
Proton Pumping and Non-Pumping Terminal Respiratory Oxidases: Active Sites Intermediates of These Molecular Machines and Their Derivatives



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction: General Properties of Terminal Respiratory Oxidases
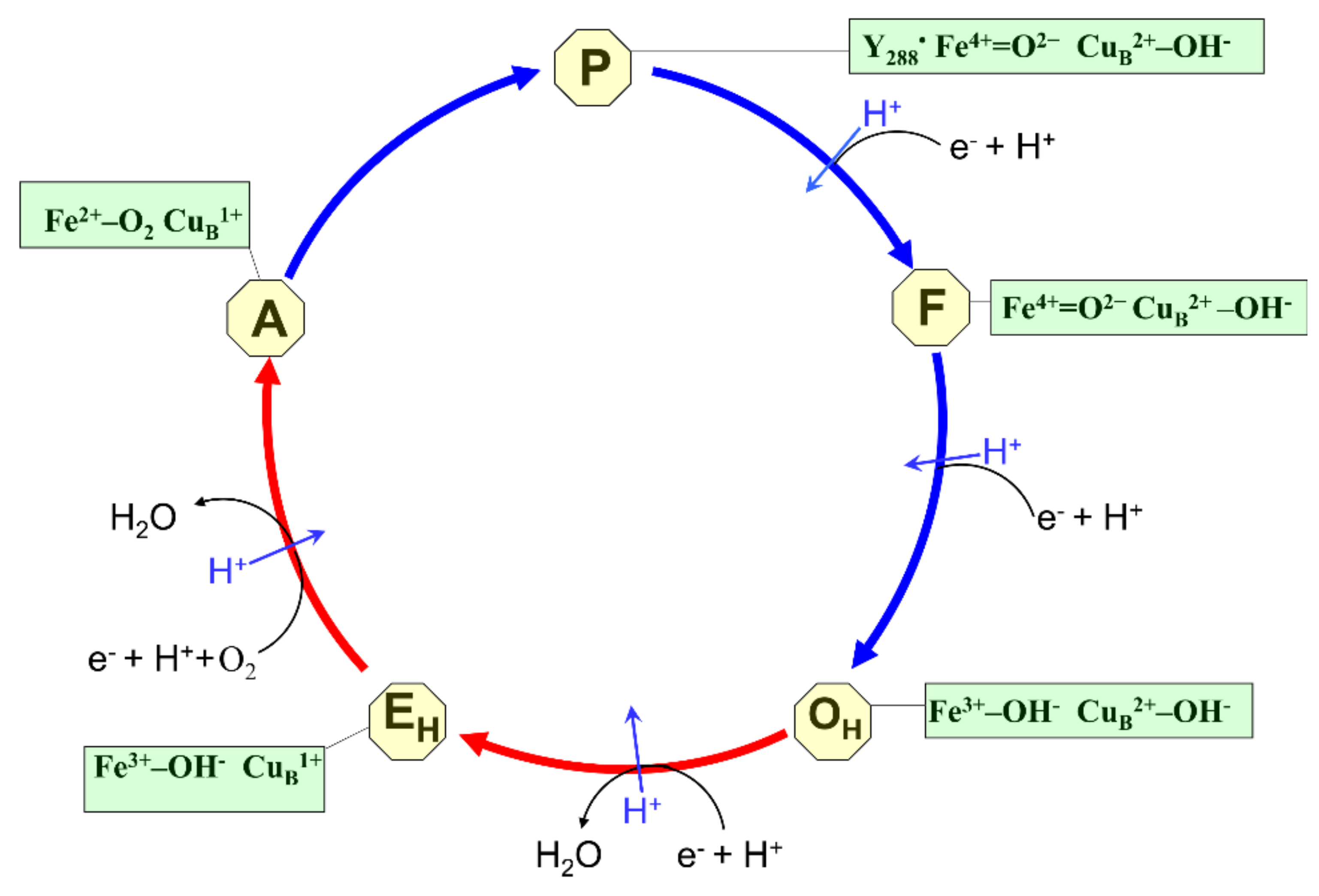
2. States of Active Sites as Intermediates of the Catalytic Cycle of Oxygen Reduction
2.1. O/OH
2.2. O/O1
2.3. E/EH
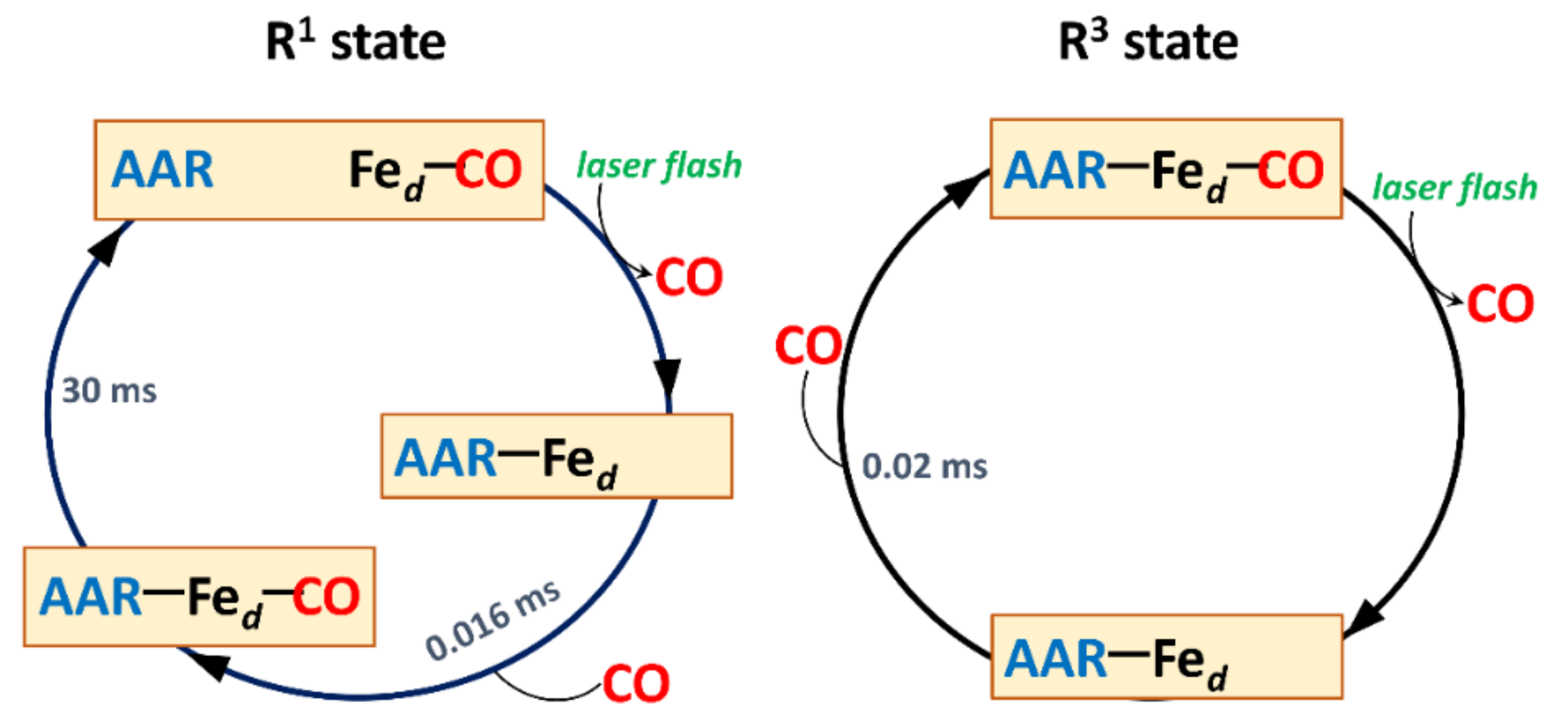
2.4. R/R3
2.5. A and Ip
2.6. A1 and A3
2.7. P and F
3. Noncatalytic States of Active Sites That Occur When External Ligands Are Added: Heme-Copper, bd
4. BNC vs. DHAS
5. Concluding Remarks
Funding
Conflicts of Interest
References
- Hemp, J.; Gennis, R.B. Diversity of the Heme–Copper Superfamily in Archaea: Insights from Genomics and Structural Modeling. Results Probl. Cell Differ. 2008, 45, 1–31. [Google Scholar] [CrossRef]
- Siletsky, S.A. Steps of the coupled charge translocation in the catalytic cycle of cytochrome c oxidase. Front. Biosci. 2013, 18, 36–57. [Google Scholar] [CrossRef]
- Borisov, V.B.; Siletsky, S.A.; Paiardini, A.; Hoogewijs, D.; Forte, E.; Giuffrè, A.; Poole, R.K. Bacterial Oxidases of the Cytochrome bd Family: Redox Enzymes of Unique Structure, Function, and Utility As Drug Targets. Antioxid. Redox Signal. 2021, 34, 1280–1318. [Google Scholar] [CrossRef]
- Murali, R.; Gennis, R.B.; Hemp, J. Evolution of the cytochrome bd oxygen reductase superfamily and the function of CydAA’ in Archaea. ISME J. 2021, 1–15. [Google Scholar] [CrossRef]
- Poole, R.K.; Cook, G.M. Redundancy of aerobic respiratory chains in bacteria? Routes, reasons and regulation. Adv. Microb. Physiol. 2000, 43, 165–224. [Google Scholar] [CrossRef]
- Ingledew, W.J.; Poole, R.K. The respiratory chains of Escherichia coli. Microbiol. Rev. 1984, 48, 222–271. [Google Scholar]
- Siletsky, S.A.; Borisov, V.B.; Mamedov, M.D. Photosystem II and terminal respiratory oxidases molecular machines operating in opposite directions. Front. Biosci. 2017, 22, 1379–1426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malatesta, F.; Antonini, G.; Sarti, P.; Brunori, M. Structure and function of a molecular machine: Cytochrome c oxidase. Biophys. Chem. 1995, 54, 1–33. [Google Scholar] [CrossRef]
- Melo, A.M.; Teixeira, M. Supramolecular organization of bacterial aerobic respiratory chains: From cells and back. Biochim. Biophys. Acta Bioenerg. 2016, 1857, 190–197. [Google Scholar] [CrossRef]
- Pereira, M.M.; Sousa, F.L.; Veríssimo, A.F.; Teixeira, M. Looking for the minimum common denominator in haem–copper oxygen reductases: Towards a unified catalytic mechanism. Biochim. Biophys. Acta Bioenerg. 2008, 1777, 929–934. [Google Scholar] [CrossRef] [Green Version]
- Papa, S.; Capitanio, N.; Capitanio, G.; Palese, L.L. Protonmotive cooperativity in cytochrome c oxidase. Biochim. Biophys. Acta Bioenerg. 2004, 1658, 95–105. [Google Scholar] [CrossRef] [Green Version]
- Borisov, V.B.; Siletsky, S.A. Features of Organization and Mechanism of Catalysis of Two Families of Terminal Oxidases: Heme-Copper and bd-Type. Biochemistry (Moscow) 2019, 84, 1390–1402. [Google Scholar] [CrossRef] [PubMed]
- Hederstedt, L. Molecular Biology of Bacillus subtilis Cytochromes anno 2020. Biochemistry (Moscow) 2021, 86, 8–21. [Google Scholar] [CrossRef]
- Borisov, V.B.; Siletsky, S.A.; Nastasi, M.R.; Forte, E. ROS Defense Systems and Terminal Oxidases in Bacteria. Antioxidants 2021, 10, 839. [Google Scholar] [CrossRef]
- Sousa, F.L.; Alves, R.J.; Ribeiro, M.A.; Pereira-Leal, J.B.; Teixeira, M.; Pereira, M.M. The superfamily of heme–copper oxygen reductases: Types and evolutionary considerations. Biochim. Biophys. Acta Bioenerg. 2011, 1817, 629–637. [Google Scholar] [CrossRef] [Green Version]
- Wikström, M.; Krab, K.; Sharma, V. Oxygen Activation and Energy Conservation by Cytochrome c Oxidase. Chem. Rev. 2018, 118, 2469–2490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Capitanio, N.; Palese, L.L.; Capitanio, G.; Martino, P.L.; Richter, O.-M.H.; Ludwig, B.; Papa, S. Allosteric interactions and proton conducting pathways in proton pumping aa3 oxidases: Heme a as a key coupling element. Biochim. Biophys. Acta Bioenerg. 2011, 1817, 558–566. [Google Scholar] [CrossRef] [Green Version]
- Maneg, O.; Malatesta, F.; Ludwig, B.; Drosou, V. Interaction of cytochrome c with cytochrome oxidase: Two different docking scenarios. Biochim. Biophys. Acta Bioenerg. 2004, 1655, 274–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Ballmoos, C.; Ädelroth, P.; Gennis, R.B.; Brzezinski, P. Proton transfer in ba3 cytochrome c oxidase from Thermus thermophilus. Biochim. Biophys. Acta Bioenerg. 2012, 1817, 650–657. [Google Scholar] [CrossRef] [Green Version]
- Rich, P.R. Mitochondrial cytochrome c oxidase: Catalysis, coupling and controversies. Biochem. Soc. Trans. 2017, 45, 813–829. [Google Scholar] [CrossRef]
- Yoshikawa, S.; Shimada, A. Reaction Mechanism of Cytochrome c Oxidase. Chem. Rev. 2015, 115, 1936–1989. [Google Scholar] [CrossRef] [PubMed]
- Forte, E.; Giuffrè, A.; Huang, L.S.; Berry, E.A.; Borisov, V.B. Nitric Oxide Does Not Inhibit but Is Metabolized by the Cytochrome bcc-aa3 Supercomplex. Int. J. Mol. Sci. 2020, 21, 8521. [Google Scholar] [CrossRef] [PubMed]
- Wikstrom, M.K.F. Proton pump coupled to cytochrome c oxidase in mitochondria. Nature 1977, 266, 271–273. [Google Scholar] [CrossRef] [PubMed]
- Ter Beek, J.; Krause, N.; Ädelroth, P. Investigating the Proton Donor in the NO Reductase from Paracoccus denitrificans. PLoS ONE 2016, 11, e0152745. [Google Scholar] [CrossRef] [PubMed]
- Borisov, V.B.; Verkhovsky, M.I. Oxygen as Acceptor. EcoSal Plus 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Siletsky, S.A.; Belevich, I.; Jasaitis, A.; Konstantinov, A.A.; Wikström, M.; Soulimane, T.; Verkhovsky, M.I. Time-resolved single-turnover of ba3 oxidase from Thermus thermophilus. Biochim. Biophys. Acta Bioenerg. 2007, 1767, 1383–1392. [Google Scholar] [CrossRef] [Green Version]
