
Empagliflozin Relaxes Resistance Mesenteric Arteries by Stimulating Multiple Smooth Muscle Cell Voltage-Gated K+ (KV) Channels


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
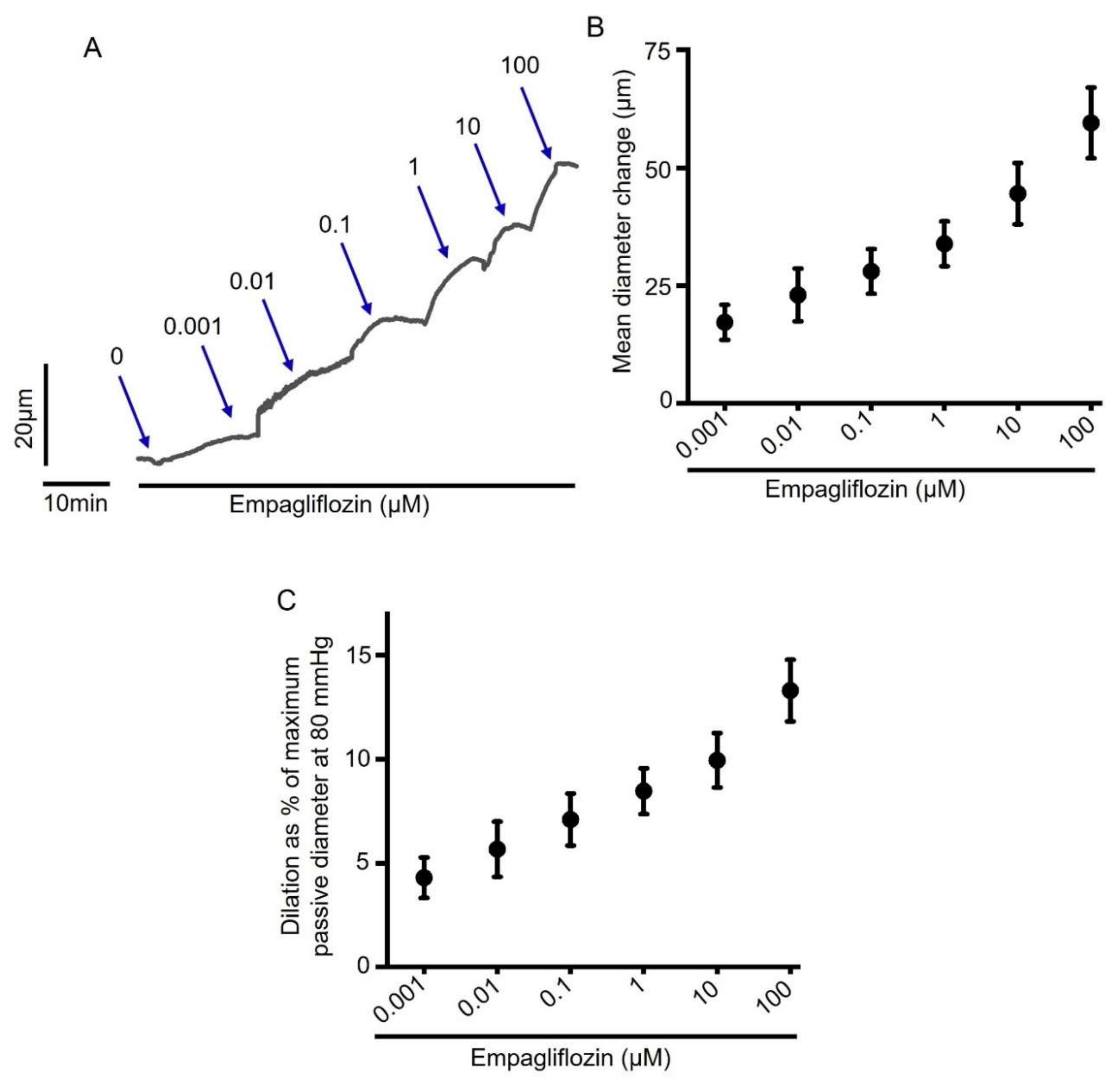
2.1. Acute Empagliflozin Application Produces Vasodilation in Resistance Mesenteric Arteries
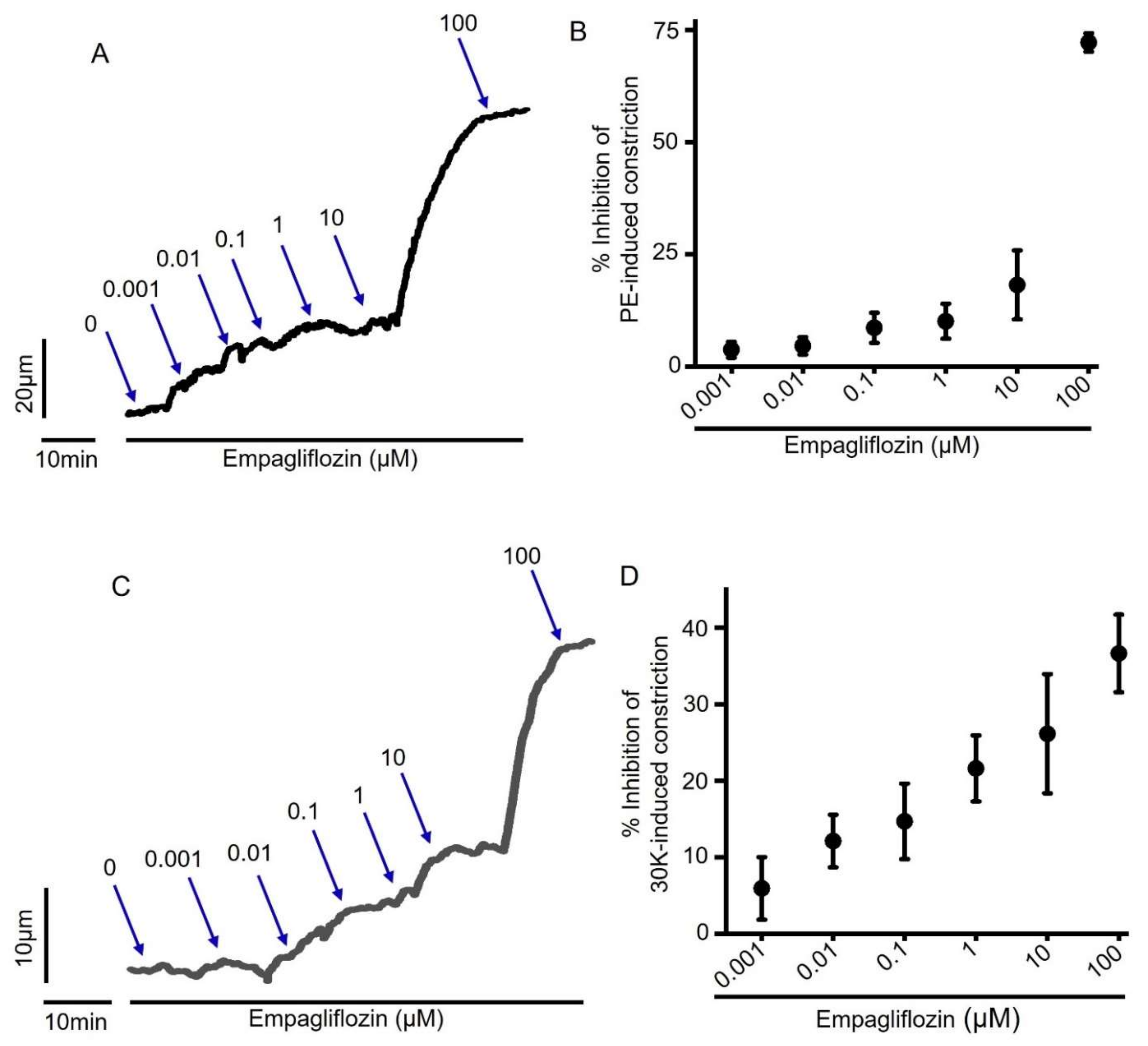
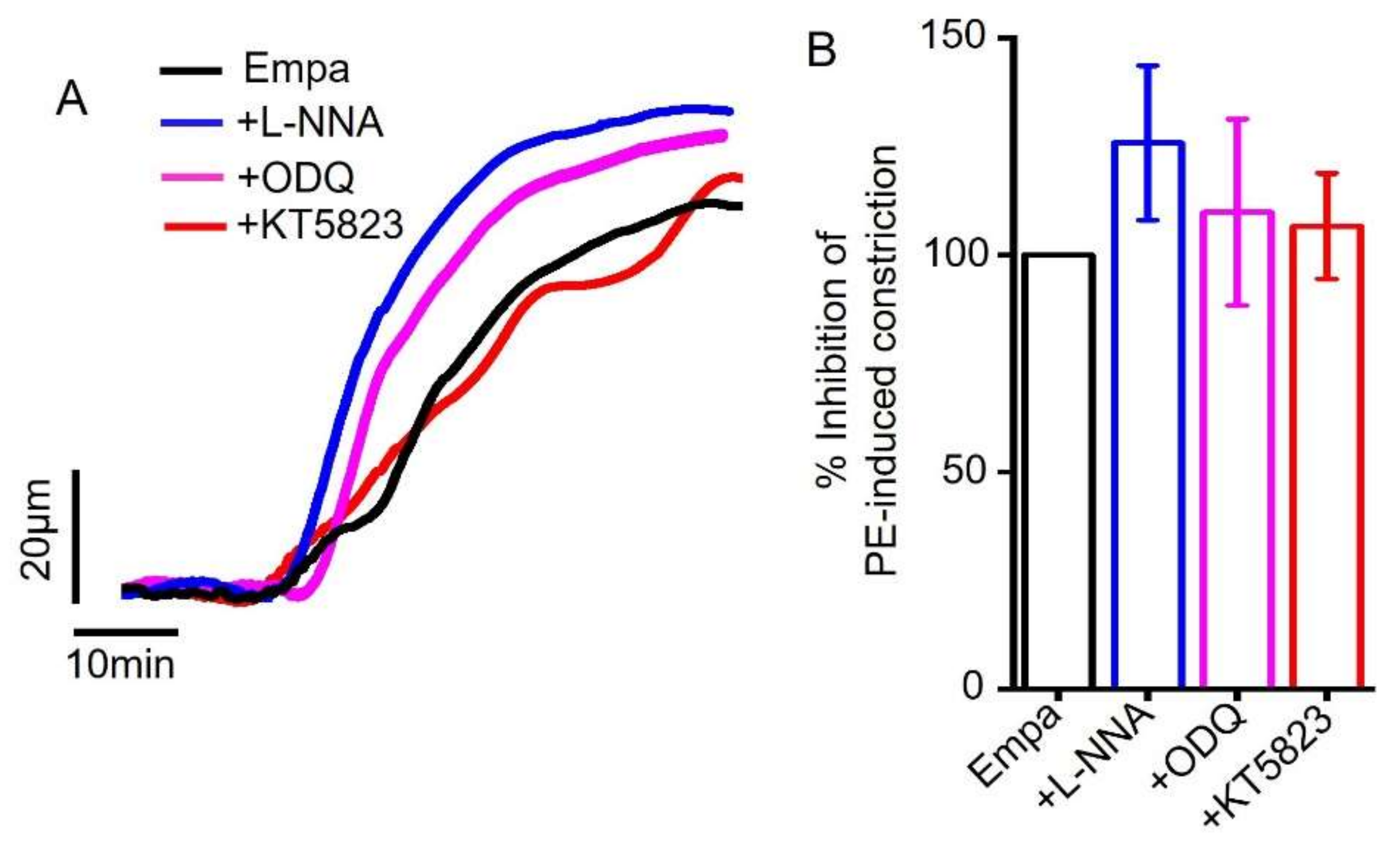
2.2. Empagliflozin-Induced Mesenteric Artery Vasodilation Is Independent of NO-sGC-PKG Signaling Axis
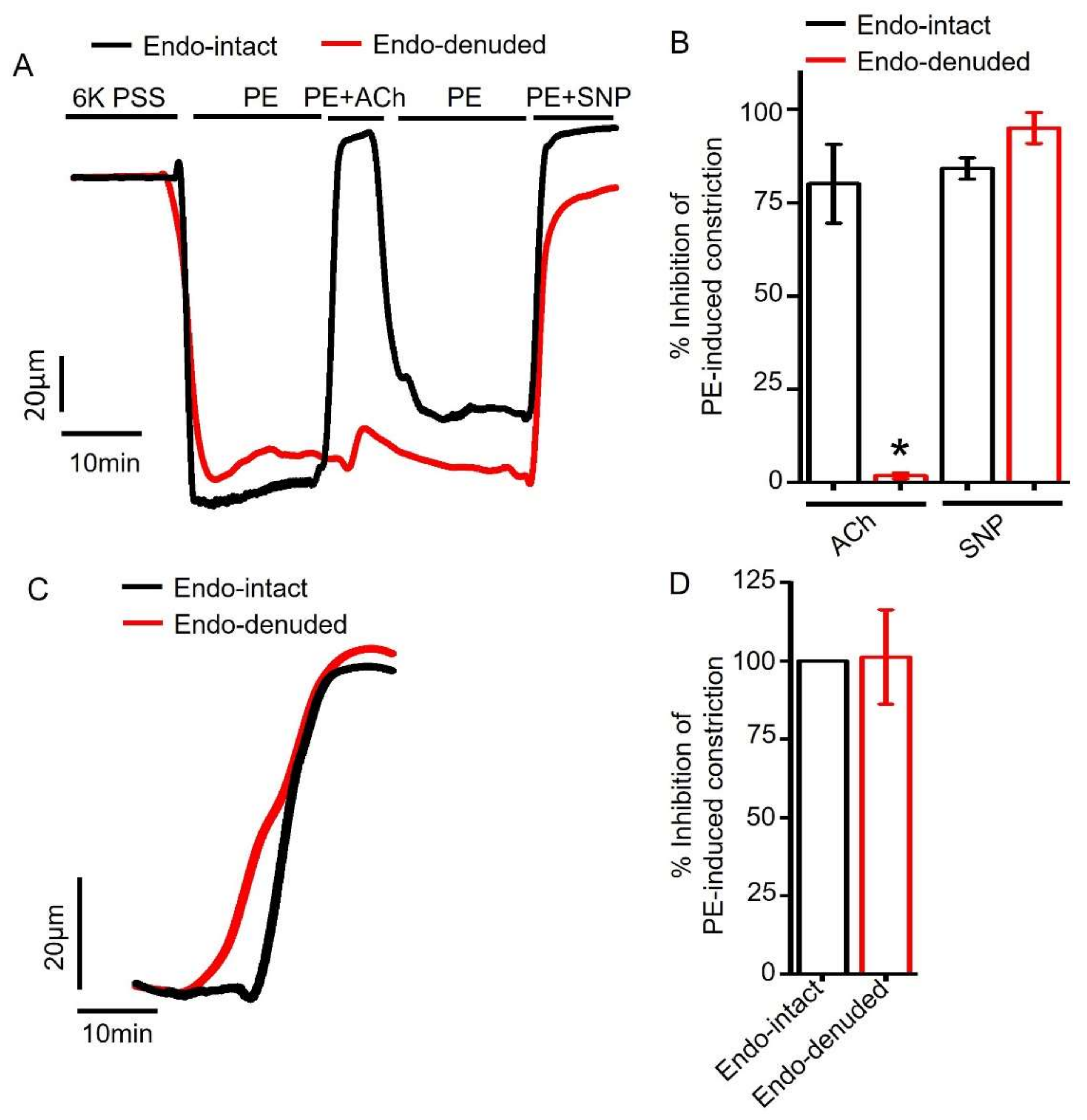
2.3. Empagliflozin-Evoked Mesenteric Artery Vasodilation Does Not Depend on Endothelial PGI2
2.4. Endothelium Denudation Does Not Alter Empagliflozin-Evoked Vasodilation
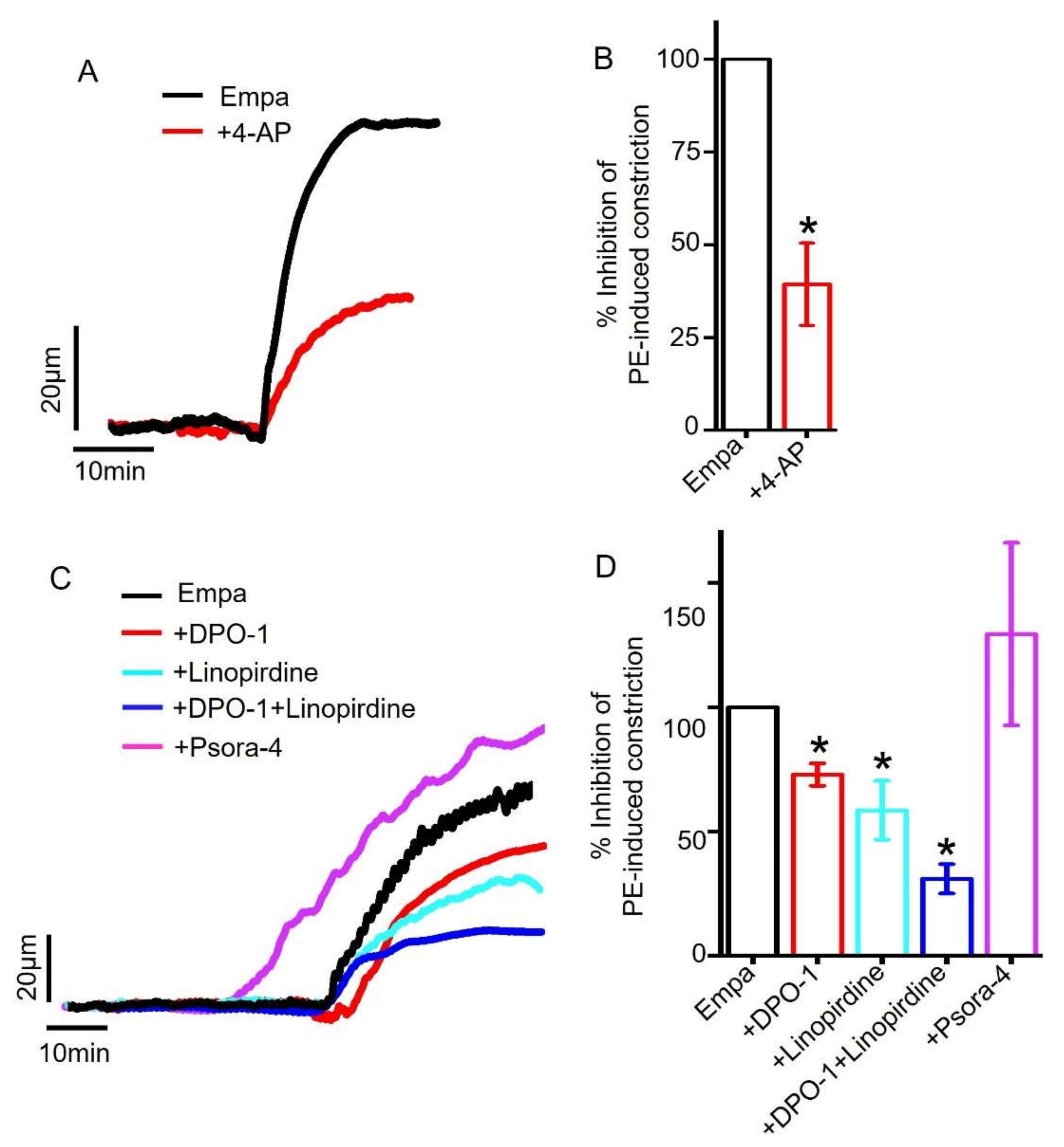
2.5. Role of Smooth Muscle Cell Voltage-Gated K+ (KV) Channels in Empagliflozin-Induced Vasodilation
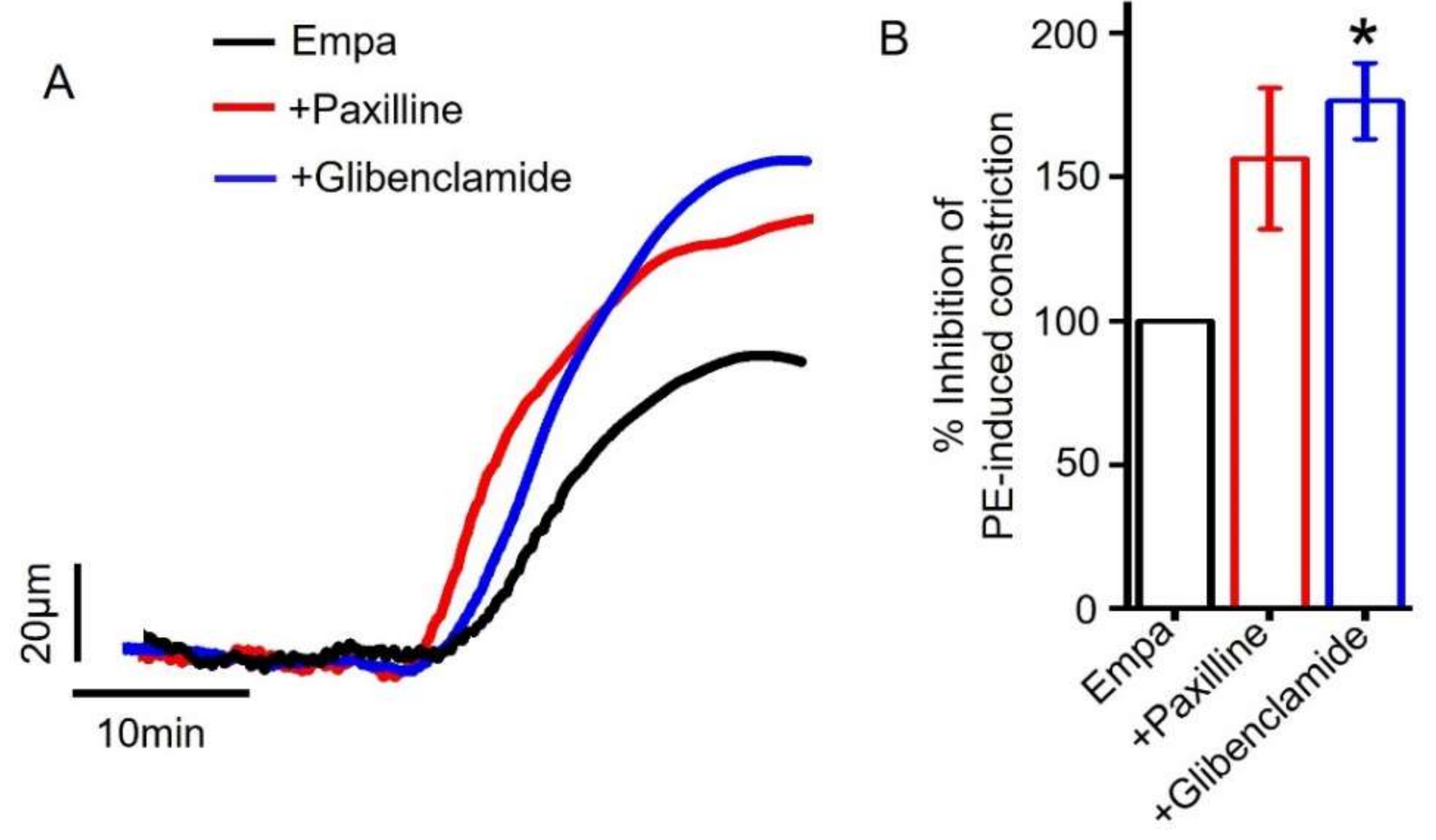
2.6. Role of Smooth Muscle Cell BKCa and KATP Channels in Empagliflozin-Induced Vasodilation
3. Discussion
4. Materials and Methods
4.1. Animals
4.2. Tissue Preparation
4.3. Solutions and Chemicals
4.4. Pressure Myography
4.5. Endothelium Denudation
4.6. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Colussi, G.; Da Porto, A.; Cavarape, A. Hypertension and type 2 diabetes: Lights and shadows about causality. J. Hum. Hypertens. 2020, 34, 91–93. [Google Scholar] [CrossRef]
- Verdecchia, P.; Reboldi, G.; Angeli, F.; Borgioni, C.; Gattobigio, R.; Filippucci, L.; Norgiolini, S.; Bracco, C.; Porcellati, C. Adverse prognostic significance of new diabetes in treated hypertensive subjects. Hypertension 2004, 43, 963–969. [Google Scholar] [CrossRef] [Green Version]
- Tatsumi, Y.; Ohkubo, T. Hypertension with diabetes mellitus: Significance from an epidemiological perspective for Japanese. Hypertens. Res. 2017, 40, 795–806. [Google Scholar] [CrossRef]
- Khangura, D.; Kurukulasuriya, L.R.; Whaley-Connell, A.; Sowers, J.R. Diabetes and Hypertension: Clinical Update. Am. J. Hypertens. 2018, 31, 515–521. [Google Scholar] [CrossRef]
- Lopaschuk, G.D.; Verma, S. Mechanisms of Cardiovascular Benefits of Sodium Glucose Co-Transporter 2 (SGLT2) Inhibitors: A State-of-the-Art Review. JACC Basic Transl. Sci. 2020, 5, 632–644. [Google Scholar] [CrossRef] [PubMed]
