tRNA Biology in the Pathogenesis of Diabetes: Role of Genetic and Environmental Factors


Abstract
:1. Introduction
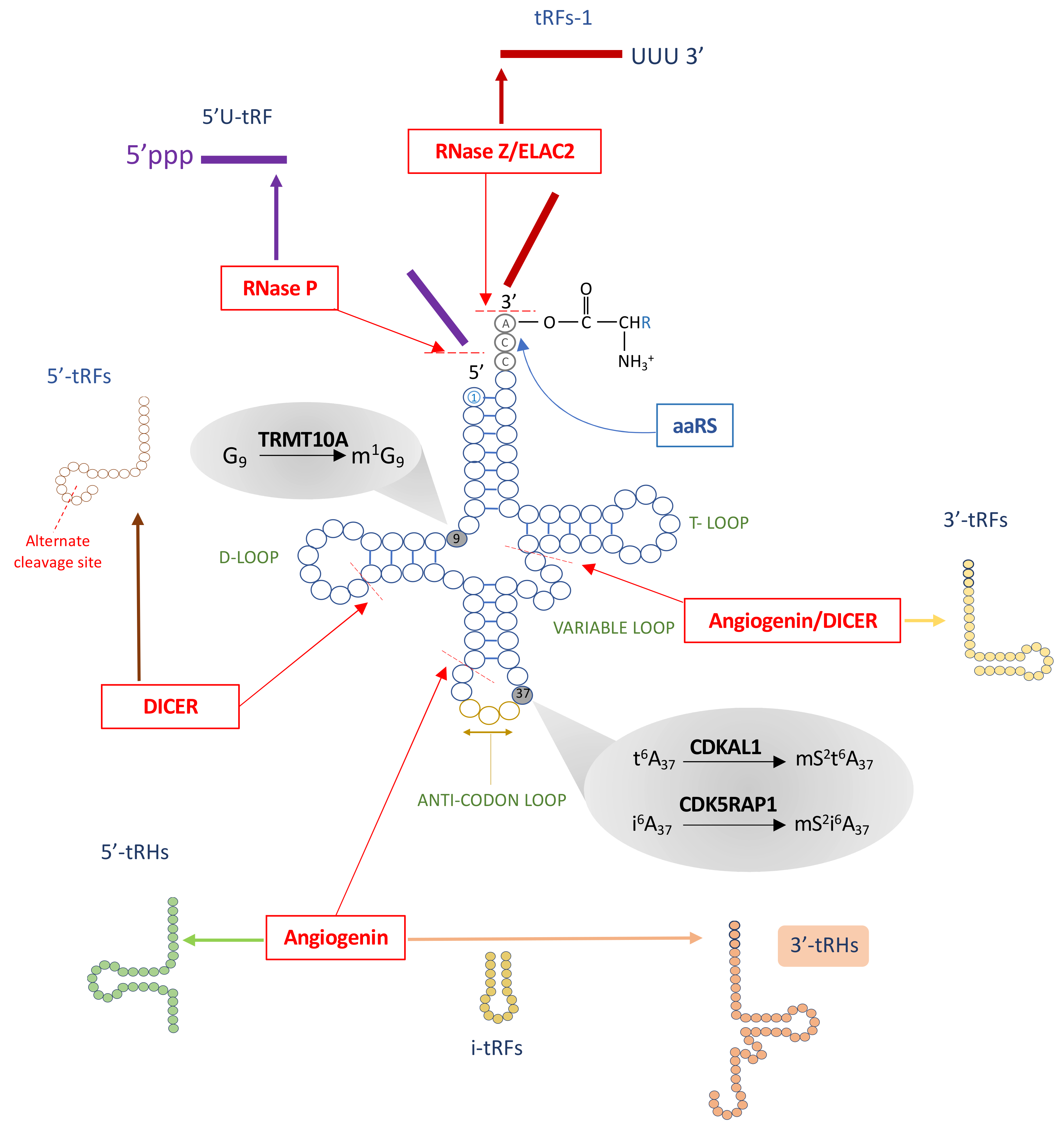
2. tRNA Biogenesis, Structure, and Function
3. Post-Transcriptional tRNA Modifications
4. tRNA Fragments, a New Class of Small Noncoding RNAs
5. Dysregulated tRNA Metabolism in Diabetes
6. Genetic Factors Affecting tRNA Biology and Their Association with Impaired Glucose Metabolism
6.1. Pathogenic Variants in CDKAL1 and TRMT10A
6.2. Pathogenic Variants in aaRSs
6.3. Regulation of aaRSs Expression by T2D Susceptibility Gene Variants
6.4. Mutations in mt-tRNA Genes
7. Environmental Factors Affecting tRNA Aminoacylation, Modification, and Fragmentation
7.1. Nongenetic Inhibition of tRNA-Modifying Enzymes
7.2. Modulation of aaRS Activity by Nutrients and tRNA Fragments
7.3. Modulation of tRNA[Ser]Sec Modifications by Selenium and Statins
7.4. tRNA Fragments in Intergenerational Inheritance of Metabolic Disorders
8. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Nathan, D.M. Diabetes. JAMA 2015, 314, 1052–1062. [Google Scholar] [CrossRef]
- Xue, A.; eQTLGen Consortium; Wu, Y.; Zhu, Z.; Zhang, F.; Kemper, K.E.; Zheng, Z.; Yengo, L.; Lloyd-Jones, L.R.; Sidorenko, J.; et al. Genome-wide association analyses identify 143 risk variants and putative regulatory mechanisms for type 2 diabetes. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Wu, L.; Wang, C.C. Genetic variants in promoter regions associated with type 2 diabetes mellitus: A large-scale meta-analysis and subgroup analysis. J. Cell. Biochem. 2019, 120, 13012–13025. [Google Scholar] [CrossRef]
- Cersosimo, E.; Triplitt, C.; Solis-Herrera, C.; Mandarino, L.J.; DeFronzo, R.A. Pathogenesis of Type 2 Diabetes Mellitus; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dungan, K., Grossman, A., Hershman, J.M., Hofland, H.J., Kaltsas, G., Eds.; Endotext [Internet]: South Dartmouth, MA, USA, 2018. [Google Scholar]
- Zhou, Z.; Sun, B.; Huang, S.; Jia, W.; Yu, D. The tRNA-associated dysregulation in diabetes mellitus. Metabolism 2019, 94, 9–17. [Google Scholar] [CrossRef]
- Wei, F.-Y.; Tomizawa, K. tRNA modifications and islet function. Diabetes Obes. Metab. 2018, 20, 20–27. [Google Scholar] [CrossRef] [Green Version]
- Cosentino, C.; Cnop, M.; Igoillo-Esteve, M. The tRNA Epitranscriptome and Diabetes: Emergence of tRNA Hypomodifications as a Cause of Pancreatic β-Cell Failure. Endocrinology 2019, 160, 1262–1274. [Google Scholar] [CrossRef]
- Goodenbour, J.M.; Pan, T. Diversity of tRNA genes in eukaryotes. Nucleic Acids Res. 2006, 34, 6137–6146. [Google Scholar] [CrossRef] [Green Version]
- Yu, Y.C.; Han, J.M.; Kim, S. Aminoacyl-tRNA Synthetases and amino acid signaling. Biochim. Biophys. Acta (BBA) Bioenerg. 2021, 1868, 118889. [Google Scholar] [CrossRef] [PubMed]
- Hardt, W.-D.; Schlegl, J.; Erdmann, V.A.; Hartmann, R.K. Role of the D arm and the anticodon arm in tRNA recognition by eubacterial and eukaryotic RNase P enzymes. Biochemistry 1993, 32, 13046–13053. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.W.; Chetnani, B.; Mondragón, A. Structure and function of the T-loop structural motif in noncoding RNAs. Wiley Interdiscip. Rev. RNA 2013, 4, 507–522. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, H.A.; Hoffer, E.D.; Dunham, C.M. Importance of a tRNA anticodon loop modification and a conserved, noncanonical anticodon stem pairing in tRNACGGPro for decoding. J. Biol. Chem. 2019, 294, 5281–5291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suzuki, T.; Yashiro, Y.; Kikuchi, I.; Ishigami, Y.; Saito, H.; Matsuzawa, I.; Okada, S.; Mito, M.; Iwasaki, S.; Ma, D.; et al. Complete chemical structures of human mitochondrial tRNAs. Nat. Commun. 2020, 11, 1–15. [Google Scholar] [CrossRef]
- Chan, C.; Pham, P.; Dedon, P.C.; Begley, T.J. Lifestyle modifications: Coordinating the tRNA epitranscriptome with codon bias to adapt translation during stress responses. Genome Biol. 2018, 19, 228. [Google Scholar] [CrossRef]
- Schmitt, B.M.; Rudolph, K.L.; Karagianni, P.; Fonseca, N.A.; White, R.J.; Talianidis, I.; Odom, D.T.; Marioni, J.C.; Kutter, C. High-resolution mapping of transcriptional dynamics across tissue development reveals a stable mRNA–tRNA interface. Genome Res. 2014, 24, 1797–1807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kutter, C.; Brown, G.D.; Gonçalves, Â.; Wilson, M.D.; Watt, S.; Brazma, A.; White, R.J.; Odom, D.T. Pol III binding in six mammals shows conservation among amino acid isotypes despite divergence among tRNA genes. Nat. Genet. 2011, 43, 948–955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, T. Modifications and functional genomics of human transfer RNA. Cell Res. 2018, 28, 395–404. [Google Scholar] [CrossRef] [PubMed]
- Chan, P.P.; Lowe, T.M. GtRNAdb 2.0: An expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res. 2016, 44, D184–D189. [Google Scholar] [CrossRef] [Green Version]
- Phizicky, E.M.; Hopper, A.K. tRNA biology charges to the front. Genes Dev. 2010, 24, 1832–1860. [Google Scholar] [CrossRef] [Green Version]
- Schimmel, P. The emerging complexity of the tRNA world: Mammalian tRNAs beyond protein synthesis. Nat. Rev. Mol. Cell Biol. 2018, 19, 45–58. [Google Scholar] [CrossRef]
- Agris, P.F. Decoding the genome: A modified view. Nucleic Acids Res. 2004, 32, 223–238. [Google Scholar] [CrossRef] [Green Version]
- Powell, C.A.; Nicholls, T.J.; Minczuk, M. Nuclear-encoded factors involved in post-transcriptional processing and modification of mitochondrial tRNAs in human disease. Front. Genet. 2015, 6, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Asano, K.; Suzuki, T.; Saito, A.; Wei, F.-Y.; Ikeuchi, Y.; Numata, T.; Tanaka, R.; Yamane, Y.; Yamamoto, T.; Goto, T.; et al. Metabolic and chemical regulation of tRNA modification associated with taurine deficiency and human disease. Nucleic Acids Res. 2018, 46, 1565–1583. [Google Scholar] [CrossRef] [Green Version]
- Brierley, I.; Meredith, M.R.; Bloys, A.J.; Hagervall, T.G. Expression of a coronavirus ribosomal frameshift signal in Escherichia coli: Influence of tRNA anticodon modification on frameshifting. J. Mol. Biol. 1997, 270, 360–373. [Google Scholar] [CrossRef]
- Tuorto, F.; Lyko, F. Genome recoding by tRNA modifications. Open Biol. 2016, 6, 160287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lorenz, C.; Lünse, C.E.; Mörl, M. tRNA Modifications: Impact on Structure and Thermal Adaptation. Biomolecules 2017, 7, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyons, S.M.; Fay, M.M.; Ivanov, P. The role of RNA modifications in the regulation of tRNA cleavage. FEBS Lett. 2018, 592, 2828–2844. [Google Scholar] [CrossRef] [Green Version]
- Emara, M.M.; Ivanov, P.; Hickman, T.; Dawra, N.; Tisdale, S.; Kedersha, N.; Hu, G.-F.; Anderson, P. Angiogenin-induced tRNA-derived Stress-induced RNAs Promote Stress-induced Stress Granule Assembly. J. Biol. Chem. 2010, 285, 10959–10968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ivanov, P.; Emara, M.M.; Villen, J.; Gygi, S.P.; Anderson, P. Angiogenin-Induced tRNA Fragments Inhibit Translation Initiation. Mol. Cell 2011, 43, 613–623. [Google Scholar] [CrossRef] [Green Version]
- Chen, Q.; Yan, M.; Cao, Z.; Li, X.; Zhang, Y.; Shi, J.; Feng, G.-H.; Peng, H.; Zhang, X.; Qian, J.; et al. Sperm tsRNAs contribute to intergenerational inheritance of an acquired metabolic disorder. Science 2016, 351, 397–400. [Google Scholar] [CrossRef] [Green Version]