- Siletsky, S.A.; Belevich, I.; Belevich, N.P.; Soulimane, T.; Wikström, M. Time-resolved generation of membrane potential by ba3 cytochrome c oxidase from Thermus thermophilus coupled to single electron injection into the O and OH states. Biochim. Biophys. Acta Bioenerg. 2017, 1858, 915–926. [Google Scholar] [CrossRef]
- Lyons, J.; Aragao, D.; Slattery, O.; Pisliakov, A.; Soulimane, T.; Caffrey, M. Structural insights into electron transfer in caa3-type cytochrome oxidase. Nature 2012, 487, 514–518. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Han, L.; Vallese, F.; Ding, Z.; Choi, S.K.; Hong, S.; Luo, Y.; Liu, B.; Chan, C.K.; Tajkhorshid, E.; et al. Cryo-EM structures of Escherichia coli cytochrome bo3 reveal bound phospholipids and ubiquinone-8 in a dynamic substrate binding site. Proc. Natl. Acad. Sci. USA 2021, 118. [Google Scholar] [CrossRef]
- Buschmann, S.; Warkentin, E.; Xie, H.; Langer, J.D.; Ermler, U.; Michel, H. The Structure of cbb3 Cytochrome Oxidase Provides Insights into Proton Pumping. Science 2010, 329, 327–330. [Google Scholar] [CrossRef]
- Andersson, R.; Safari, C.; Dods, R.; Nango, E.; Tanaka, R.; Yamashita, A.; Nakane, T.; Tono, K.; Joti, Y.; Båth, P.; et al. Serial femtosecond crystallography structure of cytochrome c oxidase at room temperature. Sci. Rep. 2017, 7, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Safarian, S.; Rajendran, C.; Müller, H.; Preu, J.; Langer, J.D.; Ovchinnikov, S.; Hirose, T.; Kusumoto, T.; Sakamoto, J.; Michel, H. Structure of a bd oxidase indicates similar mechanisms for membrane-integrated oxygen reductases. Science 2016, 352, 583–586. [Google Scholar] [CrossRef] [Green Version]
- Safarian, S.; Hahn, A.; Mills, D.J.; Radloff, M.; Eisinger, M.L.; Nikolaev, A.; Meier-Credo, J.; Melin, F.; Miyoshi, H.; Gennis, R.B.; et al. Active site rearrangement and structural divergence in prokaryotic respiratory oxidases. Science 2019, 366, 100–104. [Google Scholar] [CrossRef] [PubMed]
- Theßeling, A.; Rasmussen, T.; Burschel, S.; Wohlwend, D.; Kägi, J.; Müller, R.; Böttcher, B.; Friedrich, T. Homologous bd oxidases share the same architecture but differ in mechanism. Nat. Commun. 2019, 10, 5138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, J.J.; Alben, J.O.; Gennis, R.B. Spectroscopic evidence for a heme-heme binuclear center in the cytochrome bd ubiquinol oxidase from Escherichia coli. Proc. Natl. Acad. Sci. USA 1993, 90, 5863–5867. [Google Scholar] [CrossRef] [Green Version]
- Tsubaki, M.; Hori, H.; Mogi, T.; Anraku, Y. Cyanide-binding Site of bd-type Ubiquinol Oxidase from Escherichia coli. J. Biol. Chem. 1995, 270, 28565–28569. [Google Scholar] [CrossRef] [Green Version]
- Borisov, V.; Arutyunyan, A.M.; Osborne, J.P.; Gennis, R.B.; Konstantinov, A.A. Magnetic Circular Dichroism Used To Examine the Interaction of Escherichia coli Cytochrome bd with Ligands. Biochemistry 1998, 38, 740–750. [Google Scholar] [CrossRef]
- Vos, M.H.; Borisov, V.B.; Liebl, U.; Martin, J.L.; Konstantinov, A.A. Femtosecond resolution of ligand-heme interactions in the high-affinity quinol oxidase bd: A di-heme active site? Proc. Natl. Acad. Sci. USA 2000, 97, 1554–1559. [Google Scholar] [CrossRef] [Green Version]
- Borisov, V.B.; Sedelnikova, S.E.; Poole, R.K.; Konstantinov, A.A. Interaction of Cytochrome bd with Carbon Monoxide at Low and Room Temperatures. J. Biol. Chem. 2001, 276, 22095–22099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borisov, V.B.; Liebl, U.; Rappaport, F.; Martin, J.-L.; Zhang, J.; Gennis, R.B.; Konstantinov, A.A.; Vos, M. Interactions between Heme d and Heme b595 in Quinol Oxidase bd from Escherichia coli: A Photoselection Study Using Femtosecond Spectroscopy. Biochemistry 2002, 41, 1654–1662. [Google Scholar] [CrossRef] [Green Version]
- Arutyunyan, A.M.; Borisov, V.B.; Novoderezhkin, V.; Ghaim, J.; Zhang, J.; Gennis, R.B.; Konstantinov, A.A. Strong Excitonic Interactions in the Oxygen-Reducing Site of bd-Type Oxidase: The Fe-to-Fe Distance between Hemes d and b595 is 10 Å. Biochemistry 2008, 47, 1752–1759. [Google Scholar] [CrossRef] [PubMed]
- Borisov, V.B. Interaction of bd-type quinol oxidase from Escherichia coli and carbon monoxide: Heme d binds CO with high affinity. Biochemistry (Moscow) 2008, 73, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Bloch, D.A.; Borisov, V.B.; Mogi, T.; Verkhovsky, M.I. Heme/heme redox interaction and resolution of individual optical absorption spectra of the hemes in cytochrome bd from Escherichia coli. Biochim. Biophys. Acta Bioenerg. 2009, 1787, 1246–1253. [Google Scholar] [CrossRef] [Green Version]
- Rappaport, F.; Zhang, J.; Vos, M.H.; Gennis, R.B.; Borisov, V.B. Heme–heme and heme–ligand interactions in the di-heme oxygen-reducing site of cytochrome bd from Escherichia coli revealed by nanosecond absorption spectroscopy. Biochim. Biophys. Acta Bioenerg. 2010, 1797, 1657–1664. [Google Scholar] [CrossRef]
- Borisov, V.B.; Verkhovsky, M.I. Accommodation of CO in the di-heme active site of cytochrome bd terminal oxidase from Escherichia coli. J. Inorg. Biochem. 2013, 118, 65–67. [Google Scholar] [CrossRef]
- Siletsky, S.A.; Zaspa, A.A.; Poole, R.K.; Borisov, V.B. Microsecond Time-Resolved Absorption Spectroscopy Used to Study CO Compounds of Cytochrome bd from Escherichia coli. PLoS ONE 2014, 9, e95617. [Google Scholar] [CrossRef] [PubMed]
- Siletsky, S.A.; Rappaport, F.; Poole, R.K.; Borisov, V.B. Evidence for Fast Electron Transfer between the High-Spin Haems in Cytochrome bd-I from Escherichia coli. PLoS ONE 2016, 11, e0155186. [Google Scholar] [CrossRef]
- Siletsky, S.A.; Dyuba, A.V.; Elkina, D.A.; Monakhova, M.V.; Borisov, V.B. Spectral-kinetic analysis of recombination reaction of heme centers of bd-type quinol oxidase from Escherichia coli with carbon monoxide. Biochemistry (Moscow) 2017, 82, 1354–1366. [Google Scholar] [CrossRef]