- Juni, R.P.; Al-Shama, R.; Kuster, D.W.D.; van der Velden, J.; Hamer, H.M.; Vervloet, M.G.; Eringa, E.C.; Koolwijk, P.; van Hinsbergh, V.W.M. Empagliflozin restores chronic kidney disease-induced impairment of endothelial regulation of cardiomyocyte relaxation and contraction. Kidney Int. 2021, 99, 1088–1101. [Google Scholar] [CrossRef] [PubMed]
- Uthman, L.; Homayr, A.; Juni, R.P.; Spin, E.L.; Kerindongo, R.; Boomsma, M.; Hollmann, M.W.; Preckel, B.; Koolwijk, P.; van Hinsbergh, V.W.M.; et al. Empagliflozin and Dapagliflozin Reduce ROS Generation and Restore NO Bioavailability in Tumor Necrosis Factor α-Stimulated Human Coronary Arterial Endothelial Cells. Cell Physiol. Biochem. 2019, 53, 865–886. [Google Scholar] [CrossRef] [PubMed]
- Varzideh, F.; Kansakar, U.; Santulli, G. SGLT2 inhibitors in cardiovascular medicine. Eur. Heart J. Cardiovasc. Pharmacother. 2021, 7, e67–e68. [Google Scholar] [CrossRef]
- Takahashi, H.; Nomiyama, T.; Terawaki, Y.; Horikawa, T.; Kawanami, T.; Hamaguchi, Y.; Tanaka, T.; Motonaga, R.; Fukuda, T.; Tanabe, M.; et al. Combined treatment with DPP-4 inhibitor linagliptin and SGLT2 inhibitor empagliflozin attenuates neointima formation after vascular injury in diabetic mice. Biochem. Biophys. Rep. 2019, 18, 100640. [Google Scholar] [CrossRef]
- Griffin, M.; Riello, R.; Rao, V.S.; Ivey-Miranda, J.; Fleming, J.; Maulion, C.; McCallum, W.; Sarnak, M.; Collins, S.; Inzucchi, S.E.; et al. Sodium glucose cotransporter 2 inhibitors as diuretic adjuvants in acute decompensated heart failure: A case series. ESC Heart Fail. 2020, 7, 1966–1971. [Google Scholar] [CrossRef]
- Böhm, M.; Anker, S.D.; Butler, J.; Filippatos, G.; Ferreira, J.P.; Pocock, S.J.; Mahfoud, F.; Brueckmann, M.; Jamal, W.; Ofstad, A.P.; et al. EMPEROR-Reduced Trial Committees and Investigators. Empagliflozin Improves Cardiovascular and Renal Outcomes in Heart Failure Irrespective of Systolic Blood Pressure. J. Am. Coll. Cardiol. 2021, 78, 1337–1348. [Google Scholar] [CrossRef]