- Sharma, U.; Conine, C.C.; Shea, J.M.; Boskovic, A.; Derr, A.G.; Bing, X.Y.; Belleannee, C.; Kucukural, A.; Serra, R.W.; Sun, F.; et al. Biogenesis and function of tRNA fragments during sperm maturation and fertilization in mammals. Science 2016, 351, 391–396. [Google Scholar] [CrossRef] [Green Version]
- Eraina, M.; Ibba, M. tRNAs as regulators of biological processes. Front. Genet. 2014, 5, 171. [Google Scholar] [CrossRef] [Green Version]
- Sobala, A.; Hutvagner, G. Small RNAs derived from the 5′ end of tRNA can inhibit protein translation in human cells. RNA Biol. 2013, 10, 553–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mleczko, A.M.; Celichowski, P.; Bąkowska-Żywicka, K. Transfer RNA-derived fragments target and regulate ribosome-associated aminoacyl-transfer RNA synthetases. Biochim. Biophys. Acta (BBA) Bioenerg. 2018, 1861, 647–656. [Google Scholar] [CrossRef] [PubMed]
- Magee, R.; Rigoutsos, I. On the expanding roles of tRNA fragments in modulating cell behavior. Nucleic Acids Res. 2020, 48, 9433–9448. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Xu, Z.; Sheng, J. tRNA-Derived Small RNA: A Novel Regulatory Small Non-Coding RNA. Genes 2018, 9, 246. [Google Scholar] [CrossRef] [Green Version]
- Kuscu, C.; Kumar, P.; Kiran, M.; Su, Z.; Malik, A.; Dutta, A. tRNA fragments (tRFs) guide Ago to regulate gene expression post-transcriptionally in a Dicer-independent manner. RNA 2018, 24, 1093–1105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keam, S.P.; Sobala, A.; Have, S.T.; Hutvagner, G. tRNA-Derived RNA Fragments Associate with Human Multisynthetase Complex (MSC) and Modulate Ribosomal Protein Translation. J. Proteome Res. 2016, 16, 413–420. [Google Scholar] [CrossRef]
- Keam, S.P.; Hutvagner, G. tRNA-Derived Fragments (tRFs): Emerging New Roles for an Ancient RNA in the Regulation of Gene Expression. Life 2015, 5, 1638–1651. [Google Scholar] [CrossRef] [Green Version]
- Gebetsberger, J.; Zywicki, M.; Künzi, A.; Polacek, N. tRNA-Derived Fragments Target the Ribosome and Function as Regulatory Non-Coding RNA inHaloferax volcanii. Archaea 2012, 2012, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Gebetsberger, J.V.; Wyss, L.; Mleczko, A.M.; Reuther, J.; Polacek, N. A tRNA-derived fragment competes with mRNA for ribosome binding and regulates translation during stress. RNA Biol. 2017, 14, 1364–1373. [Google Scholar] [CrossRef] [Green Version]
- Gebetsberger, J.V.; Polacek, N. Slicing tRNAs to boost functional ncRNA diversity. RNA Biol. 2013, 10, 1798–1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Q.; Lee, I.; Ren, J.; Ajay, S.S.; Lee, Y.S.; Bao, X. Identification and Functional Characterization of tRNA-derived RNA Fragments (tRFs) in Respiratory Syncytial Virus Infection. Mol. Ther. 2013, 21, 368–379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cole, C.; Sobala, A.; Lu, C.; Thatcher, S.R.; Bowman, A.; Brown, J.W.S.; Green, P.J.; Barton, G.J.; Hutvagner, G. Filtering of deep sequencing data reveals the existence of abundant Dicer-dependent small RNAs derived from tRNAs. RNA 2009, 15, 2147–2160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maute, R.L.; Schneider, C.; Sumazin, P.; Holmes, A.; Califano, A.; Basso, K.; Dalla-Favera, R. tRNA-derived microRNA modulates proliferation and the DNA damage response and is down-regulated in B cell lymphoma. Proc. Natl. Acad. Sci. USA 2013, 110, 1404–1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, P.; Anaya, J.; Mudunuri, S.B.; Dutta, A. Meta-analysis of tRNA derived RNA fragments reveals that they are evolutionarily conserved and associate with AGO proteins to recognize specific RNA targets. BMC Biol. 2014, 12, 1–14. [Google Scholar] [CrossRef]
- Su, Z.; Kuscu, C.; Malik, A.; Shibata, E.; Dutta, A. Angiogenin generates specific stress-induced tRNA halves and is not involved in tRF-3–mediated gene silencing. J. Biol. Chem. 2019, 294, 16930–16941. [Google Scholar] [CrossRef]
- Torres, A.G.; Reina, O.; Attolini, C.S.-O.; De Pouplana, L.R. Differential expression of human tRNA genes drives the abundance of tRNA-derived fragments. Proc. Natl. Acad. Sci. USA 2019, 116, 8451–8456. [Google Scholar] [CrossRef] [Green Version]
- Yamasaki, S.; Ivanov, P.; Hu, G.-F.; Anderson, P. Angiogenin cleaves tRNA and promotes stress-induced translational repression. J. Cell Biol. 2009, 185, 35–42. [Google Scholar] [CrossRef] [Green Version]
- Thompson, D.M.; Lu, C.; Green, P.J.; Parker, R. tRNA cleavage is a conserved response to oxidative stress in eukaryotes. RNA 2008, 14, 2095–2103. [Google Scholar] [CrossRef] [Green Version]
- Oberbauer, V.; Schaefer, M.R. tRNA-Derived Small RNAs: Biogenesis, Modification, Function and Potential Impact on Human Disease Development. Genes 2018, 9, 607. [Google Scholar] [CrossRef] [Green Version]
- Blanco, S.; Dietmann, S.; Flores, J.V.; Hussain, S.; Kutter, C.; Humphreys, P.; Lukk, M.; Lombard, P.; Treps, L.; Popis, M.; et al. Aberrant methylation of t RNA s links cellular stress to neuro-developmental disorders. EMBO J. 2014, 33, 2020–2039. [Google Scholar] [CrossRef] [PubMed]
- Cosentino, C.; Toivonen, S.; Villamil, E.D.; Atta, M.; Ravanat, J.-L.; Demine, S.; Schiavo, A.A.; Pachera, N.; Deglasse, J.-P.; Jonas, J.-C.; et al. Pancreatic β-cell tRNA hypomethylation and fragmentation link TRMT10A deficiency with diabetes. Nucleic Acids Res. 2018, 46, 10302–10318. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Qi, M.; Shen, B.; Luo, G.; Wu, Y.; Li, J.; Lu, Z.; Zheng, Z.; Dai, Q.; Wang, H. Transfer RNA demethylase ALKBH3 promotes cancer progression via induction of tRNA-derived small RNAs. Nucleic Acids Res. 2019, 47, 2533–2545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Zhang, X.; Shi, J.; Tuorto, F.; Li, X.; Liu, Y.; Liebers, R.; Zhang, L.; Qu, Y.; Qian, J.; et al. Dnmt2 mediates intergenerational transmission of paternally acquired metabolic disorders through sperm small non-coding RNAs. Nat. Cell Biol. 2018, 20, 535–540. [Google Scholar] [CrossRef] [Green Version]
- Bento-Abreu, A.; Jager, G.; Swinnen, B.; Rué, L.; Hendrickx, S.; Jones, A.; Staats, K.A.; Taes, I.; Eykens, C.; Nonneman, A.; et al. Elongator subunit 3 (ELP3) modifies ALS through tRNA modification. Hum. Mol. Genet. 2018, 27, 1276–1289. [Google Scholar] [CrossRef] [Green Version]
- Orioli, A. tRNA biology in the omics era: Stress signalling dynamics and cancer progression. BioEssays 2016, 39, 39. [Google Scholar] [CrossRef]
- Close, P.; Bose, D.; Chariot, A.; Leidel, S.A. Dynamic Regulation of tRNA Modifications in Cancer. Cancer Noncoding RNAs 2018, 163–186. [Google Scholar] [CrossRef]
- Tuorto, F.; Parlato, R. rRNA and tRNA Bridges to Neuronal Homeostasis in Health and Disease. J. Mol. Biol. 2019, 431, 1763–1779. [Google Scholar] [CrossRef]
- Schaffer, A.E.; Pinkard, O.; Coller, J.M. tRNA Metabolism and Neurodevelopmental Disorders. Annu. Rev. Genom. Hum. Genet. 2019, 20, 359–387. [Google Scholar] [CrossRef]
- Liu, E.Y.; Cali, C.P.; Lee, E.B. RNA metabolism in neurodegenerative disease. Dis. Model. Mech. 2017, 10, 509–518. [Google Scholar] [CrossRef] [Green Version]
- Angelova, M.T.; Dimitrova, D.G.; Dinges, N.; Lence, T.; Worpenberg, L.; Carré, C.; Roignant, J.-Y. The Emerging Field of Epitranscriptomics in Neurodevelopmental and Neuronal Disorders. Front. Bioeng. Biotechnol. 2018, 6, 46. [Google Scholar] [CrossRef] [Green Version]
- Green, J.; Ansari, M.Y.; Ball, H.; Haqqi, T.M. tRNA-derived fragments (tRFs) regulate post-transcriptional gene expression via AGO-dependent mechanism in IL-1β stimulated chondrocytes. Osteoarthr. Cartil. 2020, 28, 1102–1110. [Google Scholar] [CrossRef] [PubMed]