- Azarkina, N.; Borisov, V.; Konstantinov, A.A. Spontaneous spectral changes of the reduced cytochrome bd. FEBS Lett. 1997, 416, 171–174. [Google Scholar] [CrossRef] [Green Version]
- Azarkina, N.; Siletsky, S.; Borisov, V.; von Wachenfeldt, C.; Hederstedt, L.; Konstantinov, A.A. A Cytochrome bb′-type Quinol Oxidase in Bacillus subtilis Strain 168. J. Biol. Chem. 1999, 274, 32810–32817. [Google Scholar] [CrossRef] [Green Version]
- Pereira, M.M.; Santana, M.; Teixeira, M. A novel scenario for the evolution of haem–copper oxygen reductases. Biochim. Biophys. Acta Bioenerg. 2001, 1505, 185–208. [Google Scholar] [CrossRef] [Green Version]
- Pereira, M.M.; Gomes, C.M.; Teixeira, M. Plasticity of proton pathways in haem-copper oxygen reductases. FEBS Lett. 2002, 522, 14–18. [Google Scholar] [CrossRef] [Green Version]
- Pereira, M.M.; Teixeira, M. Proton pathways, ligand binding and dynamics of the catalytic site in haem-copper oxygen reductases: A comparison between the three families. Biochim. Biophys. Acta Bioenerg. 2004, 1655, 340–346. [Google Scholar] [CrossRef] [Green Version]
- Tsukihara, T.; Aoyama, H.; Yamashita, E.; Tomizaki, T.; Yamaguchi, H.; Shinzawa-Itoh, K.; Nakashima, R.; Yaono, R.; Yoshikawa, S. The Whole Structure of the 13-Subunit Oxidized Cytochrome c Oxidase at 2.8 A. Science 1996, 272, 1136–1144. [Google Scholar] [CrossRef] [PubMed]
- Yano, N.; Muramoto, K.; Shimada, A.; Takemura, S.; Baba, J.; Fujisawa, H.; Mochizuki, M.; Shinzawa-Itoh, K.; Yamashita, E.; Tsukihara, T.; et al. The Mg2+-containing Water Cluster of Mammalian Cytochrome c Oxidase Collects Four Pumping Proton Equivalents in Each Catalytic Cycle. J. Biol. Chem. 2016, 291, 23882–23894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimada, A.; Etoh, Y.; Kitoh-Fujisawa, R.; Sasaki, A.; Shinzawa-Itoh, K.; Hiromoto, T.; Yamashita, E.; Muramoto, K.; Tsukihara, T.; Yoshikawa, S. X-ray structures of catalytic intermediates of cytochrome c oxidase provide insights into its O2 activation and unidirectional proton-pump mechanisms. J. Biol. Chem. 2020, 295, 5818–5833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimada, A.; Hara, F.; Shinzawa-Itoh, K.; Kanehisa, N.; Yamashita, E.; Muramoto, K.; Tsukihara, T.; Yoshikawa, S. Critical roles of the CuB site in efficient proton pumping as revealed by crystal structures of mammalian cytochrome c oxidase catalytic intermediates. J. Biol. Chem. 2021, 297, 100967. [Google Scholar] [CrossRef]
- Koepke, J.; Olkhova, E.; Angerer, H.; Müller, H.; Peng, G.; Michel, H. High resolution crystal structure of Paracoccus denitrificans cytochrome c oxidase: New insights into the active site and the proton transfer pathways. Biochim. Biophys. Acta Bioenerg. 2009, 1787, 635–645. [Google Scholar] [CrossRef] [Green Version]
- Svensson-Ek, M.; Abramson, J.; Larsson, G.; Törnroth-Horsefield, S.; Brzezinski, P.; Iwata, S. The X-ray Crystal Structures of Wild-type and EQ(I-286) Mutant Cytochrome c Oxidases from Rhodobacter sphaeroides. J. Mol. Biol. 2002, 321, 329–339. [Google Scholar] [CrossRef]
- Qin, L.; Liu, J.; Mills, D.A.; Proshlyakov, D.A.; Hiser, C.; Ferguson-Miller, S. Redox-Dependent Conformational Changes in Cytochrome c Oxidase Suggest a Gating Mechanism for Proton Uptake. Biochemistry 2009, 48, 5121–5130. [Google Scholar] [CrossRef] [Green Version]
- Abramson, J.; Riistama, S.; Larsson, G.; Jasaitis, A.; Svensson-Ek, M.; Laakkonen, L.; Puustinen, A.; Iwata, S.; Wikström, M. The structure of the ubiquinol oxidase from Escherichia coli and its ubiquinone binding site. Nat. Genet. 2000, 7, 910–917. [Google Scholar] [CrossRef]
- Kruse, F.; Nguyen, A.D.; Dragelj, J.; Heberle, J.; Hildebrandt, P.; Mroginski, M.A.; Weidinger, I.M. A Resonance Raman Marker Band Characterizes the Slow and Fast Form of Cytochrome c Oxidase. J. Am. Chem. Soc. 2021, 143, 2769–2776. [Google Scholar] [CrossRef]
- Yano, N.; Muramoto, K.; Mochizuki, M.; Shinzawa-Itoh, K.; Yamashita, E.; Yoshikawa, S.; Tsukihara, T. X-ray structure of cyanide-bound bovine heart cytochrome c oxidase in the fully oxidized state at 2.0 Å resolution. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2015, 71, 726–730. [Google Scholar] [CrossRef] [Green Version]
- Letts, J.; Fiedorczuk, K.; Sazanov, L. The architecture of respiratory supercomplexes. Nature 2016, 537, 644–648. [Google Scholar] [CrossRef] [PubMed]
- Shinzawa-Itoh, K.; Sugimura, T.; Misaki, T.; Tadehara, Y.; Yamamoto, S.; Hanada, M.; Yano, N.; Nakagawa, T.; Uene, S.; Yamada, T.; et al. Monomeric structure of an active form of bovine cytochrome c oxidase. Proc. Natl. Acad. Sci. USA 2019, 116, 19945–19951. [Google Scholar] [CrossRef] [Green Version]
- Wikström, M.; Sharma, V. Proton pumping by cytochrome c oxidase—A 40 year anniversary. Biochim. Biophys. Acta Bioenerg. 2018, 1859, 692–698. [Google Scholar] [CrossRef] [PubMed]
- Konstantinov, A.A.; Siletsky, S.; Mitchell, D.; Kaulen, A.; Gennis, R.B. The roles of the two proton input channels in cytochrome c oxidase from Rhodobacter sphaeroides probed by the effects of site-directed mutations on time-resolved electrogenic intraprotein proton transfer. Proc. Natl. Acad. Sci. USA 1997, 94, 9085–9090. [Google Scholar] [CrossRef] [Green Version]
- Siletsky, S.A.; Pawate, A.S.; Weiss, K.; Gennis, R.B.; Konstantinov, A.A. Transmembrane Charge Separation during the Ferryl-oxo → Oxidized Transition in a Nonpumping Mutant of Cytochrome c Oxidase. J. Biol. Chem. 2004, 279, 52558–52565. [Google Scholar] [CrossRef] [Green Version]
- Siletsky, S.A.; Zhu, J.; Gennis, R.B.; Konstantinov, A.A. Partial Steps of Charge Translocation in the Nonpumping N139L Mutant of Rhodobacter sphaeroides Cytochrome c Oxidase with a Blocked D-Channel. Biochemistry 2010, 49, 3060–3073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vilhjálmsdóttir, J.; Albertsson, I.; Blomberg, M.R.A.; Ädelroth, P.; Brzezinski, P. Proton transfer in uncoupled variants of cytochrome c oxidase. FEBS Lett. 2019, 594, 813–822. [Google Scholar] [CrossRef]