- Kolwelter, J.; Bosch, A.; Jung, S.; Stabel, L.; Kannenkeril, D.; Ott, C.; Bramlage, P.; Schiffer, M.; Achenbach, S.; Schmieder, R.E. Effects of the sodium-glucose cotransporter 2 inhibitor empagliflozin on vascular function in patients with chronic heart failure. ESC Heart Fail. 2021. Epub ahead of print. [Google Scholar] [CrossRef]
- Li, X.; Lu, Q.; Qiu, Y.; do Carmo, J.M.; Wang, Z.; da Silva, A.A.; Mouton, A.; Omoto, A.C.M.; Hall, M.E.; Li, J.; et al. Direct Cardiac Actions of the Sodium Glucose Co-Transporter 2 Inhibitor Empagliflozin Improve Myocardial Oxidative Phosphorylation and Attenuate Pressure-Overload Heart Failure. J. Am. Heart Assoc. 2021, 10, e018298. [Google Scholar] [CrossRef] [PubMed]
- Neal, B.; Perkovic, V.; Matthews, D.R. Canagliflozin and Cardiovascular and Renal Events in Type 2 Diabetes. N. Engl. J. Med. 2017, 377, 2099. [Google Scholar] [CrossRef] [PubMed]
- Weber, M.A.; Mansfield, T.A.; Cain, V.A.; Iqbal, N.; Parikh, S. Blood pressure and glycaemic effects of dapagliflozin versus placebo in patients with type 2 diabetes on combination antihypertensive therapy: A randomised, double-blind, placebo-controlled, phase 3 study. Lancet Diabetes Endocrinol. 2016, 4, 211–220. [Google Scholar] [CrossRef]
- Fitchett, D.; Zinman, B.; Wanner, C.; Lachin, J.M.; Hantel, S.; Salsali, A.; Johansen, O.E.; Woerle, H.J.; Broedl, U.C.; Inzucchi, S.E. Heart failure outcomes with empagliflozin in patients with type 2 diabetes at high cardiovascular risk: Results of the EMPA-REG OUTCOME® trial. Eur. Heart J. 2016, 37, 1526–1534. [Google Scholar] [CrossRef] [Green Version]
- Zinman, B.; Wanner, C.; Lachin, J.M.; Fitchett, D.; Bluhmki, E.; Hantel, S.; Mattheus, M.; Devins, T.; Johansen, O.E.; Woerle, H.J.; et al. Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. N. Engl. J. Med. 2015, 373, 2117–2128. [Google Scholar] [CrossRef]
- Teo, Y.H.; Teo, Y.N.; Syn, N.L.; Kow, C.S.; Yoong, C.S.Y.; Tan, B.Y.Q.; Yeo, T.C.; Lee, C.H.; Lin, W.; Sia, C.H. Effects of Sodium/Glucose Cotransporter 2 (SGLT2) Inhibitors on Cardiovascular and Metabolic Outcomes in Patients Without Diabetes Mellitus: A Systematic Review and Meta-Analysis of Randomized-Controlled Trials. J. Am. Heart Assoc. 2021, 10, e019463. [Google Scholar] [CrossRef]