- Gregory, R.I.; Chendrimada, T.P.; Cooch, N.; Shiekhattar, R. Human RISC Couples MicroRNA Biogenesis and Posttranscriptional Gene Silencing. Cell 2005, 123, 631–640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, P.; Tang, T.; Liu, T.; Zhou, J.; Cui, H.; He, Z.; Zhong, Y.; Hu, E.; Yang, A.; Wei, G.; et al. Systematic Analysis of tRNA-Derived Small RNAs Reveals Novel Potential Therapeutic Targets of Traditional Chinese Medicine (Buyang-Huanwu-Decoction) on Intracerebral Hemorrhage. Int. J. Biol. Sci. 2019, 15, 895–908. [Google Scholar] [CrossRef] [PubMed]
- Keam, S.P.; Young, P.E.; McCorkindale, A.L.; Dang, T.H.; Clancy, J.L.; Humphreys, D.T.; Preiss, T.; Hutvagner, G.; Martin, D.I.; Cropley, J.E.; et al. The human Piwi protein Hiwi2 associates with tRNA-derived piRNAs in somatic cells. Nucleic Acids Res. 2014, 42, 8984–8995. [Google Scholar] [CrossRef]
- Gogakos, T.; Brown, M.; Garzia, A.; Meyer, C.; Hafner, M.; Tuschl, T. Characterizing Expression and Processing of Precursor and Mature Human tRNAs by Hydro-tRNAseq and PAR-CLIP. Cell Rep. 2017, 20, 1463–1475. [Google Scholar] [CrossRef] [Green Version]
- Hafner, M.; Landthaler, M.; Burger, L.; Khorshid, M.; Hausser, J.; Berninger, P.; Rothballer, A.; Ascano, M.; Jungkamp, A.-C.; Munschauer, M.; et al. Transcriptome-wide Identification of RNA-Binding Protein and MicroRNA Target Sites by PAR-CLIP. Cell 2010, 141, 129–141. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.K.; Fuchs, G.; Wang, S.; Wei, W.; Zhang, Y.; Park, H.; Roy-Chaudhuri, B.; Cliff, Z.Q.; Xu, J.; Shengchun, W.; et al. A transfer-RNA-derived small RNA regulates ribosome biogenesis. Nat. Cell Biol. 2017, 552, 57–62. [Google Scholar] [CrossRef]
- Karaiskos, S.; Naqvi, A.S.; Swanson, K.E.; Grigoriev, A. Age-driven modulation of tRNA-derived fragments in Drosophila and their potential targets. Biol. Direct 2015, 10, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Cozen, A.E.; Liu, Y.; Chen, Q.; Lowe, T.M. Small RNA Modifications: Integral to Function and Disease. Trends Mol. Med. 2016, 22, 1025–1034. [Google Scholar] [CrossRef] [Green Version]
- Torres, A.G.; Batlle, E.; De Pouplana, L.R. Role of tRNA modifications in human diseases. Trends Mol. Med. 2014, 20, 306–314. [Google Scholar] [CrossRef] [PubMed]
- Landgraf, B.J.; McCarthy, E.L.; George, G.N. RadicalS-Adenosylmethionine Enzymes in Human Health and Disease. Annu. Rev. Biochem. 2016, 85, 485–514. [Google Scholar] [CrossRef]
- Abbott, J.A.; Francklyn, C.S.; Robey-Bond, S.M. Transfer RNA and human disease. Front. Genet. 2014, 5, 158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- M’t Hart, L.; Hansen, T.; Rietveld, I.; Dekker, J.M.; Nijpels, G.; Janssen, G.M.; Arp, P.A.; Uitterlinden, A.G.; Jørgensen, T.; Borch-Johnsen, K.; et al. Evidence that the Mitochondrial Leucyl tRNA Synthetase (LARS2) Gene Represents a Novel Type 2 Diabetes Susceptibility Gene. Diabetes 2005, 54, 1892–1895. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doumatey, A.P.; Ekoru, K.; Adeyemo, A.; Rotimi, C. Genetic Basis of Obesity and Type 2 Diabetes in Africans: Impact on Precision Medicine. Curr. Diabetes Rep. 2019, 19, 105. [Google Scholar] [CrossRef] [PubMed]
- Sahibdeen, V.; Crowther, N.J.; Soodyall, H.; Hendry, L.M.; Munthali, R.J.; Hazelhurst, S.; Choudhury, A.; Norris, S.A.; Ramsay, M.; Lombard, Z. Genetic variants in SEC16B are associated with body composition in black South Africans. Nutr. Diabetes 2018, 8, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Shungin, D.; Winkler, T.W.; Croteau-Chonka, D.C.; Ferreira, T.; Locke, A.E.; Mägi, R.; Strawbridge, R.J.; Pers, T.H.; Fischer, K.; Justice, A.E.; et al. New genetic loci link adipose and insulin biology to body fat distribution. Nat. Cell Biol. 2015, 518, 187–196. [Google Scholar] [CrossRef] [Green Version]
- Akiyama, M.; Okada, Y.; Kanai, M.; Takahashi, A.; Momozawa, Y.; Ikeda, M.; Iwata, N.; Ikegawa, S.; Hirata, M.; Matsuda, K.; et al. Genome-wide association study identifies 112 new loci for body mass index in the Japanese population. Nat. Genet. 2017, 49, 1458–1467. [Google Scholar] [CrossRef]
- Liu, L.; Fan, Q.; Zhang, F.; Guo, X.; Liang, X.; Du, Y.; Li, P.; Wen, Y.; Hao, J.; Wang, W.; et al. A Genomewide Integrative Analysis of GWAS and eQTLs Data Identifies Multiple Genes and Gene Sets Associated with Obesity. BioMed Res. Int. 2018, 2018, 1–5. [Google Scholar] [CrossRef]
- Song, Q.; Meng, X.-R.; Hinney, A.; Song, J.-Y.; Huang, T.; Ma, J.; Wang, H.-J. Waist-hip ratio related genetic loci are associated with risk of impaired fasting glucose in Chinese children: A case control study. Nutr. Metab. 2018, 15, 34. [Google Scholar] [CrossRef] [Green Version]
- Pravenec, M.; Zídek, V.; Landa, V.; Mlejnek, P.; Šilhavý, J.; Šimáková, M.; Trnovská, J.; Škop, V.; Marková, I.; Malínská, H.; et al. Mutant Wars2 Gene in Spontaneously Hypertensive Rats Impairs Brown Adipose Tissue Function and Predisposes to Visceral Obesity. Physiol. Res. 2017, 66, 917–924. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.Y.; Tomizawa, K. Functional loss of Cdkal1, a novel tRNA modification enzyme, causes the develop-ment of type 2 diabetes. Endocr. J. 2011, 58, 819–825. [Google Scholar] [CrossRef] [Green Version]
- Wei, F.-Y.; Suzuki, T.; Watanabe, S.; Kimura, S.; Kaitsuka, T.; Fujimura, A.; Matsui, H.; Atta, M.G.; Michiue, H.; Fontecave, M.; et al. Deficit of tRNALys modification by Cdkal1 causes the development of type 2 diabetes in mice. J. Clin. Investig. 2011, 121, 3598–3608. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmer, C.J.; Bruckner, R.J.; Paulo, J.A.; Kazak, L.; Long, J.Z.; Mina, A.I.; Deng, Z.; LeClair, K.B.; Hall, J.A.; Hong, S.; et al. Cdkal1, a type 2 diabetes susceptibility gene, regulates mitochondrial function in adipose tissue. Mol. Metab. 2017, 6, 1212–1225. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, Y.; Momoi, M.Y.; Tominaga, K.; Momoi, T.; Nihei, K.; Yanagisawa, M.; Kagawa, Y.; Ohta, S. A point mutation in the mitochondrial tRNA(Leu)(UUR) gene in MELAS (mitochondrial myopathy, encephalopa-thy, lactic acidosis and stroke-like episodes). Biochem. Biophys. Res. Commun. 1990, 173, 816–822. [Google Scholar] [CrossRef]
- Kirino, Y.; Suzuki, T. Human Mitochondrial Diseases Associated with tRNA Wobble Modification Deficiency. RNA Biol. 2005, 2, 41–44. [Google Scholar] [CrossRef] [Green Version]
- Finsterer, J.; Frank, M. Maternally inherited diabetes and deafness is a mitochondrial multiorgan disorder syndrome (MIMODS). Acta Diabetol. 2017, 54, 979–980. [Google Scholar] [CrossRef]
- Wang, M.; Liu, H.; Zheng, J.; Chen, B.; Zhou, M.; Fan, W.; Wang, H.; Liang, X.; Zhou, X.; Eriani, G.; et al. A Deafness- and Diabetes-associated tRNA Mutation Causes Deficient Pseudouridinylation at Position 55 in tRNAGlu and Mitochondrial Dysfunction*. J. Biol. Chem. 2016, 291, 21029–21041. [Google Scholar] [CrossRef] [Green Version]
- Ding, Y.; Xia, B.-H.; Zhang, C.-J.; Zhuo, G.-C. Mutations in mitochondrial tRNA genes may be related to insulin resistance in women with polycystic ovary syndrome. Am. J. Transl. Res. 2017, 9, 2984–2996. [Google Scholar]
- Igoillo-Esteve, M.; Genin, A.; Lambert, N.; Désir, J.; Pirson, I.; Abdulkarim, B.; Simonis, N.; Drielsma, A.; Marselli, L.; Marchetti, P.; et al. tRNA Methyltransferase Homolog Gene TRMT10A Mutation in Young Onset Diabetes and Primary Microcephaly in Humans. PLoS Genet. 2013, 9, e1003888. [Google Scholar] [CrossRef] [Green Version]
- Gillis, D.; Krishnamohan, A.; Yaacov, B.; Shaag, A.; E Jackman, J.; Elpeleg, O. TRMT10A dysfunction is associated with abnormalities in glucose homeostasis, short stature and microcephaly. J. Med Genet. 2014, 51, 581–586. [Google Scholar] [CrossRef]