- Berg, J.; Liu, J.; Svahn, E.; Ferguson-Miller, S.; Brzezinski, P. Structural changes at the surface of cytochrome c oxidase alter the proton-pumping stoichiometry. Biochim. Biophys. Acta Bioenerg. 2020, 1861, 148116. [Google Scholar] [CrossRef]
- Siletsky, S.A.; Soulimane, T.; Belevich, I.; Gennis, R.B.; Wikström, M. Specific inhibition of proton pumping by the T315V mutation in the K channel of cytochrome ba3 from Thermus thermophilus. Biochim. Biophys. Acta Bioenerg. 2021, 1862, 148450. [Google Scholar] [CrossRef]
- Blomberg, M.R.A. Role of the Two Metals in the Active Sites of Heme Copper Oxidases–A Study of NO Reduction in cbb3 Cytochrome c Oxidase. Inorg. Chem. 2020, 59, 11542–11553. [Google Scholar] [CrossRef]
- Maréchal, A.; Xu, J.-Y.; Genko, N.; Hartley, A.M.; Haraux, F.; Meunier, B.; Rich, P.R. A common coupling mechanism for A-type heme-copper oxidases from bacteria to mitochondria. Proc. Natl. Acad. Sci. USA 2020, 117, 9349–9355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borisov, V.B.; Gennis, R.B.; Hemp, J.; Verkhovsky, M.I. The cytochrome bd respiratory oxygen reductases. Biochim. Biophys. Acta Bioenerg. 2011, 1807, 1398–1413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arutyunyan, A.M.; Sakamoto, J.; Inadome, M.; Kabashima, Y.; Borisov, V.B. Optical and magneto-optical activity of cytochrome bd from Geobacillus thermodenitrificans. Biochim. Biophys. Acta Bioenerg. 2012, 1817, 2087–2094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borisov, V.B. Cytochrome bd: Structure and properties. Biochemistry (Moscow) 1996, 61, 565–574. [Google Scholar]
- Jünemann, S. Cytochrome bd terminal oxidase. Biochim. Biophys. Acta Bioenerg. 1997, 1321, 107–127. [Google Scholar] [CrossRef] [Green Version]
- Forte, E.; Borisov, V.B.; Vicente, J.B.; Giuffrè, A. Cytochrome bd and Gaseous Ligands in Bacterial Physiology. Adv. Microb. Physiol. 2017, 71, 171–234. [Google Scholar] [CrossRef]
- Wang, W.; Gao, Y.; Tang, Y.; Zhou, X.; Lai, Y.; Zhou, S.; Zhang, Y.; Yang, X.; Liu, F.; Guddat, L.W.; et al. Cryo-EM structure of mycobacterial cytochrome bd reveals two oxygen access channels. Nat. Commun. 2021, 12, 1–8. [Google Scholar] [CrossRef]
- Jasaitis, A.; Borisov, V.B.; Belevich, N.P.; Morgan, J.E.; Konstantinov, A.A.; Verkhovsky, M.I. Electrogenic Reactions of Cytochrome bd. Biochemistry 2000, 39, 13800–13809. [Google Scholar] [CrossRef] [PubMed]
- Belevich, I.; Borisov, V.B.; Zhang, J.; Yang, K.; Konstantinov, A.A.; Gennis, R.B.; Verkhovsky, M.I. Time-resolved electrometric and optical studies on cytochrome bd suggest a mechanism of electron-proton coupling in the di-heme active site. Proc. Natl. Acad. Sci. USA 2005, 102, 3657–3662. [Google Scholar] [CrossRef] [Green Version]
- Forte, E.; Borisov, V.B.; Konstantinov, A.A.; Brunori, M.; Giuffrè, A.; Sarti, P. Cytochrome bd, a key oxidase in bacterial survival and tolerance to nitrosative stress. Ital. J. Biochem. 2007, 56. [Google Scholar]
- Borisov, V.B.; Forte, E.; Siletsky, S.A.; Arese, M.; Davletshin, A.I.; Sarti, P.; Giuffrè, A. Cytochrome bd protects bacteria against oxidative and nitrosative stress: A potential target for next-generation antimicrobial agents. Biochemistry (Moscow) 2015, 80, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Giuffrè, A.; Borisov, V.B.; Mastronicola, D.; Sarti, P.; Forte, E. Cytochrome bd oxidase and nitric oxide: From reaction mechanisms to bacterial physiology. FEBS Lett. 2011, 586, 622–629. [Google Scholar] [CrossRef]
- Giuffrè, A.; Borisov, V.B.; Arese, M.; Sarti, P.; Forte, E. Cytochrome bd oxidase and bacterial tolerance to oxidative and nitrosative stress. Biochim. Biophys. Acta Bioenerg. 2014, 1837, 1178–1187. [Google Scholar] [CrossRef] [Green Version]
- Gavrikova, E.V.; Grivennikova, V.G.; Borisov, V.B.; Cecchini, G.; Vinogradov, A.D. Assembly of a chimeric respiratory chain from bovine heart submitochondrial particles and cytochrome bd terminal oxidase of Escherichia coli. FEBS Lett. 2009, 583, 1287–1291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Belevich, I.; Borisov, V.B.; Verkhovsky, M.I. Discovery of the True Peroxy Intermediate in the Catalytic Cycle of Terminal Oxidases by Real-time Measurement. J. Biol. Chem. 2007, 282, 28514–28519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borisov, V.B.; Belevich, I.; Bloch, D.A.; Mogi, T.; Verkhovsky, M.I. Glutamate 107 in Subunit I of Cytochrome bd from Escherichia coli Is Part of a Transmembrane Intraprotein Pathway Conducting Protons from the Cytoplasm to the Heme b595/Heme d Active Site. Biochemistry 2008, 47, 7907–7914. [Google Scholar] [CrossRef]
- Borisov, V.B.; Murali, R.; Verkhovskaya, M.; Bloch, D.A.; Han, H.; Gennis, R.B.; Verkhovsky, M.I. Aerobic respiratory chain of Escherichia coli is not allowed to work in fully uncoupled mode. Proc. Natl. Acad. Sci. USA 2011, 108, 17320–17324. [Google Scholar] [CrossRef] [Green Version]
- Siletsky, S.A.; Gennis, R.B. Time-Resolved Electrometric Study of the F→O Transition in Cytochrome c Oxidase. The Effect of Zn2+ Ions on the Positive Side of the Membrane. Biochemistry (Moscow) 2021, 86, 105–122. [Google Scholar] [CrossRef]
- Borisov, V.B.; Forte, E.; Konstantinov, A.A.; Poole, R.K.; Sarti, P.; Giuffrè, A. Interaction of the bacterial terminal oxidase cytochrome bd with nitric oxide. FEBS Lett. 2004, 576, 201–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borisov, V.B.; Forte, E.; Sarti, P.; Brunori, M.; Konstantinov, A.A.; Giuffrè, A. Nitric oxide reacts with the ferryl-oxo catalytic intermediate of the CuB-lacking cytochrome bd terminal oxidase. FEBS Lett. 2006, 580, 4823–4826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borisov, V.B.; Forte, E.; Sarti, P.; Brunori, M.; Konstantinov, A.A.; Giuffrè, A. Redox control of fast ligand dissociation from Escherichia coli cytochrome bd. Biochem. Biophys. Res. Commun. 2007, 355, 97–102. [Google Scholar] [CrossRef]
- Mason, M.G.; Shepherd, M.; Nicholls, P.J.; Dobbin, P.S.; Dodsworth, K.S.; Poole, R.K.; Cooper, C. Cytochrome bd confers nitric oxide resistance to Escherichia coli. Nat. Chem. Biol. 2008, 5, 94–96. [Google Scholar] [CrossRef]