- Cosentino, F.; Grant, P.J.; Aboyans, V.; Bailey, C.J.; Ceriello, A.; Delgado, V.; Federici, M.; Filippatos, G.; Grobbee, D.E.; Hansen, T.B.; et al. 2019 ESC Guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD. Eur. Heart J. 2020, 41, 255–323. [Google Scholar] [CrossRef] [Green Version]
- Oliva, R.V.; Barkis, G.L. Blood pressure effects of sodium-glucose co-transport 2 (SGLT2) inhibitors. J. Am. Soc. Hypertens. 2014, 5, 330–339. [Google Scholar] [CrossRef]
- Li, H.; Shin, S.E.; Seo, M.S.; An, J.R.; Choi, I.W.; Jung, W.K.; Firth, A.L.; Lee, D.S.; Yim, M.J.; Choi, G.; et al. The anti-diabetic drug dapagliflozin induces vasodilation via activation of PKG and Kv channels. Life Sci. 2018, 197, 46–55. [Google Scholar] [CrossRef]
- Gaspari, T.; Spizzo, I.; Liu, H.; Hu, Y.; Simpson, R.W.; Widdop, R.E.; Dear, A.E. Dapagliflozin attenuates human vascular endothelial cell activation and induces vasorelaxation: A potential mechanism for inhibition of atherogenesis. Diab. Vasc. Dis. Res. 2018, 15, 64–73. [Google Scholar] [CrossRef] [Green Version]
- Fala, L. Jardiance (Empagliflozin), an SGLT2 Inhibitor, Receives FDA Approval for the Treatment of Patients with Type 2 Diabetes. Am. Health Drug Benefits 2015, 8, 92–95. [Google Scholar]
- Bosch, A.; Ott, C.; Jung, S.; Striepe, K.; Karg, M.V.; Kannenkeril, D.; Dienemann, T.; Schmieder, R.E. How does empagliflozin improve arterial stiffness in patients with type 2 diabetes mellitus? Sub analysis of a clinical trial. Cardiovasc. Diabetol. 2019, 18, 44. [Google Scholar] [CrossRef] [PubMed]
- Chilton, R.; Tikkanen, I.; Cannon, C.P.; Crowe, S.; Woerle, H.J.; Broedl, U.C.; Johansen, O.E. Effects of empagliflozin on blood pressure and markers of arterial stiffness and vascular resistance in patients with type 2 diabetes. Diabetes Obes. Metab. 2015, 17, 1180–1193. [Google Scholar] [CrossRef]
- Tikkanen, I.; Narko, K.; Zeller, C.; Green, A.; Salsali, A.; Broedl, U.C.; Woerle, H.J. Empagliflozin reduces blood pressure in patients with type 2 diabetes and hypertension. Diabetes Care. 2015, 38, 420–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seo, M.S.; Jung, H.S.; An, J.R.; Kang, M.; Heo, R.; Li, H.; Han, E.T.; Yang, S.R.; Cho, E.H.; Bae, Y.M.; et al. Empagliflozin dilates the rabbit aorta by activating PKG and voltage-dependent K(+) channels. Toxicol. Appl. Pharmacol. 2020, 403, 115153. [Google Scholar] [CrossRef] [PubMed]
