lipoxygenase: Identification of a Novel Pathway for Regulating Allergic Inflammation. J. Immunol. 2009, 182, 5393–5399. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, M.; Yokomizo, T. The role of leukotrienes in allergic diseases. Allergol. Int. 2015, 64, 17–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, H.W.; Liu, G.Y.; Zhao, C.F.; Li, X.F.; Yang, X.Y. Differential expression of COX-2 in osteoarthritis and rheumatoid arthritis. Genet. Mol. Res. 2015, 14, 12872–12879. [Google Scholar] [CrossRef]
- Zhang, L.; Bertucci, A.M.; Smith, K.A.; Xu, L.; Datta, S.K. Hyperexpression of cyclooxygenase 2 in the lupus immune system and effect of cyclooxygenase 2 inhibitor diet therapy in a murine model of systemic lupus erythematosus. Arthritis Rheum. 2007, 56, 4132–4141. [Google Scholar] [CrossRef] [Green Version]
- Pellefigues, C.; Dema, B.; Lamri, Y.; Saidoune, F.; Chavarot, N.; Lohéac, C.; Pacreau, E.; Dussiot, M.; Bidault, C.; Marquet, F.; et al. Prostaglandin D2 amplifies lupus disease through basophil accumulation in lymphoid organs. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Fattahi, M.J.; Mirshafiey, A. Prostaglandins and Rheumatoid Arthritis. Arthritis 2012, 2012, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Clark, P.; Rowland, S.E.; Denis, D.; Mathieu, M.-C.; Stocco, R.; Poirier, H.; Burch, J.; Han, Y.; Audoly, L.; Therien, A.G.; et al. MF498 [N-{[4-(5,9-Diethoxy-6-oxo-6,8-dihydro-7H-pyrrolo[3,4-g]quinolin-7-yl)-3-methylbenzyl]sulfonyl}-2-(2-methoxyphenyl)acetamide], a Selective E Prostanoid Receptor 4 Antagonist, Relieves Joint Inflammation and Pain in Rodent Models of Rheumatoid and Osteoarthritis. J. Pharmacol. Exp. Ther. 2008, 325, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.-Y.; Lin, T.-H.; Chiu, Y.-C.; Liou, H.-C.; Yang, R.-S.; Fu, W.-M. Involvement of 15-lipoxygenase in the inflammatory arthritis. J. Cell. Biochem. 2012, 113, 2279–2289. [Google Scholar] [CrossRef]
- Gheorghe, K.R.; Korotkova, M.; Catrina, A.I.; Backman, L.; Klint, E.A.; Claesson, H.-E.; Rådmark, O.; Jakobsson, P.-J. Expression of 5-lipoxygenase and 15-lipoxygenase in rheumatoid arthritis synovium and effects of intraarticular glucocorticoids. Arthritis Res. Ther. 2009, 11, R83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauer, J.; Ripperger, A.; Frantz, S.; Ergün, S.; Schwedhelm, E.; Benndorf, R.A. Pathophysiology of isoprostanes in the cardiovascular system: Implications of isoprostane-mediated thromboxane A2 receptor activation. Br. J. Pharmacol. 2014, 171, 3115–3131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Łuczaj, W.; Gęgotek, A.; Skrzydlewska, E. Antioxidants and HNE in redox homeostasis. Free Radic. Biol. Med. 2017, 111, 87–101. [Google Scholar] [CrossRef]
- Gęgotek, A.; Skrzydlewska, E. Biological effect of protein modifications by lipid peroxidation products. Chem. Phys. Lipids 2019, 221, 46–52. [Google Scholar] [CrossRef]
- Ye, Y.; Wu, T.; Zhang, T.; Han, J.; Habazi, D.; Saxena, R.; Mohan, C. Elevated oxidized lipids, anti-lipid autoantibodies and oxidized lipid immune complexes in active SLE. J. Clin. Immunol. 2019, 205, 43–48. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, W.; Yang, H.; Shao, D.; Zhao, X.; Zhang, G. Intraperitoneal injection of 4-hydroxynonenal (4-HNE), a lipid peroxidation product, exacerbates colonic inflammation through activation of Toll-like receptor 4 signaling. Free Radic. Biol. Med. 2019, 131, 237–242. [Google Scholar] [CrossRef] [PubMed]
- Jang, E.J.; Kim, D.H.; Lee, B.; Lee, E.K.; Chung, K.W.; Moon, K.M.; Kim, M.J.; An, H.J.; Jeong, J.W.; Kim, Y.R.; et al. Activation of proinflammatory signaling by 4-hydroxynonenal-Src adducts in aged kidneys. Oncotarget 2016, 7, 50864–50874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, G.; Wang, Y.; Cen, X.-M.; Yang, M.; Liang, Y.; Xie, Q.-B. Lipid Peroxidation-Mediated Inflammation Promotes Cell Apoptosis through Activation of NF-κB Pathway in Rheumatoid Arthritis Synovial Cells. Mediat. Inflamm. 2015, 2015, 460310. [Google Scholar] [CrossRef] [Green Version]