- Borisov, V.B.; Forte, E.; Giuffrè, A.; Konstantinov, A.; Sarti, P. Reaction of nitric oxide with the oxidized di-heme and heme–copper oxygen-reducing centers of terminal oxidases: Different reaction pathways and end-products. J. Inorg. Biochem. 2009, 103, 1185–1187. [Google Scholar] [CrossRef] [PubMed]
- Shepherd, M.; Achard, M.E.S.; Idris, A.; Totsika, M.; Phan, M.-D.; Peters, K.M.; Sarkar, S.; Ribeiro, C.; Holyoake, L.V.; Ladakis, D.; et al. The cytochrome bd-I respiratory oxidase augments survival of multidrug-resistant Escherichia coli during infection. Sci. Rep. 2016, 6, 35285. [Google Scholar] [CrossRef]
- Holyoake, L.V.; Hunt, S.; Sanguinetti, G.; Cook, G.M.; Howard, M.; Rowe, M.L.; Poole, R.K.; Shepherd, M. CydDC-mediated reductant export in Escherichia coli controls the transcriptional wiring of energy metabolism and combats nitrosative stress. Biochem. J. 2016, 473, 693–701. [Google Scholar] [CrossRef] [Green Version]
- Jones-Carson, J.; HUSA in, M.; Liu, L.; Orlicky, D.J.; Vázquez-Torres, A. Cytochrome bd-Dependent Bioenergetics and Antinitrosative Defenses in Salmonella Pathogenesis. mBio 2016, 7, e02052-16. [Google Scholar] [CrossRef] [Green Version]
- Meng, Q.; Yin, J.; Jin, M.; Gao, H. Distinct Nitrite and Nitric Oxide Physiologies in Escherichia coli and Shewanella oneidensis. Appl. Environ. Microbiol. 2018, 84. [Google Scholar] [CrossRef] [Green Version]
- Beebout, C.; Eberly, A.R.; Werby, S.H.; Reasoner, S.; Brannon, J.; De, S.; Fitzgerald, M.; Huggins, M.M.; Clayton, D.B.; Cegelski, L.; et al. Respiratory Heterogeneity Shapes Biofilm Formation and Host Colonization in Uropathogenic Escherichia coli. mBio 2019, 10, e02400-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borisov, V.B.; Forte, E.; Siletsky, S.A.; Sarti, P.; Giuffrè, A. Cytochrome bd from Escherichia coli catalyzes peroxynitrite decomposition. Biochim. Biophys. Acta Bioenerg. 2015, 1847, 182–188. [Google Scholar] [CrossRef] [Green Version]
- Borisov, V.B.; Davletshin, A.I.; Konstantinov, A.A. Peroxidase activity of cytochrome bd from Escherichia coli. Biochemistry (Moscow) 2010, 75, 428–436. [Google Scholar] [CrossRef]
- Borisov, V.B.; Forte, E.; Davletshin, A.; Mastronicola, D.; Sarti, P.; Giuffrè, A. Cytochrome bd oxidase from Escherichia coli displays high catalase activity: An additional defense against oxidative stress. FEBS Lett. 2013, 587, 2214–2218. [Google Scholar] [CrossRef] [PubMed]
- Forte, E.; Borisov, V.B.; Davletshin, A.; Mastronicola, D.; Sarti, P.; Giuffrè, A.; Planet, P.J.; LaRussa, S.J.; Dana, A.; Smith, H.; et al. Cytochrome bd Oxidase and Hydrogen Peroxide Resistance in Mycobacterium tuberculosis. mBio 2013, 4, e01006-13. [Google Scholar] [CrossRef] [Green Version]
- Al-Attar, S.; Yu, Y.; Pinkse, M.; Hoeser, J.; Friedrich, T.; Bald, D.; De Vries, S. Cytochrome bd Displays Significant Quinol Peroxidase Activity. Sci. Rep. 2016, 6, 27631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kita, K.; Konishi, K.; Anraku, Y. Terminal oxidases of Escherichia coli aerobic respiratory chain. II. Purification and properties of cytochrome b558-d complex from cells grown with limited oxygen and evidence of branched electron-carrying systems. J. Biol. Chem. 1984, 259, 3375–3381. [Google Scholar] [CrossRef]
- Sakamoto, J.; Koga, E.; Mizuta, T.; Sato, C.; Noguchi, S.; Sone, N. Gene structure and quinol oxidase activity of a cytochrome bd-type oxidase from Bacillus stearothermophilus. Biochim. Biophys. Acta Bioenerg. 1999, 1411, 147–158. [Google Scholar] [CrossRef] [Green Version]
- Forte, E.; Borisov, V.B.; Falabella, M.; Colaço, H.; Tinajero-Trejo, M.; Poole, R.K.; Vicente, J.; Sarti, P.; Giuffrè, A. The Terminal Oxidase Cytochrome bd Promotes Sulfide-resistant Bacterial Respiration and Growth. Sci. Rep. 2016, 6, 23788. [Google Scholar] [CrossRef] [Green Version]
- Korshunov, S.; Imlay, K.R.C.; Imlay, J.A. The cytochrome bd oxidase of Escherichia coli prevents respiratory inhibition by endogenous and exogenous hydrogen sulfide. Mol. Microbiol. 2016, 101, 62–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forte, E.; Giuffrè, A. How bacteria breathe in hydrogen sulfide-rich environments. Biochemist 2016, 38, 8–11. [Google Scholar] [CrossRef]
- Borisov, V.B.; Forte, E. Terminal Oxidase Cytochrome bd Protects Bacteria Against Hydrogen Sulfide Toxicity. Biochemistry (Moscow) 2021, 86, 22–32. [Google Scholar] [CrossRef]
- Forte, E.; Siletsky, S.A.; Borisov, V.B. In Escherichia coli Ammonia Inhibits Cytochrome bo3 But Activates Cytochrome bd-I. Antioxidants 2020, 10, 13. [Google Scholar] [CrossRef]
- Mascolo, L.; Bald, D. Cytochrome bd in Mycobacterium tuberculosis: A respiratory chain protein involved in the defense against antibacterials. Prog. Biophys. Mol. Biol. 2019, 152, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.S.; Sviriaeva, E.; Pethe, K. Targeting the cytochrome oxidases for drug development in mycobacteria. Prog. Biophys. Mol. Biol. 2020, 152, 45–54. [Google Scholar] [CrossRef]
- Cook, G.M.; Hards, K.; Dunn, E.; Heikal, A.; Nakatani, Y.; Greening, C.; Crick, D.C.; Fontes, F.L.; Pethe, K.; Hasenoehrl, E.; et al. Oxidative Phosphorylation as a Target Space for Tuberculosis: Success, Caution, and Future Directions. Microbiol. Spectr. 2017, 5. [Google Scholar] [CrossRef] [Green Version]
- Bald, D.; Villellas, C.; Lu, P.; Koul, A. Targeting Energy Metabolism in Mycobacterium tuberculosis, a New Paradigm in Antimycobacterial Drug Discovery. mBio 2017, 8, e00272-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hards, K.; Cook, G.M. Targeting bacterial energetics to produce new antimicrobials. Drug Resist. Updat. 2018, 36, 1–12. [Google Scholar] [CrossRef]
- Winstedt, L.; Frankenberg, L.; Hederstedt, L.; von Wachenfeldt, C. Enterococcus faecalis V583 Contains a Cytochrome bd-Type Respiratory Oxidase. J. Bacteriol. 2000, 182, 3863–3866. [Google Scholar] [CrossRef] [Green Version]