- Hasan, R.; Lasker, S.; Hasan, A.; Zerin, F.; Zamila, M.; Parvez, F.; Rahman, M.M.; Khan, F.; Subhan, N.; Alam, M.A. Canagliflozin ameliorates renal oxidative stress and inflammation by stimulating AMPK-Akt-eNOS pathway in the isoprenaline-induced oxidative stress model. Sci. Rep. 2020, 10, 14659. [Google Scholar] [CrossRef] [PubMed]
- Hasan, R.; Lasker, S.; Hasan, A.; Zerin, F.; Zamila, M.; Parvez, F.; Rahman, M.M.; Khan, F.; Subhan, N.; Alam, M.A. Canagliflozin attenuates isoprenaline-induced cardiac oxidative stress by stimulating multiple antioxidant and anti-inflammatory signaling pathways. Sci. Rep. 2020, 10, 14459. [Google Scholar] [CrossRef] [PubMed]
- Sayour, A.A.; Korkmaz-Icöz, S.; Loganathan, S.; Ruppert, M.; Sayour, V.N.; Oláh, A.; Benke, K.; Brune, M.; Benkő, R.; Horváth, E.M.; et al. Acute canagliflozin treatment protects against in vivo myocardial ischemia-reperfusion injury in non-diabetic male rats and enhances endothelium-dependent vasorelaxation. J. Transl. Med. 2019, 17, 127. [Google Scholar] [CrossRef] [PubMed]
- Aragón-Herrera, A.; Feijóo-Bandín, S.; Otero Santiago, M.; Barral, L.; Campos-Toimil, M.; Gil-Longo, J.; Costa Pereira, T.M.; García-Caballero, T.; Rodríguez-Segade, S.; Rodríguez, J.; et al. Empagliflozin reduces the levels of CD36 and cardiotoxic lipids while improving autophagy in the hearts of Zucker diabetic fatty rats. Biochem. Pharmacol. 2019, 170, 113677. [Google Scholar] [CrossRef] [PubMed]
- Nasiri-Ansari, Ν.; Dimitriadis, G.K.; Agrogiannis, G.; Perrea, D.; Kostakis, I.D.; Kaltsas, G.; Papavassiliou, A.G.; Randeva, H.S.; Kassi, E. Canagliflozin attenuates the progression of atherosclerosis and inflammation process in APOE knockout mice. Cardiovasc. Diabetol. 2018, 17, 106. [Google Scholar] [CrossRef] [Green Version]
