
Pseudomonas Phage MD8: Genetic Mosaicism and Challenges of Taxonomic Classification of Lambdoid Bacteriophages

 ,
,  , , ,
, , ,
Abstract
:1. Introduction
2. Results
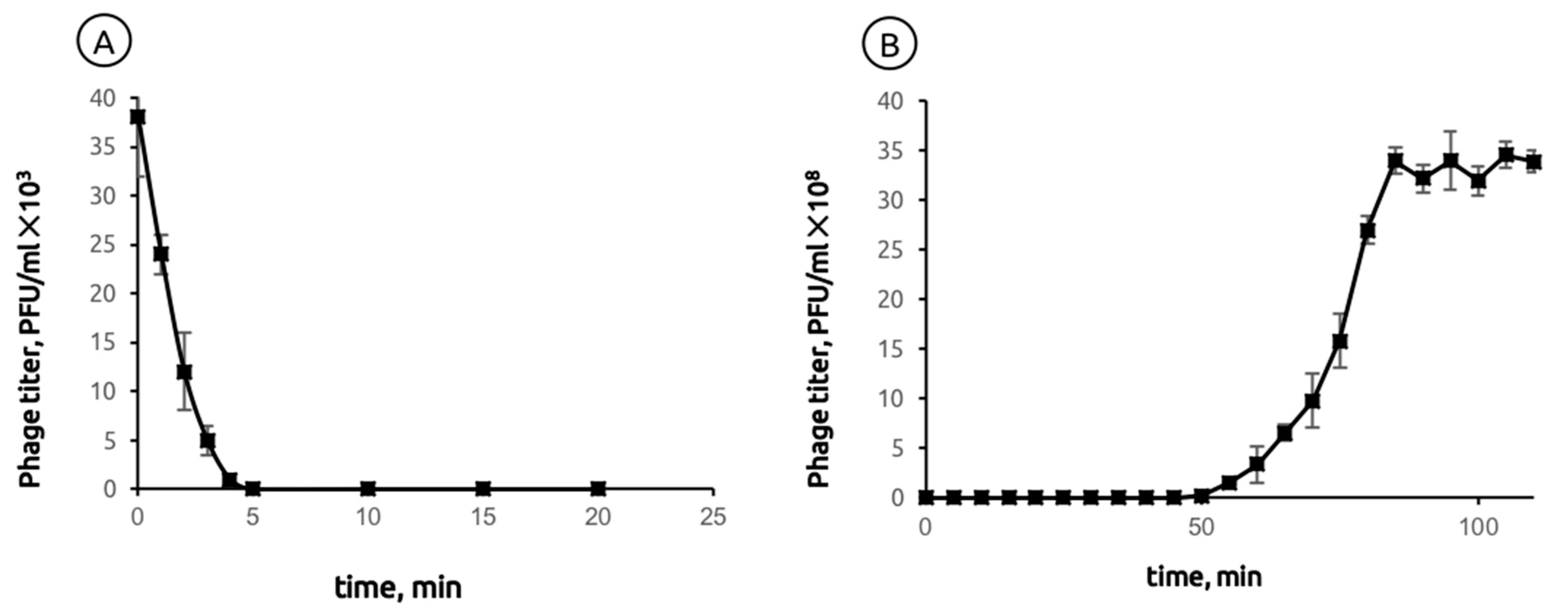
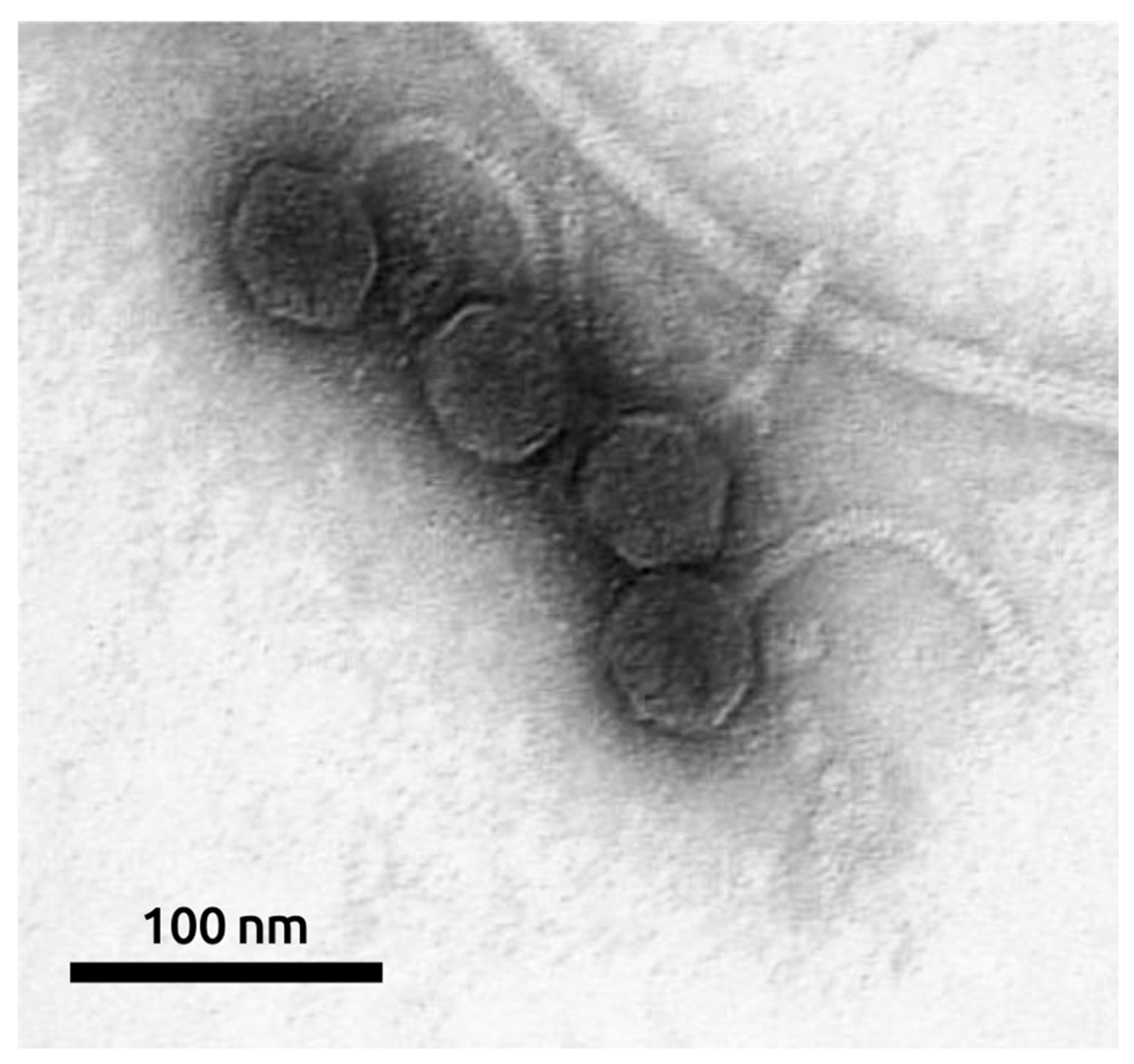
2.1. General Biological Features of Phage MD8