- Gęgotek, A.; Domingues, P.; Wroński, A.; Wójcik, P.; Skrzydlewska, E. Proteomic plasma profile of psoriatic patients. J. Pharm. Biomed. Anal. 2018, 155, 185–193. [Google Scholar] [CrossRef]
- Gęgotek, A.; Domingues, P.; Wroński, A.; Ambrożewicz, E.; Skrzydlewska, E. The Proteomic Profile of Keratinocytes and Lymphocytes in Psoriatic Patients. Proteom. Clin. Appl. 2019, 13, 1800119. [Google Scholar] [CrossRef]
- Karabowicz, P.; Wroński, A.; Ostrowska, H.; Waeg, G.; Zarkovic, N.; Skrzydlewska, E. Reduced Proteasome Activity and Enhanced Autophagy in Blood Cells of Psoriatic Patients. Int. J. Mol. Sci. 2020, 21, 7608. [Google Scholar] [CrossRef]
- Akgöl, G.; Ulusoy, H.; Telo, S.; Gülkesen, A.; Yildirim, T.; Poyraz, A.K.; Kaya, A. Is 4-Hydroxynonenal a Predictive Parameter for the Development of Joint Erosion in Patients With Rheumatoid Arthritis? Arch. Rheumatol. 2016, 31, 76–81. [Google Scholar] [CrossRef] [Green Version]
- Luczaj, W.; Jarocka-Karpinska, I.; Sierakowski, S.; Andrisic, L.; Zarkovic, N.; Skrzydlewska, E. Lipid peroxidation in Rheumatoid arthritis; consequences and monitoring. Free Radic. Biol. Med. 2014, 75 (Suppl. 1), S49. [Google Scholar] [CrossRef]
- Shah, D.; Mahajan, N.; Sah, S.; Nath, S.K.; Paudyal, B. Oxidative stress and its biomarkers in systemic lupus erythematosus. J. Biomed. Sci. 2014, 21, 23. [Google Scholar] [CrossRef] [Green Version]
- Grönwall, C.; Amara, K.; Hardt, U.; Krishnamurthy, A.; Steen, J.; Engström, M.; Sun, M.; Ytterberg, A.J.; Zubarev, R.A.; Scheel-Toellner, D.; et al. Autoreactivity to malondialdehyde-modifications in rheumatoid arthritis is linked to disease activity and synovial pathogenesis. J. Autoimmun. 2017, 84, 29–45. [Google Scholar] [CrossRef] [Green Version]
- Leitinger, N.; Huber, J.; Rizza, C.; Mechtcheriakova, D.; Bochkov, V.; Koshelnick, Y.; Berliner, J.A.; Binder, B.R. The isoprostane 8-iso-PGF(2alpha) stimulates endothelial cells to bind monocytes: Differences from thromboxane-mediated endothelial activation. FASEB J. 2001, 15, 1254–1256. [Google Scholar] [CrossRef] [PubMed]
- Zahler, S.; Becker, B.F. Indirect enhancement of neutrophil activity and adhesion to cultured human umbilical vein endothelial cells by isoprostanes (iPF2alpha-III and iPE2-III). Prostaglandins Other Lipid Mediat. 1999, 57, 319–331. [Google Scholar] [CrossRef]
- Scholz, H.; Yndestad, A.; Damås, J.K.; Wæhre, T.; Tonstad, S.; Aukrust, P.; Halvorsen, B. 8-Isoprostane increases expression of interleukin-8 in human macrophages through activation of mitogen-activated protein kinases. Cardiovasc. Res. 2003, 59, 945–954. [Google Scholar] [CrossRef] [Green Version]
- Milne, G.L.; Yin, H.; Hardy, K.D.; Davies, S.S.; Roberts, L.J. Isoprostane Generation and Function. Chem. Rev. 2011, 111, 5973–5996. [Google Scholar] [CrossRef] [Green Version]
- Carlström, K.E.; Ewing, E.; Granqvist, M.; Gyllenberg, A.; Aeinehband, S.; Enoksson, S.L.; Checa, A.; Badam, T.V.S.; Huang, J.; Gomez-Cabrero, D.; et al. Therapeutic efficacy of dimethyl fumarate in relapsing-remitting multiple sclerosis associates with ROS pathway in monocytes. Nat. Commun. 2019, 10, 3081. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wójcik, P.; Gęgotek, A.; Žarković, N.; Skrzydlewska, E. Oxidative Stress and Lipid Mediators Modulate Immune Cell Functions in Autoimmune Diseases. Int. J. Mol. Sci. 2021, 22, 723. https://doi.org/10.3390/ijms22020723
Wójcik P, Gęgotek A, Žarković N, Skrzydlewska E. Oxidative Stress and Lipid Mediators Modulate Immune Cell Functions in Autoimmune Diseases. International Journal of Molecular Sciences. 2021; 22(2):723. https://doi.org/10.3390/ijms22020723
Chicago/Turabian StyleWójcik, Piotr, Agnieszka Gęgotek, Neven Žarković, and Elżbieta Skrzydlewska. 2021. "Oxidative Stress and Lipid Mediators Modulate Immune Cell Functions in Autoimmune Diseases" International Journal of Molecular Sciences 22, no. 2: 723. https://doi.org/10.3390/ijms22020723