- Lahnwong, S.; Chattipakorn, S.C.; Chattipakorn, N. Potential mechanisms responsible for cardioprotective effects of sodium-glucose co-transporter 2 inhibitors. Cardiovasc. Diabetol. 2018, 17, 101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steven, S.; Oelze, M.; Hanf, A.; Kröller-Schön, S.; Kashani, F.; Roohani, S.; Welschof, P.; Kopp, M.; Gödtel-Armbrust, U.; Xia, N.; et al. The SGLT2 inhibitor empagliflozin improves the primary diabetic complications in ZDF rats. Redox Biol. 2017, 13, 370–385. [Google Scholar] [CrossRef]
- Han, J.H.; Oh, T.J.; Lee, G.; Maeng, H.J.; Lee, D.H.; Kim, K.M.; Choi, S.H.; Jang, H.C.; Lee, H.S.; Park, K.S.; et al. The beneficial effects of empagliflozin, an SGLT2 inhibitor, on atherosclerosis in ApoE (-/-) mice fed a western diet. Diabetologia 2017, 60, 364–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francis, S.H.; Busch, J.L.; Corbin, J.D.; Sibley, D. cGMP-dependent protein kinases and cGMP phosphodiesterases in nitric oxide and cGMP action. Pharmacol. Rev. 2010, 62, 525–563. [Google Scholar] [CrossRef]
- Majed, B.H.; Khalil, R.A. Molecular mechanisms regulating the vascular prostacyclin pathways and their adaptation during pregnancy and in the newborn. Pharmacol. Rev. 2012, 64, 540–582. [Google Scholar] [CrossRef] [Green Version]
- Vane, J.; Corin, R.E. Prostacyclin: A vascular mediator. Eur. J. Vasc Endovasc. Surg. 2003, 26, 571–578. [Google Scholar] [CrossRef] [Green Version]
- Hasan, R.; Leo, M.D.; Muralidharan, P.; Mata-Daboin, A.; Yin, W.; Bulley, S.; Fernandez-Peña, C.; MacKay, C.E.; Jaggar, J.H. SUMO1 modification of PKD2 channels regulates arterial contractility. Proc. Natl. Acad. Sci. USA 2019, 116, 27095–27104. [Google Scholar] [CrossRef]
- Bulley, S.; Fernández-Peña, C.; Hasan, R.; Leo, M.D.; Muralidharan, P.; Mackay, C.E.; Evanson, K.W.; Moreira-Junior, L.; Mata-Daboin, A.; Burris, S.K.; et al. Arterial smooth muscle cell PKD2 (TRPP1) channels regulate systemic blood pressure. Elife 2018, 7, e42628. [Google Scholar] [CrossRef] [PubMed]
- Kidd, M.W.; Leo, M.D.; Bannister, J.P.; Jaggar, J.H. Intravascular pressure enhances the abundance of functional Kv1.5 channels at the surface of arterial smooth muscle cells. Sci. Signal. 2015, 8, ra83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tykocki, N.R.; Boerman, E.M.; Jackson, W.F. Smooth Muscle Ion Channels and Regulation of Vascular Tone in Resistance Arteries and Arterioles. Compr. Physiol. 2017, 7, 485–581. [Google Scholar] [CrossRef] [Green Version]