- Siletsky, S.A.; Konstantinov, A.A. Cytochrome c oxidase: Charge translocation coupled to single-electron partial steps of the catalytic cycle. Biochim. Biophys. Acta Bioenerg. 2012, 1817, 476–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshikawa, S.; Shinzawa-Itoh, K.; Nakashima, R.; Yaono, R.; Yamashita, E.; Inoue, N.; Yao, M.; Fei, M.J.; Libeu, C.P.; Mizushima, T.; et al. Redox-Coupled Crystal Structural Changes in Bovine Heart Cytochrome c Oxidase. Science 1998, 280, 1723–1729. [Google Scholar] [CrossRef] [Green Version]
- Buse, G.; Soulimane, T.; Dewor, M.; Meyer, H.E.; Blüggel, M. Evidence for a copper-coordinated histidine-tyrosine cross-link in the active site of cytochrome oxidase. Protein Sci. 1999, 8, 985–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rauhamaki, V.; Baumann, M.; Soliymani, R.; Puustinen, A.; Wikstrom, M. Identification of a histidine-tyrosine cross-link in the active site of the cbb3-type cytochrome c oxidase from Rhodobacter sphaeroides. Proc. Natl. Acad. Sci. USA 2006, 103, 16135–16140. [Google Scholar] [CrossRef] [Green Version]
- Borisov, V.B.; Forte, E.; Sarti, P.; Giuffrè, A. Catalytic intermediates of cytochrome bd terminal oxidase at steady-state: Ferryl and oxy-ferrous species dominate. Biochim. Biophys. Acta Bioenerg. 2011, 1807, 503–509. [Google Scholar] [CrossRef] [PubMed]
- Paulus, A.; Rossius, S.G.H.; Dijk, M.; de Vries, S. Oxoferryl-Porphyrin Radical Catalytic Intermediate in Cytochrome bd Oxidases Protects Cells from Formation of Reactive Oxygen Species. J. Biol. Chem. 2012, 287, 8830–8838. [Google Scholar] [CrossRef] [Green Version]
- Belevich, I.; Bloch, D.A.; Wikstrom, M.; Verkhovsky, M.I. Exploring the proton pump mechanism of cytochrome c oxidase in real time. Proc. Natl. Acad. Sci. USA 2007, 104, 2685–2690. [Google Scholar] [CrossRef] [Green Version]
- Bloch, D.; Belevich, I.; Jasaitis, A.; Ribacka, C.; Puustinen, A.; Verkhovsky, M.I.; Wikstrom, M. The catalytic cycle of cytochrome c oxidase is not the sum of its two halves. Proc. Natl. Acad. Sci. USA 2003, 101, 529–533. [Google Scholar] [CrossRef] [Green Version]
- Jancura, D.; Berka, V.; Antalik, M.; Bagelova, J.; Gennis, R.B.; Palmer, G.; Fabian, M. Spectral and Kinetic Equivalence of Oxidized Cytochrome c Oxidase as Isolated and “Activated” by Reoxidation. J. Biol. Chem. 2006, 281, 30319–30325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brand, S.E.; Rajagukguk, S.; Ganesan, K.; Geren, L.; Fabian, M.; Han, D.; Gennis, R.B.; Durham, B.; Millett, F. A New Ruthenium Complex To Study Single-Electron Reduction of the Pulsed OH State of Detergent-Solubilized Cytochrome Oxidase. Biochemistry 2007, 46, 14610–14618. [Google Scholar] [CrossRef] [PubMed]
- Vilhjálmsdóttir, J.; Gennis, R.B.; Brzezinski, P. The electron distribution in the “activated” state of cytochrome c oxidase. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Siletskiy, S.; Soulimane, T.; Azarkina, N.; Vygodina, T.; Buse, G.; Kaulen, A.; Konstantinov, A. Time-resolved generation of a membrane potential by ba3 cytochrome coxidase from Thermus thermophilus. FEBS Lett. 1999, 457, 98–102. [Google Scholar] [CrossRef] [Green Version]
- Siletsky, S.A.; Belevich, I.; Wikström, M.; Soulimane, T.; Verkhovsky, M.I. Time-resolved OH→EH transition of the aberrant ba3 oxidase from Thermus thermophilus. Biochim. Biophys. Acta Bioenerg. 2009, 1787, 201–205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siletsky, S.A.; Belevich, I.; Belevich, N.P.; Soulimane, T.; Verkhovsky, M.I. Time-resolved single-turnover of caa3 oxidase from Thermus thermophilus. Fifth electron of the fully reduced enzyme converts OH into EH state. Biochim. Biophys. Acta Bioenerg. 2011, 1807, 1162–1169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siletsky, S.A.; Belevich, I.; Soulimane, T.; Verkhovsky, M.I.; Wikström, M. The fifth electron in the fully reduced caa3 from Thermus thermophilus is competent in proton pumping. Biochim. Biophys. Acta Bioenerg. 2013, 1827, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Yoshikawa, S.; Muramoto, K.; Shinzawa-Itoh, K. Proton-Pumping Mechanism of Cytochrome c Oxidase. Annu. Rev. Biophys. 2011, 40, 205–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, V.; Karlin, K.D.; Wikström, M. Computational study of the activated OH state in the catalytic mechanism of cytochrome c oxidase. Proc. Natl. Acad. Sci. USA 2013, 110, 16844–16849. [Google Scholar] [CrossRef] [Green Version]
- Luo, F.; Shinzawa-Itoh, K.; Hagimoto, K.; Shimada, A.; Shimada, S.; Yamashita, E.; Yoshikawa, S.; Tsukihara, T. Structure of bovine cytochrome c oxidase crystallized at a neutral pH using a fluorinated detergent. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2017, 73, 416–422. [Google Scholar] [CrossRef] [Green Version]
- Sharma, V.; Wikström, M. The role of the K-channel and the active-site tyrosine in the catalytic mechanism of cytochrome c oxidase. Biochim. Biophys. Acta Bioenerg. 2016, 1857, 1111–1115. [Google Scholar] [CrossRef]
- Blomberg, M.R. Active Site Midpoint Potentials in Different Cytochrome c Oxidase Families: A Computational Comparison. Biochemistry 2019, 58, 2028–2038. [Google Scholar] [CrossRef]
- Yang, K.; Borisov, V.B.; Konstantinov, A.A.; Gennis, R.B. The fully oxidized form of the cytochrome bd quinol oxidase from E. coli does not participate in the catalytic cycle: Direct evidence from rapid kinetics studies. FEBS Lett. 2008, 582, 3705–3709. [Google Scholar] [CrossRef] [Green Version]
- Borisov, V.B.; Smirnova, I.A.; Krasnosel’skaya, I.A.; Konstantinov, A.A. Oxygenated cytochrome bd from Escherichia coli can be converted into the oxidized form by lipophilic electron acceptors. Biochemistry (Moscow) 1994, 59, 437–443. [Google Scholar]
- Blomberg, M.R. The structure of the oxidized state of cytochrome c oxidase-experiments and theory compared. J. Inorg. Biochem. 2020, 206, 111020. [Google Scholar] [CrossRef] [PubMed]