- Zung, A.; Kori, M.; Burundukov, E.; Ben-Yosef, T.; Tatoor, Y.; Granot, E. Homozygous deletion ofTRMT10Aas part of a contiguous gene deletion in a syndrome of failure to thrive, delayed puberty, intellectual disability and diabetes mellitus. Am. J. Med Genet. Part A 2015, 167, 3167–3173. [Google Scholar] [CrossRef] [PubMed]
- Narayanan, M.; Ramsey, K.; Grebe, T.; Schrauwen, I.; Szelinger, S.; Huentelman, M.; Craig, D.; Narayanan, V. Case Report: Compound heterozygous nonsense mutations in TRMT10A are associated with microcephaly, delayed development, and periventricular white matter hyperintensities. F1000Research 2015, 4, 912. [Google Scholar] [CrossRef] [Green Version]
- Yew, T.W.; McCreight, L.; Colclough, K.; Ellard, S.; Pearson, E.R. tRNA methyltransferase homologue gene TRMT10A mutation in young adult-onset diabetes with intellectual disability, microcephaly and epilepsy. Diabet. Med. 2016, 33, e21–e25. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.; Zhou, X.; Chen, X.; Huang, K.; Wu, W.; Fu, J.; Li, Y.; Polychronakos, C.; Dong, G. tRNA methyltransferase 10 homologue A (TRMT10A) mutation in a Chinese patient with diabetes, insulin resistance, intellectual deficiency and microcephaly. BMJ Open Diabetes Res. Care 2020, 8, e001601. [Google Scholar] [CrossRef] [PubMed]
- Wei, F.-Y.; Zhou, B.; Suzuki, T.; Miyata, K.; Ujihara, Y.; Horiguchi, H.; Takahashi, N.; Xie, P.; Michiue, H.; Fujimura, A.; et al. Cdk5rap1-Mediated 2-Methylthio Modification of Mitochondrial tRNAs Governs Protein Translation and Contributes to Myopathy in Mice and Humans. Cell Metab. 2015, 21, 428–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, N.; Wei, F.-Y.; Watanabe, S.; Hirayama, M.; Ohuchi, Y.; Fujimura, A.; Kaitsuka, T.; Ishii, I.; Sawa, T.; Nakayama, H.; et al. Reactive sulfur species regulate tRNA methylthiolation and contribute to insulin secretion. Nucleic Acids Res. 2017, 45, 435–445. [Google Scholar] [CrossRef] [PubMed]
- Pascoe, L.; Tura, A.; Patel, S.K.; Ibrahim, I.M.; Ferrannini, E.; Zeggini, E.; Weedon, M.N.; Mari, A.; Hattersley, A.T.; McCarthy, M.I.; et al. Common Variants of the Novel Type 2 Diabetes Genes CDKAL1 and HHEX/IDE Are Associated With Decreased Pancreatic -Cell Function. Diabetes 2007, 56, 3101–3104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, B.; Wei, F.-Y.; Kanai, N.; Fujimura, A.; Kaitsuka, T.; Tomizawa, K. Identification of a splicing variant that regulates type 2 diabetes risk factor CDKAL1 level by a coding-independent mechanism in human. Hum. Mol. Genet. 2014, 23, 4639–4650. [Google Scholar] [CrossRef] [Green Version]
- Steinthorsdottir, V.; Thorleifsson, G.; Reynisdóttir, I.; Benediktsson, R.; Jonsdottir, T.; Walters, G.B.; Styrkarsdottir, U.; Gretarsdottir, S.; Emilsson, V.; Ghosh, S.; et al. A variant in CDKAL1 influences insulin response and risk of type 2 diabetes. Nat. Genet. 2007, 39, 770–775. [Google Scholar] [CrossRef] [Green Version]
- Kirchhoff, K.; Machicao, F.; Haupt, A.; Schäfer, S.A.; Tschritter, O.; Staiger, H.; Stefan, N.; Häring, H.-U.; Fritsche, A. Polymorphisms in the TCF7L2, CDKAL1 and SLC30A8 genes are associated with impaired proinsulin conversion. Diabetol. 2008, 51, 597–601. [Google Scholar] [CrossRef] [Green Version]
- Saxena, R.; Voight, B.F.; Lyssenko, V.; Burtt, N.P.; De Bakker, P.I.W.; Chen, H.; Roix, J.J.; Kathiresan, S.; Hirschhorn, J.N.; Daly, M.J.; et al. Genome-Wide Association Analysis Identifies Loci for Type 2 Diabetes and Triglyceride Levels. Science 2007, 316, 1331–1336. [Google Scholar] [CrossRef] [PubMed]
- Dehwah, M.; Wang, M.; Huang, Q.-Y. CDKAL1 and type 2 diabetes: A global meta-analysis. Genet. Mol. Res. 2010, 9, 1109–1120. [Google Scholar] [CrossRef] [PubMed]
- Zeggini, E.; Weedon, M.N.; Lindgren, C.M.; Frayling, T.M.; Elliott, K.S.; Lango, H.; Timpson, N.J.; Perry, J.R.B.; Rayner, N.W.; Freathy, R.M.; et al. Replication of Genome-Wide Association Signals in UK Samples Reveals Risk Loci for Type 2 Diabetes. Science 2007, 316, 1336–1341. [Google Scholar] [CrossRef] [Green Version]
- Liu, X.; Liang, J.; Geng, H.; Xu, W.; Teng, F.; Yang, M. Association of the CDKAL1 polymorphism rs10946398 with type 2 diabetes mellitus in adults. Medicine 2020, 99, e21383. [Google Scholar] [CrossRef] [PubMed]
- Tian, Y.; Xu, J.; Huang, T.; Cui, J.; Zhang, W.; Song, W.; Chen, H.; Huang, P.; Yang, S.; Wang, L.; et al. A Novel Polymorphism (rs35612982) in CDKAL1 Is a Risk Factor of Type 2 Diabetes: A Case-Control Study. Kidney Blood Press. Res. 2019, 44, 1313–1326. [Google Scholar] [CrossRef]
- Wood, A.R.; The GIANT consortium; Tyrrell, J.; Beaumont, R.; Jones, S.E.; Tuke, M.A.; Ruth, K.S.; Yaghootkar, H.; Freathy, R.M.; Murray, A.; et al. Variants in the FTO and CDKAL1 loci have recessive effects on risk of obesity and type 2 diabetes, respectively. Diabetol. 2016, 59, 1214–1221. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, J.; Pei, Y.; Liu, X.; Qiu, Q.; Sun, Y.; Zhu, Y.; Yang, M.; Qi, L. The CDKAL1 gene is associated with impaired insulin secretion and glucose-related traits: The Cardiometabolic Risk in Chinese (CRC) study. Clin. Endocrinol. 2015, 83, 651–655. [Google Scholar] [CrossRef]
- Plengvidhya, N.; Chanprasert, C.; Chongjaroen, N.; Yenchitsomanus, P.-T.; Homsanit, M.; Tangjittipokin, W. Impact of KCNQ1, CDKN2A/2B, CDKAL1, HHEX, MTNR1B, SLC30A8, TCF7L2, and UBE2E2 on risk of developing type 2 diabetes in Thai population. BMC Med. Genet. 2018, 19, 93. [Google Scholar] [CrossRef]
- Locke, J.M.; Wei, F.-Y.; Tomizawa, K.; Weedon, M.N.; Harries, L.W. A cautionary tale: The non-causal association between type 2 diabetes risk SNP, rs7756992, and levels of non-coding RNA, CDKAL1-v1. Diabetologia 2015, 58, 745–748. [Google Scholar] [CrossRef] [Green Version]
- Brambillasca, S.; Altkrueger, A.; Colombo, S.F.; Friederich, A.; Eickelmann, P.; Mark, M.; Borgese, N.; Solimena, M. CDK5 Regulatory Subunit-associated Protein 1-Like 1 (CDKAL1) Is a Tail-anchored Protein in the Endoplasmic Reticulum (ER) of Insulinoma Cells. J. Biol. Chem. 2012, 287, 41808–41819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arragain, S.; Handelman, S.K.; Forouhar, F.; Wei, F.-Y.; Tomizawa, K.; Hunt, J.F.; Douki, T.; Fontecave, M.; Mulliez, E.; Atta, M. Identification of Eukaryotic and Prokaryotic Methylthiotransferase for Biosynthesis of 2-Methylthio-N6-threonylcarbamoyladenosine in tRNA. J. Biol. Chem. 2010, 285, 28425–28433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, P.; Wei, F.-Y.; Hirata, S.; Kaitsuka, T.; Suzuki, T.; Suzuki, T.; Tomizawa, K. Quantitative PCR Measurement of tRNA 2-Methylthio Modification for Assessing Type 2 Diabetes Risk. Clin. Chem. 2013, 59, 1604–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McCrate, N.E.; Varner, M.E.; Kim, K.I.; Nagan, M.C. Molecular dynamics simulations of human Formula: The role of modified bases in mRNA recognition. Nucleic Acids Res. 2006, 34, 5361–5368. [Google Scholar] [CrossRef] [Green Version]
- Stančáková, A.; Pihlajamäki, J.; Kuusisto, J.; Stefan, N.; Fritsche, A.; Häring, H.; Andreozzi, F.; Succurro, E.; Sesti, G.; Boesgaard, T.W.; et al. Single-Nucleotide Polymorphism rs7754840 ofCDKAL1Is Associated with Impaired Insulin Secretion in Nondiabetic Offspring of Type 2 Diabetic Subjects and in a Large Sample of Men with Normal Glucose Tolerance. J. Clin. Endocrinol. Metab. 2008, 93, 1924–1930. [Google Scholar] [CrossRef] [Green Version]