- Jackson, W.F. K(V) channels and the regulation of vascular smooth muscle tone. Microcirculation 2018, 25, e12421. [Google Scholar] [CrossRef]
- Hasan, R.; Jaggar, J.H. K(V) channel trafficking and control of vascular tone. Microcirculation 2018, 25. [Google Scholar] [CrossRef]
- Tamargo, J.; Caballero, R.; Gómez, R.; Valenzuela, C.; Delpón, E. Pharmacology of cardiac potassium channels. Cardiovasc. Res. 2004, 62, 9–33. [Google Scholar] [CrossRef] [Green Version]
- Lagrutta, A.; Wang, J.; Fermini, B.; Salata, J.J. Novel, potent inhibitors of human Kv1.5 K+ channels and ultrarapidly activating delayed rectifier potassium current. J. Pharmacol. Exp. Ther. 2006, 317, 1054–1063. [Google Scholar] [CrossRef] [Green Version]
- Stump, G.L.; Wallace, A.A.; Regan, C.P.; Lynch, J.J., Jr. In vivo antiarrhythmic and cardiac electrophysiologic effects of a novel diphenylphosphine oxide IKur blocker (2-isopropyl-5-methylcyclohexyl) diphenylphosphine oxide. J. Pharmacol. Exp. Ther. 2005, 315, 1362–1367. [Google Scholar] [CrossRef] [Green Version]
- Søgaard, R.; Ljungstrøm, T.; Pedersen, K.A.; Olesen, S.P.; Jensen, B.S. KCNQ4 channels expressed in mammalian cells: Functional characteristics and pharmacology. Am. J. Physiol. Cell Physiol. 2001, 280, C859–C866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greene, D.L.; Kang, S.; Hoshi, N. XE991 and Linopirdine Are State-Dependent Inhibitors for Kv7/KCNQ Channels that Favor Activated Single Subunits. J. Pharmacol. Exp. Ther. 2017, 362, 177–185. [Google Scholar] [CrossRef]
- Ko, E.A.; Han, J.; Jung, I.D.; Park, W.S. Physiological roles of K+ channels in vascular smooth muscle cells. J. Smooth Muscle Res. 2008, 44, 65–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, Y.; Lingle, C.J. Paxilline inhibits BK channels by an almost exclusively closed-channel block mechanism. J. Gen. Physiol. 2014, 144, 415–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Teramoto, N.; Zhu, H.L.; Ito, Y. Blocking actions of glibenclamide on ATP-sensitive K+ channels in pig urethral myocytes. J. Pharm. Pharmacol. 2004, 56, 395–399. [Google Scholar] [CrossRef]