- Blomberg, M.R.A. Mechanism of Oxygen Reduction in Cytochrome c Oxidase and the Role of the Active Site Tyrosine. Biochemistry 2016, 55, 489–500. [Google Scholar] [CrossRef]
- Belevich, I.; Borisov, V.B.; Konstantinov, A.A.; Verkhovsky, M.I. Oxygenated complex of cytochrome bd from Escherichia coli: Stability and photolability. FEBS Lett. 2005, 579, 4567–4570. [Google Scholar] [CrossRef] [Green Version]
- Belevich, I.; Borisov, V.B.; Bloch, D.A.; Konstantinov, A.A.; Verkhovsky, M.I. Cytochrome bd from Azotobacter vinelandii: Evidence for High-Affinity Oxygen Binding. Biochemistry 2007, 46, 11177–11184. [Google Scholar] [CrossRef]
- Szundi, I.; Kittredge, C.; Choi, S.K.; McDonald, W.; Ray, J.; Gennis, R.B.; Einarsdóttir, Ó. Kinetics and Intermediates of the Reaction of Fully Reduced Escherichia coli bo3 Ubiquinol Oxidase with O2. Biochemistry 2014, 53, 5393–5404. [Google Scholar] [CrossRef]
- Koutsoupakis, C.; Soulimane, T.; Varotsis, C. Spectroscopic and Kinetic Investigation of the Fully Reduced and Mixed Valence States of ba3-Cytochrome c Oxidase from Thermus thermophilus. J. Biol. Chem. 2012, 287, 37495–37507. [Google Scholar] [CrossRef] [Green Version]
- Muramoto, K.; Ohta, K.; Shinzawa-Itoh, K.; Kanda, K.; Taniguchi, M.; Nabekura, H.; Yamashita, E.; Tsukihara, T.; Yoshikawa, S. Bovine cytochrome c oxidase structures enable O2 reduction with minimization of reactive oxygens and provide a proton-pumping gate. Proc. Natl. Acad. Sci. USA 2010, 107, 7740–7745. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blomberg, M.R.A.; Siegbahn, P.E.M.; Wikström, M. Metal-Bridging Mechanism for O−O Bond Cleavage in Cytochrome c Oxidase. Inorg. Chem. 2003, 42, 5231–5243. [Google Scholar] [CrossRef] [PubMed]
- Du, W.-G.H.; Götz, A.W.; Yang, L.; Walker, R.C.; Noodleman, L. A broken-symmetry density functional study of structures, energies, and protonation states along the catalytic O–O bond cleavage pathway in ba3 cytochrome c oxidase from Thermus thermophilus. Phys. Chem. Chem. Phys. 2016, 18, 21162–21171. [Google Scholar] [CrossRef] [Green Version]
- Poiana, F.; von Ballmoos, C.; Gonska, N.; Blomberg, M.R.A.; Ädelroth, P.; Brzezinski, P. Splitting of the O–O bond at the heme-copper catalytic site of respiratory oxidases. Sci. Adv. 2017, 3, e1700279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blomberg, M.R.A. Activation of O2 and NO in heme-copper oxidases–mechanistic insights from computational modelling. Chem. Soc. Rev. 2020, 49, 7301–7330. [Google Scholar] [CrossRef]
- Jasaitis, A.; Verkhovsky, M.I.; Morgan, J.E.; Verkhovskaya, M.; Wikström, M. Assignment and Charge Translocation Stoichiometries of the Major Electrogenic Phases in the Reaction of Cytochrome c Oxidase with Dioxygen. Biochemistry 1999, 38, 2697–2706. [Google Scholar] [CrossRef]
- Szundi, I.; Funatogawa, C.; Soulimane, T.; Einarsdóttir, Ó. The Reactions of O2 and NO with Mixed-Valence ba3 Cytochrome c Oxidase from Thermus thermophilus. Biophys. J. 2019, 118, 386–395. [Google Scholar] [CrossRef] [PubMed]
- Poole, R.K.; Kumar, C.; Salmon, I.; Chance, B. The 650 nm Chromophore in Escherichia coli is an ‘Oxy-’ or Oxygenated Compound, Not the Oxidized Form of Cytochrome Oxidase d: An Hypothesis. Microbiology 1983, 129, 1335–1344. [Google Scholar] [CrossRef] [Green Version]
- Kahlow, M.A.; Loehr, T.M.; Zuberi, T.M.; Gennis, R.B. The oxygenated complex of cytochrome d terminal oxidase: Direct evidence for iron-oxygen coordination in a chlorin-containing enzyme by resonance Raman spectroscopy. J. Am. Chem. Soc. 1993, 115, 5845–5846. [Google Scholar] [CrossRef]
- Hill, B.C.; Hill, J.J.; Gennis, R.B. The room temperature reaction of carbon monoxide and oxygen with the cytochrome bd quinol oxidase from Escherichia coli. Biochemistry 1994, 33, 15110–15115. [Google Scholar] [CrossRef]
- Mamedov, M.D.; Tyunyatkina, A.A.; Siletsky, S.A.; Semenov, A.Y. Voltage changes involving photosystem II quinone–iron complex turnover. Eur. Biophys. J. 2006, 35, 647–654. [Google Scholar] [CrossRef]
- Siletsky, S.A.; Mamedov, M.D.; Lukashev, E.P.; Balashov, S.P.; Dolgikh, D.A.; Rubin, A.B.; Kirpichnikov, M.P.; Petrovskaya, L.E. Electrogenic steps of light-driven proton transport in ESR, a retinal protein from Exiguobacterium sibiricum. Biochim. Biophys. Acta Bioenerg. 2016, 1857, 1741–1750. [Google Scholar] [CrossRef]
- Siletsky, S.A.; Mamedov, M.D.; Lukashev, E.P.; Balashov, S.P.; Dolgikh, D.A.; Rubin, A.B.; Kirpichnikov, M.; Petrovskaya, L.E. Elimination of proton donor strongly affects directionality and efficiency of proton transport in ESR, a light-driven proton pump from Exiguobacterium sibiricum. Biochim. Biophys. Acta Bioenerg. 2019, 1860, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Siletsky, S.A.; Lukashev, E.P.; Mamedov, M.D.; Borisov, V.B.; Balashov, S.P.; Dolgikh, D.A.; Rubin, A.B.; Kirpichnikov, M.P.; Petrovskaya, L.E. His57 controls the efficiency of ESR, a light-driven proton pump from Exiguobacterium sibiricum at low and high pH. Biochim. Biophys. Acta Bioenerg. 2021, 1862, 148328. [Google Scholar] [CrossRef] [PubMed]
- Kovalev, K.; Volkov, D.; Astashkin, R.; Alekseev, A.; Gushchin, I.; Haro-Moreno, J.M.; Chizhov, I.; Siletsky, S.; Mamedov, M.; Rogachev, A.; et al. High-resolution structural insights into the heliorhodopsin family. Proc. Natl. Acad. Sci. USA 2020, 117, 4131–4141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siletsky, S.A.; Han, D.; Brand, S.; Morgan, J.E.; Fabian, M.; Geren, L.; Millett, F.; Durham, B.; Konstantinov, A.A.; Gennis, R.B. Single-electron photoreduction of the PM intermediate of cytochrome c oxidase. Biochim. Biophys. Acta Bioenerg. 2006, 1757, 1122–1132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ribacka, C.; Verkhovsky, M.I.; Belevich, I.; Bloch, D.A.; Puustinen, A.A.; Wikström, M. An Elementary Reaction Step of the Proton Pump Is Revealed by Mutation of Tryptophan-164 to Phenylalanine in Cytochrome c Oxidase from Paracoccus denitrificans. Biochemistry 2005, 44, 16502–16512. [Google Scholar] [CrossRef]