- Groenewoud, M.J.; Dekker, J.M.; Fritsche, A.; Reiling, E.; Nijpels, G.; Heine, R.J.; Maassen, J.A.; Machicao, F.; Schäfer, S.A.; Häring, H.U.; et al. Variants of CDKAL1 and IGF2BP2 affect first-phase insulin secretion during hyperglycaemic clamps. Diabetol. 2008, 51, 1659–1663. [Google Scholar] [CrossRef] [Green Version]
- Ohara-Imaizumi, M.; Yoshida, M.; Aoyagi, K.; Saito, T.; Okamura, T.; Takenaka, H.; Akimoto, Y.; Nakamichi, Y.; Takanashi-Yanobu, R.; Nishiwaki, C.; et al. Deletion of CDKAL1 Affects Mitochondrial ATP Generation and First-Phase Insulin Exocytosis. PLOS ONE 2010, 5, e15553. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.; Weiss, M.A.; Arunagiri, A.; Yong, J.; Rege, N.; Sun, J.; Haataja, L.; Kaufman, R.J.; Arvan, P. Biosynthesis, structure, and folding of the insulin precursor protein. Diabetes Obes. Metab. 2018, 20, 28–50. [Google Scholar] [CrossRef]
- Zeng, H.; Guo, M.; Zhou, T.; Tan, L.; Chong, C.N.; Zhang, T.; Dong, X.; Xiang, J.Z.; Yu, A.S.; Yue, L.; et al. An Isogenic Human ESC Platform for Functional Evaluation of Genome-wide-Association-Study-Identified Diabetes Genes and Drug Discovery. Cell Stem Cell 2016, 19, 326–340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Howell, N.W.; Jora, M.; Jepson, B.F.; Limbach, P.A.; Jackman, J.E. Distinct substrate specificities of the human tRNA methyltransferases TRMT10A and TRMT10B. RNA 2019, 25, 1366–1376. [Google Scholar] [CrossRef] [PubMed]
- Vilardo, E.; Nachbagauer, C.; Buzet, A.; Taschner, A.; Holzmann, J.; Rossmanith, W. A subcomplex of human mitochondrial RNase P is a bifunctional methyltransferase-extensive moonlighting in mitochondrial tRNA biogenesis. Nucleic Acids Res. 2018, 46, 11126–11127. [Google Scholar] [CrossRef] [PubMed]
- Vilardo, E.; Nachbagauer, C.; Buzet, A.; Taschner, A.; Holzmann, J.; Rossmanith, W. A subcomplex of human mitochondrial RNase P is a bifunctional methyltransferase—extensive moonlighting in mitochondrial tRNA biogenesis. Nucleic Acids Res. 2012, 40, 11583–11593. [Google Scholar] [CrossRef]
- Vilardo, E.; Amman, F.; Toth, U.; Kotter, A.; Helm, M.; Rossmanith, W. Functional characterization of the human tRNA methyltransferases TRMT10A and TRMT10B. Nucleic Acids Res. 2020, 48, 6157–6169. [Google Scholar] [CrossRef]
- Ontiveros, R.J.; Shen, H.; Stoute, J.; Yanas, A.; Cui, Y.; Zhang, Y.; Liu, K.F. Coordination of mRNA and tRNA methylations by TRMT10A. Proc. Natl. Acad. Sci. USA 2020, 117, 7782–7791. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Lu, Z.; Gomez, A.M.; Hon, G.C.; Yue, Y.; Han, D.; Fu, Y.; Parisien, M.; Dai, Q.; Jia, G.; et al. N6-methyladenosine-dependent regulation of messenger RNA stability. Nat. Cell Biol. 2014, 505, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.; Ieong, K.-W.; Demirci, H.; Chen, J.C.J.; Petrov, A.; Prabhakar, A.; O’Leary, S.E.; Dominissini, D.; Rechavi, D.D.G.; Soltis, H.D.S.M.; et al. N6-methyladenosine in mRNA disrupts tRNA selection and translation-elongation dynamics. Nat. Struct. Mol. Biol. 2016, 23, 110–115. [Google Scholar] [CrossRef] [Green Version]
- Ghafarian-Alipour, F.; Ziaee, S.; Ashoori, M.R.; Zakeri, M.S.; Boroumand, M.A.; Aghamohammadzadeh, N.; Abbasi-Majdi, M.; Shool, F.; Asbaghi, N.S.; Mohammadi, A.; et al. Association between FTO gene polymorphisms and type 2 diabetes mellitus, serum levels of apelin and androgen hormones among Iranian obese women. Gene 2018, 641, 361–366. [Google Scholar] [CrossRef]
- Frayling, T.M.; Timpson, N.J.; Weedon, M.N.; Zeggini, E.; Freathy, R.M.; Lindgren, C.M.; Perry, J.R.B.; Elliott, K.S.; Lango, H.; Rayner, N.W.; et al. A Common Variant in the FTO Gene Is Associated with Body Mass Index and Predisposes to Childhood and Adult Obesity. Science 2007, 316, 889–894. [Google Scholar] [CrossRef] [Green Version]
- Russell, M.A.; Redick, S.D.; Blodgett, D.M.; Richardson, S.J.; Leete, P.; Krogvold, L.; Dahl-Jørgensen, K.; Bottino, R.; Brissova, M.; Spaeth, J.M.; et al. HLA Class II Antigen Processing and Presentation Pathway Components Demonstrated by Transcriptome and Protein Analyses of Islet β-Cells from Donors With Type 1 Diabetes. Diabetes 2019, 68, 988–1001. [Google Scholar] [CrossRef] [Green Version]
- Park, S.-J.; Ahn, H.-S.; Kim, J.S.; Lee, C.-J. Evaluation of Multi-tRNA Synthetase Complex by Multiple Reaction Monitoring Mass Spectrometry Coupled with Size Exclusion Chromatography. PLoS ONE 2015, 10, e0142253. [Google Scholar] [CrossRef]
- Wang, X.; Pan, T. Stress Response and Adaptation Mediated by Amino Acid Misincorporation during Protein Synthesis. Adv. Nutr. 2016, 7, 773S–779S. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Netzer, N.; Goodenbour, J.M.; David, A.; Dittmar, K.A.; Jones, R.B.; Schneider, J.R.; Boone, D.L.; Eves, E.M.; Rosner, M.R.; Gibbs, J.S.; et al. Innate immune and chemically triggered oxidative stress modifies translational fidelity. Nat. Cell Biol. 2009, 462, 522–526. [Google Scholar] [CrossRef]
- Yao, P.; Fox, P.L. Aminoacyl-tRNA synthetases in medicine and disease. EMBO Mol. Med. 2013, 5, 332–343. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Satz, J.S.; Vo, M.-N.; Nangle, L.A.; Schimmel, P.; Ackerman, S.L. Deficiencies in tRNA synthetase editing activity cause cardioproteinopathy. Proc. Natl. Acad. Sci. USA 2014, 111, 17570–17575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, J.W.; Beebe, K.; Nangle, L.A.; Jang, J.; Longo-Guess, C.M.; Cook, S.A.; Davisson, M.T.; Sundberg, J.P.; Schimmel, P.; Ackerman, S.L. Editing-defective tRNA synthetase causes protein misfolding and neurodegeneration. Nat. Cell Biol. 2006, 443, 50–55. [Google Scholar] [CrossRef]
- Reiling, E.; Jafar-Mohammadi, B.; van’t Riet, E.; Weedon, M.N.; Van Vliet-Ostaptchouk, J.V.; Hansen, T.; Saxena, R.; Van Haeften, T.W.; Arp, P.A.; Das, S.; et al. Genetic association analysis of LARS2 with type 2 diabetes. Diabetologia 2009, 53, 103–110. [Google Scholar] [CrossRef] [Green Version]
- Yasukawa, T.; Suzuki, T.; Suzuki, T.; Ueda, T.; Ohta, S.; Watanabe, K. Modification Defect at Anticodon Wobble Nucleotide of Mitochondrial tRNAsLeu(UUR) with Pathogenic Mutations of Mitochondrial Myopathy, Encephalopathy, Lactic Acidosis, and Stroke-like Episodes. J. Biol. Chem. 2000, 275, 4251–4257. [Google Scholar] [CrossRef] [Green Version]
- Maassen, J.A.; M’t Hart, L.; Van Essen, E.; Heine, R.J.; Nijpels, G.; Tafrechi, R.S.J.; Raap, A.K.; Janssen, G.M.; Lemkes, H.H. Mitochondrial Diabetes: Molecular Mechanisms and Clinical Presentation. Diabetes 2004, 53, S103–S109. [Google Scholar] [CrossRef] [Green Version]
- Park, H.; Davidson, E.; King, M.P. The Pathogenic A3243G Mutation in Human Mitochondrial tRNALeu(UUR)Decreases the Efficiency of Aminoacylation. Biochemistry. 2003, 42, 958–964. [Google Scholar] [CrossRef]
- Ouweland, J.V.D.; Lemkes, H.; Ruitenbeek, W.; Sandkuijl, L.; De Vijlder, M.; Struyvenberg, P.; Van De Kamp, J.; Maassen, J. Mutation in mitochondrial tRNALeu(UUR) gene in a large pedigree with maternally transmitted type II diabetes mellitus and deafness. Nat. Genet. 1992, 1, 368–371. [Google Scholar] [CrossRef]
- Ohkubo, K.; Yamano, A.; Nagashima, M.; Mori, Y.; Anzai, K.; Akehi, Y.; Nomiyama, R.; Asano, T.; Urae, A.; Ono, J. Mitochondrial gene mutations in the tRNA(Leu(UUR)) region and diabetes: Prevalence and clinical phenotypes in Japan. Clin. Chem. 2001, 47, 1641–1648. [Google Scholar] [CrossRef] [Green Version]