- Harper, D.; Chandler, B. Splanchnic circulation. BJA Education. 2015, 16, 66–71. [Google Scholar] [CrossRef] [Green Version]
- Brozovich, F.V.; Nicholson, C.J.; Degen, C.V.; Gao, Y.Z.; Aggarwal, M.; Morgan, K.G. Mechanisms of Vascular Smooth Muscle Contraction and the Basis for Pharmacologic Treatment of Smooth Muscle Disorders. Pharmacol. Rev. 2016, 68, 476–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, L.J. Empagliflozin: A review of its use in patients with type 2 diabetes mellitus. Drugs. 2014, 74, 1769–1784. [Google Scholar] [CrossRef]
- Rydén, L.; Grant, P.J.; Anker, S.D.; Berne, C.; Cosentino, F.; Danchin, N.; Deaton, C.; Escaned, J.; Hammes, H.P.; Huikuri, H.; et al. ESC Guidelines on diabetes, pre-diabetes, and cardiovascular diseases developed in collaboration with the EASD: The Task Force on diabetes, pre-diabetes, and cardiovascular diseases of the European Society of Cardiology (ESC) and developed in collaboration with the European Association for the Study of Diabetes (EASD). Eur. Heart J. 2013, 34, 3035–3087. [Google Scholar] [CrossRef] [Green Version]
- MacKay, C.E.; Leo, M.D.; Fernández-Peña, C.; Hasan, R.; Yin, W.; Mata-Daboin, A.; Bulley, S.; Gammons, J.; Mancarella, S.; Jaggar, J.H. Correction: Intravascular flow stimulates PKD2 (polycystin-2) channels in endothelial cells to reduce blood pressure. Elife 2020, 9, e60401. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hasan, A.; Hasan, R. Empagliflozin Relaxes Resistance Mesenteric Arteries by Stimulating Multiple Smooth Muscle Cell Voltage-Gated K+ (KV) Channels. Int. J. Mol. Sci. 2021, 22, 10842. https://doi.org/10.3390/ijms221910842
Hasan A, Hasan R. Empagliflozin Relaxes Resistance Mesenteric Arteries by Stimulating Multiple Smooth Muscle Cell Voltage-Gated K+ (KV) Channels. International Journal of Molecular Sciences. 2021; 22(19):10842. https://doi.org/10.3390/ijms221910842
Chicago/Turabian StyleHasan, Ahasanul, and Raquibul Hasan. 2021. "Empagliflozin Relaxes Resistance Mesenteric Arteries by Stimulating Multiple Smooth Muscle Cell Voltage-Gated K+ (KV) Channels" International Journal of Molecular Sciences 22, no. 19: 10842. https://doi.org/10.3390/ijms221910842