- Siletsky, S.; Kaulen, A.D.; Konstantinov, A.A. Resolution of Electrogenic Steps Coupled to Conversion of Cytochrome c Oxidase from the Peroxy to the Ferryl−Oxo State. Biochemistry 1999, 38, 4853–4861. [Google Scholar] [CrossRef]
- Belevich, I.; Verkhovsky, M.I.; Wikström, M. Proton-coupled electron transfer drives the proton pump of cytochrome c oxidase. Nature 2006, 440, 829–832. [Google Scholar] [CrossRef]
- Belevich, I.; Gorbikova, E.; Belevich, N.P.; Rauhamaki, V.; Wikstrom, M.; Verkhovsky, M.I. Initiation of the proton pump of cytochrome c oxidase. Proc. Natl. Acad. Sci. USA 2010, 107, 18469–18474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Faxén, K.; Gilderson, G.; Ädelroth, P.; Brzezinski, P. A mechanistic principle for proton pumping by cytochrome c oxidase. Nature 2005, 437, 286–289. [Google Scholar] [CrossRef]
- Kuznetsova, S.S.; Azarkina, N.V.; Vygodina, T.V.; Siletsky, S.A.; Konstantinov, A.A. Zinc ions as cytochrome c oxidase inhibitors: Two sites of action. Biochemistry (Moscow) 2005, 70, 128–136. [Google Scholar] [CrossRef]
- Verkhovsky, M.I.; Jasaitis, A.; Verkhovskaya, M.; Morgan, J.E.; Wikström, M. Proton translocation by cytochrome c oxidase. Nature 1999, 400, 480–483. [Google Scholar] [CrossRef] [PubMed]
- Zaslavsky, D.; Smirnova, I.; Siletsky, S.; Kaulen, A.; Millett, F.; Konstantinov, A. Rapid kinetics of membrane potential generation by cytochrome c oxidase with the photoactive Ru(II)-tris-bipyridyl derivative of cytochrome c as electron donor. FEBS Lett. 1995, 359, 27–30. [Google Scholar] [CrossRef] [Green Version]
- Ostendorff, H.; Peirano, R.I.; Peters, M.A.; Schlüter, A.; Bossenz, M.; Scheffner, M.; Bach, I. Ubiquitination-dependent cofactor exchange on LIM homeodomain transcription factors. Nature 2002, 416, 99–103. [Google Scholar] [CrossRef] [PubMed]
- Svahn, E.; Faxén, K.; Gennis, R.B.; Brzezinski, P. Proton pumping by an inactive structural variant of cytochrome c oxidase. J. Inorg. Biochem. 2014, 140, 6–11. [Google Scholar] [CrossRef]
- Kahlow, M.A.; Zuberi, T.M.; Gennis, R.B.; Loehr, T.M. Identification of a ferryl intermediate of Escherichia coli cytochrome d terminal oxidase by resonance Raman spectroscopy. Biochemistry 1991, 30, 11485–11489. [Google Scholar] [CrossRef]
- Sztachova, T.; Pechova, I.; Mikulova, L.; Stupak, M.; Jancura, D.; Fabian, M. Peroxide stimulated transition between the ferryl intermediates of bovine cytochrome c oxidase. Biochim. Biophys. Acta Bioenerg. 2021, 1862, 148447. [Google Scholar] [CrossRef]
- Jünemann, S.; Wrigglesworth, A.J.M.; Rich, P.R. Effects of Decyl-aurachin D and Reversed Electron Transfer in Cytochrome bd. Biochemistry 1997, 36, 9323–9331. [Google Scholar] [CrossRef]
- Von der Hocht, I.; van Wonderen, J.H.; Hilbers, F.; Angerer, H.; MacMillan, F.; Michel, H. Interconversions of P and F intermediates of cytochrome c oxidase from Paracoccus denitrificans. Proc. Natl. Acad. Sci. USA 2011, 108, 3964–3969. [Google Scholar] [CrossRef] [Green Version]
- Borisov, V.B.; Gennis, R.B.; Konstantinov, A.A. Interaction of cytochrome bd from Escherichia coli with hydrogen peroxide. Biochemistry (Moscow) 1995, 60, 231–239. [Google Scholar]
- Borisov, V.; Gennis, R.; Konstantinov, A.A. Peroxide complex of cytochrome bd: Kinetics of generation and stability. Biochem. Mol. Boil. Int. 1995, 37, 975–982. [Google Scholar]
- Borisov, V.B. Effect of Membrane Environment on the Ligand-Binding Properties of the Terminal Oxidase Cytochrome bd-I from Escherichia coli. Biochemistry (Moscow) 2020, 85, 1603–1612. [Google Scholar] [CrossRef]
- Sun, J.; Osborne, J.P.; Kahlow, M.A.; Kaysser, T.M.; Gennis, R.B.; Loehr, T.M. Resonance Raman studies of Escherichia coli cytochrome bd oxidase. Selective enhancement of the three heme chromophores of the “as-isolated” enzyme and characterization of the cyanide adduct. Biochemistry 1995, 34, 12144–12151. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Kahlow, M.A.; Kaysser, T.M.; Osborne, J.P.; Hill, J.J.; Rohlfs, R.J.; Hille, R.; Gennis, R.B.; Loehr, T.M. Resonance Raman Spectroscopic Identification of a Histidine Ligand of b595 and the Nature of the Ligation of Chlorin d in the Fully Reduced Escherichia coli Cytochrome bd Oxidase. Biochemistry 1996, 35, 2403–2412. [Google Scholar] [CrossRef] [PubMed]
- Lorence, R.M.; Koland, J.; Gennis, R.B. Coulometric and spectroscopic analysis of the purified cytochrome d complex of Escherichia coli: Evidence for the identification of “cytochrome a1” as cytochrome b595. Biochemistry 1986, 25, 2314–2321. [Google Scholar] [CrossRef] [PubMed]
- Jɒnemann, S.; Wrigglesworth, J.M. Cytochrome bd Oxidase from Azotobacter vinelandii. J. Biol. Chem. 1995, 270, 16213–16220. [Google Scholar] [CrossRef] [Green Version]
- Forte, E.; Borisov, V.B.; Siletsky, S.A.; Petrosino, M.; Giuffrè, A. In the respiratory chain of Escherichia coli cytochromes bd-I and bd-II are more sensitive to carbon monoxide inhibition than cytochrome bo3. Biochim. Biophys. Acta Bioenerg. 2019, 1860, 148088. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Siletsky, S.A.; Borisov, V.B. Proton Pumping and Non-Pumping Terminal Respiratory Oxidases: Active Sites Intermediates of These Molecular Machines and Their Derivatives. Int. J. Mol. Sci. 2021, 22, 10852. https://doi.org/10.3390/ijms221910852
Siletsky SA, Borisov VB. Proton Pumping and Non-Pumping Terminal Respiratory Oxidases: Active Sites Intermediates of These Molecular Machines and Their Derivatives. International Journal of Molecular Sciences. 2021; 22(19):10852. https://doi.org/10.3390/ijms221910852
Chicago/Turabian StyleSiletsky, Sergey A., and Vitaliy B. Borisov. 2021. "Proton Pumping and Non-Pumping Terminal Respiratory Oxidases: Active Sites Intermediates of These Molecular Machines and Their Derivatives" International Journal of Molecular Sciences 22, no. 19: 10852. https://doi.org/10.3390/ijms221910852