- Chomyn, A.; Meola, G.; Bresolin, N.; Lai, S.T.; Scarlato, G.; Attardi, G. In vitro genetic transfer of protein synthesis and respiration defects to mitochondrial DNA-less cells with myopathy-patient mitochondria. Mol. Cell. Biol. 1991, 11, 2236–2244. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Guan, M.-X. Human Mitochondrial Leucyl-tRNA Synthetase Corrects Mitochondrial Dysfunctions Due to the tRNALeu(UUR) A3243G Mutation, Associated with Mitochondrial Encephalomyopathy, Lactic Acidosis, and Stroke-Like Symptoms and Diabetes. Mol. Cell. Biol. 2010, 30, 2147–2154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.C.; Park, A.; Oh, K.-J.; Kim, W.K.; Bae, K.-H. The Role of Adipose Tissue Mitochondria: Regulation of Mitochondrial Function for the Treatment of Metabolic Diseases. Int. J. Mol. Sci. 2019, 20, 4924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bournat, J.C.; Brown, C.W. Mitochondrial dysfunction in obesity. Curr. Opin. Endocrinol. Diabetes Obes. 2010, 17, 446–452. [Google Scholar] [CrossRef] [Green Version]
- Cypess, A.M.; Lehman, S.; Williams, G.; Tal, I.; Rodman, D.; Goldfine, A.B.; Kuo, F.C.; Palmer, E.L.; Tseng, Y.-H.; Doria, A.; et al. Identification and Importance of Brown Adipose Tissue in Adult Humans. N. Engl. J. Med. 2009, 360, 1509–1517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpentier, A.C.; Blondin, D.P.; Virtanen, K.A.; Richard, D.; Haman, F.; Turcotte, É.E. Brown Adipose Tissue Energy Metabolism in Humans. Front. Endocrinol. 2018, 9, 447. [Google Scholar] [CrossRef] [Green Version]
- Kobiita, A.; Godbersen, S.; Araldi, E.; Ghoshdastider, U.; Schmid, M.W.; Spinas, G.; Moch, H.; Stoffel, M. The Diabetes Gene JAZF1 Is Essential for the Homeostatic Control of Ribosome Biogenesis and Function in Metabolic Stress. Cell Rep. 2020, 32, 107846. [Google Scholar] [CrossRef]
- Antonellis, A.; Ellsworth, R.E.; Sambuughin, N.; Puls, I.; Abel, A.; Lee-Lin, S.-Q.; Jordanova, A.; Kremensky, I.; Christodoulou, K.; Middleton, L.T.; et al. Glycyl tRNA Synthetase Mutations in Charcot-Marie-Tooth Disease Type 2D and Distal Spinal Muscular Atrophy Type V. Am. J. Hum. Genet. 2003, 72, 1293–1299. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Li, R.; Li, W.; Wang, M.; Ji, J.; Zheng, J.; Mao, Z.; Mo, J.Q.; Jiang, P.-P.; Lu, J.; et al. Maternally inherited diabetes is associated with a homoplasmic T10003C mutation in the mitochondrial tRNAGly gene. Mitochondrion 2015, 21, 49–57. [Google Scholar] [CrossRef]
- Crispim, D.; Estivalet, A.A.F.; Roisenberg, I.; Gross, J.L.; Canani, L.H. Prevalence of 15 mitochondrial DNA mutations among type 2 diabetic patients with or without clinical characteristics of maternally inherited diabetes and deafness. Arq. Bras. Endocrinol. Metabol. 2008, 52, 1228–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancuso, M.; Ferraris, S.; Nishigaki, Y.; Azan, G.; Mauro, A.; Sammarco, P.; Krishna, S.; Tay, S.K.H.; Bonilla, E.; Romansky, S.G.; et al. Congenital or late-onset myopathy in patients with the T14709C mtDNA mutation. J. Neurol. Sci. 2005, 228, 93–97. [Google Scholar] [CrossRef] [PubMed]
- Ding, Y.; Xia, B.-H.; Zhang, C.-J.; Zhuo, G.-C. Mitochondrial tRNALeu(UUR) C3275T, tRNAGln T4363C and tRNALys A8343G mutations may be associated with PCOS and metabolic syndrome. Gene 2018, 642, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, G.; Ding, Y.; Feng, G.; Yu, L.; Jiang, Y. Analysis of mitochondrial DNA sequence variants in patients with polycystic ovary syndrome. Arch. Gynecol. Obstet. 2012, 286, 653–659. [Google Scholar] [CrossRef]
- Jaswa, E.G.; Pasch, L.A.; Shinkai, K.; Cedars, M.I.; Huddleston, H.G. Putative role for insulin resistance in depression risk in polycystic ovary syndrome. Fertil. Steril. 2015, 104, 707–714.e1. [Google Scholar] [CrossRef]
- Legro, R.S.; Kunselman, A.R.; Dodson, W.C.; Dunaif, A. Prevalence and Predictors of Risk for Type 2 Diabetes Mellitus and Impaired Glucose Tolerance in Polycystic Ovary Syndrome: A Prospective, Controlled Study in 254 Affected Women1. J. Clin. Endocrinol. Metab. 1999, 84, 165–169. [Google Scholar] [CrossRef]
- Finsterer, J.; Zarrouk-Mahjoub, S. Polycystic ovary syndrome in mitochondrial disorders due mtDNA or nDNA variants. Am. J. Transl. Res. 2018, 10, 13–15. [Google Scholar]
- Zillikens, M.C.; Yazdanpanah, M.; Pardo, L.M.; Rivadeneira, F.; Aulchenko, Y.S.; Oostra, B.A.; Uitterlinden, A.G.; Pols, H.A.P.; Van Duijn, C.M. Sex-specific genetic effects influence variation in body composition. Diabetologia 2008, 51, 2233–2241. [Google Scholar] [CrossRef] [Green Version]
- Zhao, L.; Lian, J.; Tian, J.; Shen, Y.; Ping, Z.; Fang, X.; Min, J.; Wang, F. Dietary intake of heme iron and body iron status are associated with the risk of gestational diabetes mellitus: A systematic review and meta-analysis. Asia Pac. J. Clin. Nutr. 2017, 26, 1092–1106. [Google Scholar]
- Simcox, J.A.; McClain, D.A. Iron and Diabetes Risk. Cell Metab. 2013, 17, 329–341. [Google Scholar] [CrossRef] [Green Version]
- Meidtner, K.; Podmore, C.; Kröger, J.; Van Der Schouw, Y.T.; Bendinelli, B.; Agnoli, C.; Arriola, L.; Barricarte, A.; Boeing, H.; Cross, A.J.; et al. Interaction of Dietary and Genetic Factors Influencing Body Iron Status and Risk of Type 2 Diabetes Within the EPIC-InterAct Study. Diabetes Care 2018, 41, 277–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernández-Cao, J.C.; Aranda, N.; Ribot, B.; Tous, M.; Arija, V. Elevated iron status and risk of gestational diabetes mellitus: A systematic review and meta-analysis. Matern. Child Nutr. 2016, 13, 13. [Google Scholar] [CrossRef] [PubMed]
- Lane, D.M. Iron stores as a risk factor for diabetes in women. JAMA 2004, 291, 2428–2429. [Google Scholar] [CrossRef] [PubMed]
- Jiang, R.; Manson, J.E.; Meigs, J.B.; Ma, J.; Rifai, N.; Hu, F.B. Body Iron Stores in Relation to Risk of Type 2 Diabetes in Apparently Healthy Women. JAMA 2004, 291, 711–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tiongco, R.E.; Arceo, E.; Clemente, B.; Pineda-Cortel, M.R.B. Association of maternal iron deficiency anemia with the risk of gestational diabetes mellitus: A meta-analysis. Arch. Gynecol. Obstet. 2018, 299, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, M.C.F.; Anderson, C.P.; Neschen, S.; Zumbrennen-Bullough, K.B.; Romney, S.J.; Kahle-Stephan, M.; Rathkolb, B.; Gailus-Durner, V.; Fuchs, H.; Wolf, E.; et al. Irp2 regulates insulin production through iron-mediated Cdkal1-catalyzed tRNA modification. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef]
- Hansen, J.B.J.; Tonnesen, M.F.M.; Madsen, A.N.A.; Hagedorn, P.H.; Friberg, J.; Grunnet, L.G.L.; Heller, R.S.R.; Nielsen, A.O.A.; Størling, J.; Baeyens, L.; et al. Divalent Metal Transporter 1 Regulates Iron-Mediated ROS and Pancreatic β Cell Fate in Response to Cytokines. Cell Metab. 2012, 16, 449–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooksey, R.C.; Jouihan, H.A.; Ajioka, R.S.; Hazel, M.W.; Weiss, S.M.; Kushner, J.P.; McClain, D.A. Oxidative Stress, β-Cell Apoptosis, and Decreased Insulin Secretory Capacity in Mouse Models of Hemochromatosis. Endocrinology 2004, 145, 5305–5312. [Google Scholar] [CrossRef]
- Paul, B.T.; Manz, D.H.; Torti, F.M.; Torti, S.V. Mitochondria and Iron: Current questions. Expert Rev. Hematol. 2017, 10, 65–79. [Google Scholar] [CrossRef] [Green Version]
- Rouault, T.A. Mammalian iron–sulphur proteins: Novel insights into biogenesis and function. Nat. Rev. Mol. Cell Biol. 2014, 16, 45–55. [Google Scholar] [CrossRef]
- Braymer, J.J.; Lill, R. Iron–sulfur cluster biogenesis and trafficking in mitochondria. J. Biol. Chem. 2017, 292, 12754–12763. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pain, D.; Dancis, A. Roles of Fe–S proteins: From cofactor synthesis to iron homeostasis to protein synthesis. Curr. Opin. Genet. Dev. 2016, 38, 45–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rouault, T.A. The role of iron regulatory proteins in mammalian iron homeostasis and disease. Nat. Chem. Biol. 2006, 2, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Anderson, C.P.; Shen, M.; Eisenstein, R.S.; Leibold, E.A. Mammalian iron metabolism and its control by iron regulatory proteins. Biochim. Biophys. Acta (BBA) Bioenerg. 2012, 1823, 1468–1483. [Google Scholar] [CrossRef] [Green Version]
- Ida, T.; Sawa, T.; Ihara, H.; Tsuchiya, Y.; Watanabe, Y.; Kumagai, Y.; Suematsu, M.; Motohashi, H.; Fujii, S.; Matsunaga, T.; et al. Reactive cysteine persulfides and S-polythiolation regulate oxidative stress and redox signaling. Proc. Natl. Acad. Sci. USA 2014, 111, 7606–7611. [Google Scholar] [CrossRef] [Green Version]
- Condon, K.J.; Sabatini, D.M. Nutrient regulation of mTORC1 at a glance. J. Cell Sci. 2019, 132, jcs222570. [Google Scholar] [CrossRef]
- Laplante, M.; Sabatini, D.M. mTOR Signaling in Growth Control and Disease. Cell 2012, 149, 274–293. [Google Scholar] [CrossRef] [Green Version]
- Han, J.M.; Jeong, S.J.; Park, M.C.; Kim, G.; Kwon, N.H.; Kim, H.K.; Ha, S.H.; Ryu, S.H.; Kim, S. Leucyl-tRNA Synthetase Is an Intracellular Leucine Sensor for the mTORC1-Signaling Pathway. Cell 2012, 149, 410–424. [Google Scholar] [CrossRef] [Green Version]
- Yoon, I.; Nam, M.; Kim, H.K.; Moon, H.-S.; Kim, S.; Jang, J.; Song, J.A.; Jeong, S.J.; Kim, S.B.; Cho, S.; et al. Glucose-dependent control of leucine metabolism by leucyl-tRNA synthetase 1. Science 2019, 367, 205–210. [Google Scholar] [CrossRef]
- Suzuki, T.; Kelly, V.P.; Motohashi, H.; Nakajima, O.; Takahashi, S.; Nishimura, S.; Yamamoto, M. Deletion of the Selenocysteine tRNA Gene in Macrophages and Liver Results in Compensatory Gene Induction of Cytoprotective Enzymes by Nrf2. J. Biol. Chem. 2008, 283, 2021–2030. [Google Scholar] [CrossRef] [Green Version]
- Moghadaszadeh, B.; Beggs, A.H. Selenoproteins and Their Impact on Human Health Through Diverse Physiological Pathways. Physiology 2006, 21, 307–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shchedrina, V.A.; Zhang, Y.; Labunskyy, V.M.; Hatfield, D.L.; Gladyshev, V.N. Structure–Function Relations, Physiological Roles, and Evolution of Mammalian ER-Resident Selenoproteins. Antioxid. Redox Signal. 2010, 12, 839–849. [Google Scholar] [CrossRef] [PubMed]
- Labunskyy, V.M.; Lee, B.C.; Handy, D.E.; Loscalzo, J.; Hatfield, D.L.; Gladyshev, V.N. Both Maximal Expression of Selenoproteins and Selenoprotein Deficiency Can Promote Development of Type 2 Diabetes-Like Phenotype in Mice. Antioxid. Redox Signal. 2011, 14, 2327–2336. [Google Scholar] [CrossRef] [PubMed]
- Moustafa, M.E.; Carlson, B.A.; El-Saadani, M.A.; Kryukov, G.V.; Sun, Q.-A.; Harney, J.W.; Hill, K.E.; Combs, G.F.; Feigenbaum, L.; Mansur, D.B.; et al. Selective Inhibition of Selenocysteine tRNA Maturation and Selenoprotein Synthesis in Transgenic Mice Expressing Isopentenyladenosine-Deficient Selenocysteine tRNA. Mol. Cell. Biol. 2001, 21, 3840–3852. [Google Scholar] [CrossRef] [Green Version]
- Fradejas, N.; Carlson, B.A.; Rijntjes, E.; Becker, N.-P.; Tobe, R.; Schweizer, U. Mammalian Trit1 is a tRNA[Ser]Sec-isopentenyl transferase required for full selenoprotein expression. Biochem. J. 2013, 450, 427–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schweizer, U.; Bohleber, S.; Fradejas-Villar, N. The modified base isopentenyladenosine and its derivatives in tRNA. RNA Biol. 2017, 14, 1197–1208. [Google Scholar] [CrossRef] [Green Version]
- Kim, L.K.; Matsufuji, T.; Matsufuji, S.; Carlson, B.A.; Kim, S.S.; Hatfield, L.L.; Lee, B.J. Methylation of the ribosyl moiety at position 34 of selenocysteine tRNA[Ser]Sec is governed by both primary and tertiary structure. RNA 2000, 6, 1306–1315. [Google Scholar] [CrossRef] [Green Version]
- Diamond, A.M.; Choi, I.S.; Crain, P.F.; Hashizume, T.; Pomerantz, S.C.; Cruz, R.; Steer, C.J.; Hill, K.E.; Burk, R.F.; McCloskey, J.A.; et al. Dietary selenium affects methylation of the wobble nucleoside in the anti-codon of selenocysteine tRNA([Ser]Sec). J. Biol. Chem. 1993, 268, 14215–14223. [Google Scholar]
- Ogawa-Wong, A.N.; Berry, M.J.; Seale, L.A. Selenium and Metabolic Disorders: An Emphasis on Type 2 Diabetes Risk. Nutrients 2016, 8, 80. [Google Scholar] [CrossRef] [Green Version]
- Stranges, S.; Marshall, J.R.; Natarajan, R.; Donahue, R.P.; Trevisan, M.; Combs, G.F.; Cappuccio, F.P.; Ceriello, A.; Reid, M.E. Effects of Long-Term Selenium Supplementation on the Incidence of Type 2 Diabetes. Ann. Intern. Med. 2007, 147, 217–223. [Google Scholar] [CrossRef]
- Yagishita, Y.; Uruno, A.; Fukutomi, T.; Saito, R.; Saigusa, D.; Pi, J.; Fukamizu, A.; Sugiyama, F.; Takahashi, S.; Yamamoto, M. Nrf2 Improves Leptin and Insulin Resistance Provoked by Hypothalamic Oxidative Stress. Cell Rep. 2017, 18, 2030–2044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warner, G.J. Inhibition of Selenoprotein Synthesis by Selenocysteine tRNA super[Ser]Sec Lacking Isopentenyladenosine. J. Biol. Chem. 2000, 276, 28110–28119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moosmann, B.; Behl, C. Selenoprotein synthesis and side-effects of statins. Lancet 2004, 363, 892–894. [Google Scholar] [CrossRef]
- Moosmann, B.; Behl, C. Selenoproteins, Cholesterol-Lowering Drugs, and the Consequences Revisiting of the Mevalonate Pathway. Trends Cardiovasc. Med. 2004, 14, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Maki, K.; Diwadkar-Navsariwala, V.; Kramer, M.W. Statin use and risk for type 2 diabetes: What clinicians should know. Postgrad. Med. 2017, 130, 166–172. [Google Scholar] [CrossRef]
- Veenendaal, M.V.E.; Painter, R.C.; De Rooij, S.R.; Bossuyt, P.M.M.; Post, J.A.M.V.D.; Gluckman, P.D.; A Hanson, M.; Roseboom, T.J. Transgenerational effects of prenatal exposure to the 1944-45 Dutch famine. BJOG: Int. J. Obstet. Gynaecol. 2013, 120, 548–554. [Google Scholar] [CrossRef]
- Painter, R.C.; Osmond, C.; Gluckman, P.; Hanson, M.; Phillips, D.I.W.; Roseboom, T.J. Transgenerational effects of prenatal exposure to the Dutch famine on neonatal adiposity and health in later life. BJOG: Int. J. Obstet. Gynaecol. 2008, 115, 1243–1249. [Google Scholar] [CrossRef]
- Kaati, G.; Bygren, L.O.; Pembrey, M.; Sjöström, M. Transgenerational response to nutrition, early life circumstances and longevity. Eur. J. Hum. Genet. 2007, 15, 784–790. [Google Scholar] [CrossRef]
- Kaspar, D.; Hastreiter, S.; Irmler, M.; De Angelis, M.H.; Beckers, J. Nutrition and its role in epigenetic inheritance of obesity and diabetes across generations. Mamm. Genome 2020, 31, 119–133. [Google Scholar] [CrossRef]
- Ng, S.-F.; Lin, R.C.Y.; Laybutt, D.R.; Barres, R.; Owens, J.A.; Morris, M.J. Chronic high-fat diet in fathers programs β-cell dysfunction in female rat offspring. Nat. Cell Biol. 2010, 467, 963–966. [Google Scholar] [CrossRef]
- Anderson, L.M.; Riffle, L.; Wilson, R.; Travlos, G.S.; Lubomirski, M.S.; Alvord, W.G. Preconceptional fasting of fathers alters serum glucose in offspring of mice. Nutrients 2006, 22, 327–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linn, T.; Loewk, E.; Schneider, K.; Federlin, K. Spontaneous glucose intolerance in the progeny of low dose streptozotocin-induced diabetic mice. Diabetologia 1993, 36, 1245–1251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wei, Y.; Yang, C.-R.; Wei, Y.-P.; Zhao, Z.-A.; Hou, Y.; Schatten, H.; Sun, Q.-Y. Paternally induced transgenerational inheritance of susceptibility to diabetes in mammals. Proc. Natl. Acad. Sci. USA 2014, 111, 1873–1878. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huypens, P.; Sass, S.; Wu, M.; Dyckhoff, D.; Tschöp, M.; Theis, F.J.; Marschall, S.; De Angelis, M.H.; Beckers, J. Epigenetic germline inheritance of diet-induced obesity and insulin resistance. Nat. Genet. 2016, 48, 497–499. [Google Scholar] [CrossRef]
- Peng, H.; Shi, J.; Zhang, Y.; Zhang, H.; Liao, S.; Li, W.; Lei, L.; Han, C.; Ning, L.; Cao, Y.; et al. A novel class of tRNA-derived small RNAs extremely enriched in mature mouse sperm. Cell Res. 2012, 22, 1609–1612. [Google Scholar] [CrossRef]
- Schaefer, M.; Pollex, T.; Hanna, K.; Tuorto, F.; Meusburger, M.; Helm, M.; Lyko, F. RNA methylation by Dnmt2 protects transfer RNAs against stress-induced cleavage. Genes Dev. 2010, 24, 1590–1595. [Google Scholar] [CrossRef] [Green Version]
- Cropley, J.E.; Eaton, S.A.; Aiken, A.; Young, P.E.; Giannoulatou, E.; Ho, J.W.; Buckland, M.E.; Keam, S.P.; Hutvagner, G.; Humphreys, D.T.; et al. Male-lineage transmission of an acquired metabolic phenotype induced by grand-paternal obesity. Mol. Metab. 2016, 5, 699–708. [Google Scholar] [CrossRef]
- de Castro Barbosa, T.; Ingerslev, L.R.; Alm, P.S.; Versteyhe, S.; Massart, J.; Rasmussen, M.; Donkin, I.; Sjogren, R.; Mudry, J.M.; Vetterli, L.; et al. High-fat diet reprograms the epige-nome of rat spermatozoa and transgenerationally affects metabolism of the offspring. Mol. Metab. 2016, 5, 184–197. [Google Scholar] [CrossRef]
- Peleg-Raibstein, D.; Sarker, G.; Litwan, K.; Krämer, S.D.; Ametamey, S.M.; Schibli, R.; Wolfrum, C. Enhanced sensitivity to drugs of abuse and palatable foods following maternal overnutrition. Transl. Psychiatry 2016, 6, e911. [Google Scholar] [CrossRef]
- Sarker, G.; Berrens, R.V.; Von Arx, J.; Pelczar, P.; Reik, W.; Wolfrum, C.; Peleg-Raibstein, D. Transgenerational transmission of hedonic behaviors and metabolic phenotypes induced by maternal overnutrition. Transl. Psychiatry 2018, 8, 195. [Google Scholar] [CrossRef]
- Sarker, G.; Sun, W.; Rosenkranz, D.; Pelczar, P.; Opitz, L.; Efthymiou, V.; Wolfrum, C.; Peleg-Raibstein, D. Maternal overnutrition programs hedonic and metabolic phenotypes across generations through sperm tsRNAs. Proc. Natl. Acad. Sci. USA 2019, 116, 10547–10556. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nätt, D.; Öst, A. Male reproductive health and intergenerational metabolic responses from a small RNA perspective. J. Intern. Med. 2020, 288, 305–320. [Google Scholar] [CrossRef] [PubMed]
- Krawetz, S.A.; Kruger, A.; Lalancette, C.; Tagett, R.; Anton, E.; Draghici, S.; Diamond, M.P. A survey of small RNAs in human sperm. Hum. Reprod. 2011, 26, 3401–3412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nätt, D.; Kugelberg, U.; Casas, E.; Nedstrand, E.; Zalavary, S.; Henriksson, P.; Nijm, C.; Jäderquist, J.; Sandborg, J.; Flinke, E.; et al. Human sperm displays rapid responses to diet. PLoS Biol. 2019, 17, e3000559. [Google Scholar] [CrossRef] [Green Version]
- Donkin, I.; Versteyhe, S.; Ingerslev, L.R.; Qian, K.; Mechta, M.; Nordkap, L.; Mortensen, B.; Appel, E.V.R.; Jørgensen, N.; Kristiansen, V.B.; et al. Obesity and Bariatric Surgery Drive Epigenetic Variation of Spermatozoa in Humans. Cell Metab. 2016, 23, 369–378. [Google Scholar] [CrossRef] [Green Version]
- Eizirik, D.L.; Pasquali, L.; Cnop, M. Pancreatic β-cells in type 1 and type 2 diabetes mellitus: Different pathways to failure. Nat. Rev. Endocrinol. 2020, 16, 349–362. [Google Scholar] [CrossRef]
- Lytrivi, M.; Castell, A.-L.; Poitout, V.; Cnop, M. Recent Insights Into Mechanisms of β-Cell Lipo- and Glucolipotoxicity in Type 2 Diabetes. J. Mol. Biol. 2020, 432, 1514–1534. [Google Scholar] [CrossRef]
- Yu, M.; Lu, B.; Zhang, J.; Ding, J.; Liu, P.; Lu, Y. tRNA-derived RNA fragments in cancer: Current status and future perspectives. J. Hematol. Oncol. 2020, 13, 1–14. [Google Scholar] [CrossRef]
- Cnop, M.; Abdulkarim, B.; Bottu, G.; Da Cunha, D.A.; Igoillo-Esteve, M.; Masini, M.; Turatsinze, J.-V.; Griebel, T.; Villate, O.; Santin, I.; et al. RNA Sequencing Identifies Dysregulation of the Human Pancreatic Islet Transcriptome by the Saturated Fatty Acid Palmitate. Diabetes 2013, 63, 1978–1993. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| F0 (Diet or Genotype) | Offspring Phenotype | |||||
|---|---|---|---|---|---|---|
| Rodent Model and Breeding Method | F0 ♂ | F0 ♀ | F1 | F2 | Mechanism Unveiled | Reference Number |
| Rat/Natural mating | HFD | NC | ♀ β -cell dysfunction (glucose intolerance, ↓ insulin secretion) | - | 642 differentially expressed genes in F1 islets. Hypomethylation of the Il13ra2 gene | [201] |
| Mouse/Natural mating | 24 h premating fasting | NC | ♂and♀ ↓ non fasting glycemia | - | - | [202] |
| Mouse/Natural mating | Multiple low dose streptozotocin | NC | ♂ Insulitis ↓ insulin secretion | - | - | [203] |
| Mouse/Natural mating | HFD | NC | ♂and♀ Glucose intolerance Insulin resistance | - | Altered expression of genes involved in glucose homeostasis and insulin secretion in F1 islets. Altered cytosine DNA methylation in F1 islets and F0 sperm. | [204] |
| Mouse/IVF (using all gamete combinations) | 6 weeks on HFD or NC | 6 weeks on NC or HFD | ♂and♀ ↑ susceptivility to develop obesity and diabetes in progeny from HFD-fed F0 respect to progeny from NC-fed F0 | - | - | [205] |
| Mouse/ IVF or zygote microinjection of total RNA or tRHs isolated from sperm from HFD-fed males | HFD | NC | ♂and♀ Glucose intolerance Insulin resistance (only after IVF) | - | ↑ m5C and m5G in F0 sperm tRNAs. ↑ 5′-tRH in F0 sperm. ↓ expression of genes involved in metabolic regulation pathways in 8 cell F1 embryos. | [30,55] |
| Mouse/Natural mating | Congenic model of obesity and pre-diabetes Avy/a mouse) NC | NC (a/a) | ♂and♀ ↑ obesity, glucose intolerance and insulin resistance in HFD-fed offspring of Avy/a mice respect to control mice | ♂and♀ ↑ obesity, glucose intolerance and insulin resistance in the progeny of Avy/a grandfathers (even if F1 males were fed NC) | miRNA and tRH changes in F1 sperm from Avy/a fathers respect to F1 from control fathers: ↑ 5′-tRHGly(CCC) and ↓ 5′-tRHGlu(CTC).5′-tRHsGlu(CTC) binds to Argonaute and may act as miRNA regulating gene expression. | [208] |
| Rat/ Natural mating | HFD for 12 weeks | NC | ♀↓ β-cell mass Glucose intolerance | ♀ ↓ insulin secretion Glucose intolerance | DNA methylation changes in sperm from F0 and F1. Differential expression of miRNAs and 41 tRNA fragments. | [209] |
| Mouse/ Natural mating in F0 (the F2 generation was obtained by microinjection of total RNA or 5-tRHs from F1 sperm into normal zygotes) | NC | HFD for 9 weeks | ♂and ♀ ↑ weight gain ↑ blood glucose ↑ insulin secretion ↑ obesogenic eating phenotype ↑addictive-like behaviour | Identification of 13 differentially expressed 5′-tRHs in sperm from F1 derived from HFD-mothers. | [210] | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arroyo, M.N.; Green, J.A.; Cnop, M.; Igoillo-Esteve, M. tRNA Biology in the Pathogenesis of Diabetes: Role of Genetic and Environmental Factors. Int. J. Mol. Sci. 2021, 22, 496. https://doi.org/10.3390/ijms22020496
Arroyo MN, Green JA, Cnop M, Igoillo-Esteve M. tRNA Biology in the Pathogenesis of Diabetes: Role of Genetic and Environmental Factors. International Journal of Molecular Sciences. 2021; 22(2):496. https://doi.org/10.3390/ijms22020496
Chicago/Turabian StyleArroyo, Maria Nicol, Jonathan Alex Green, Miriam Cnop, and Mariana Igoillo-Esteve. 2021. "tRNA Biology in the Pathogenesis of Diabetes: Role of Genetic and Environmental Factors" International Journal of Molecular Sciences 22, no. 2: 496. https://doi.org/10.3390/ijms22020496