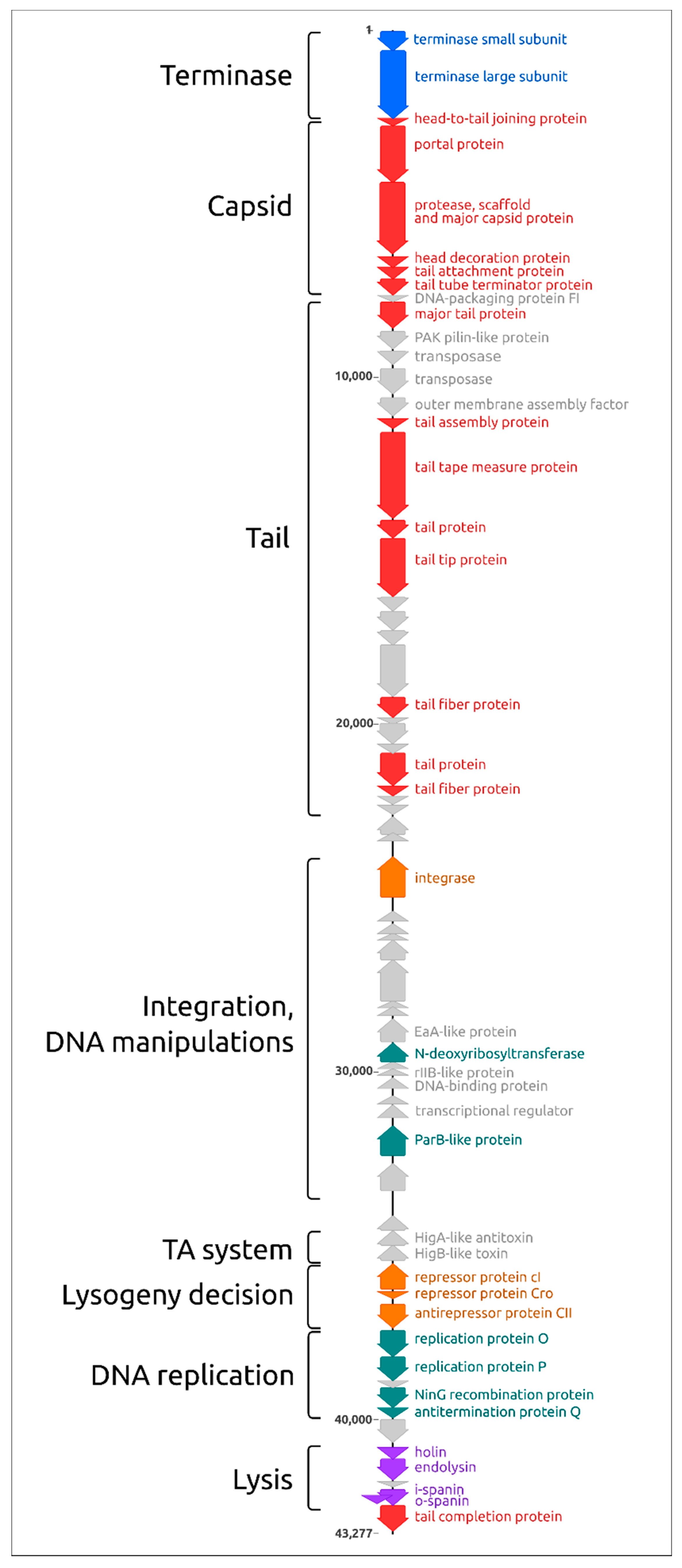
2.2. General Features of Genome and Proteome
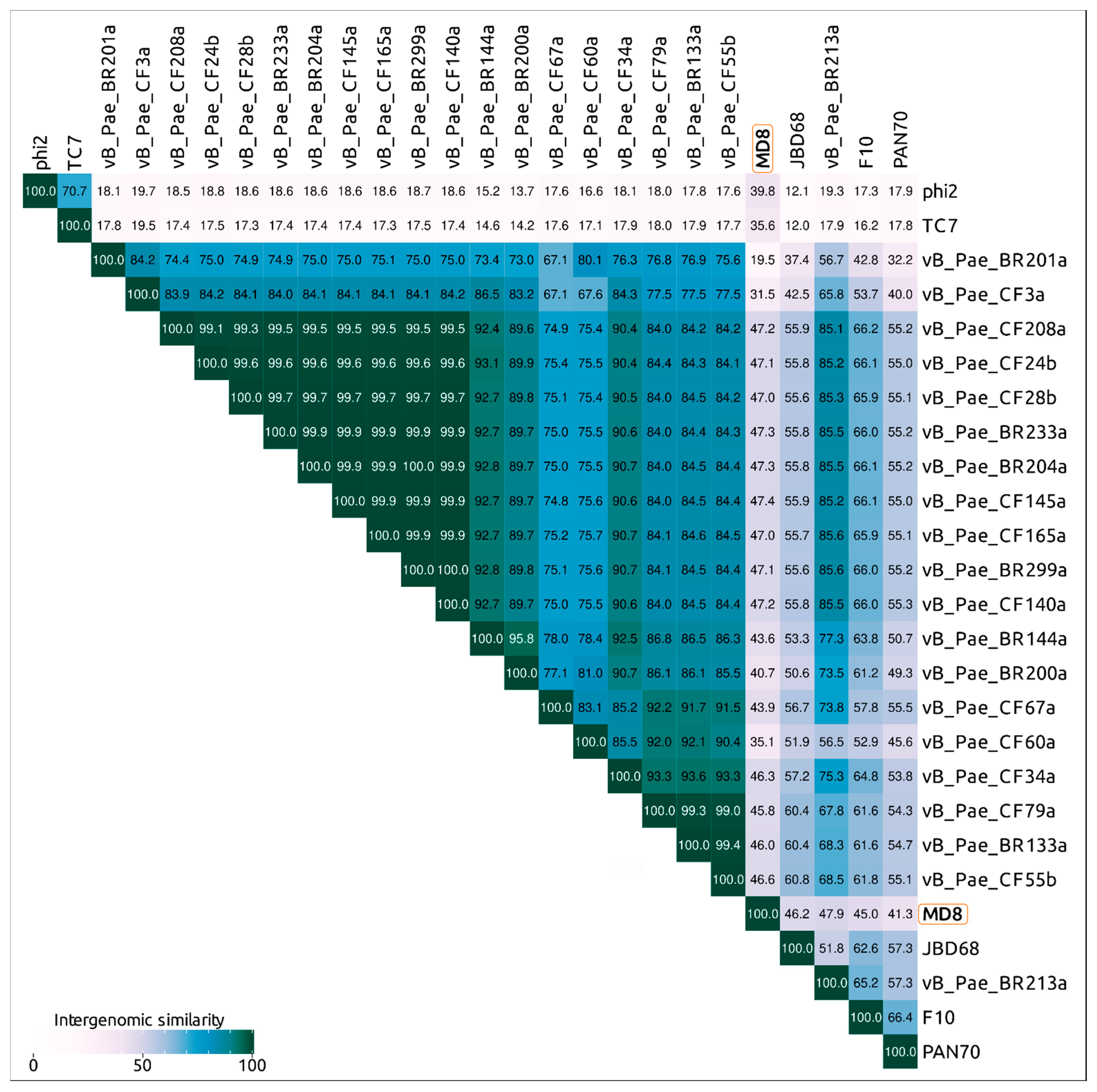
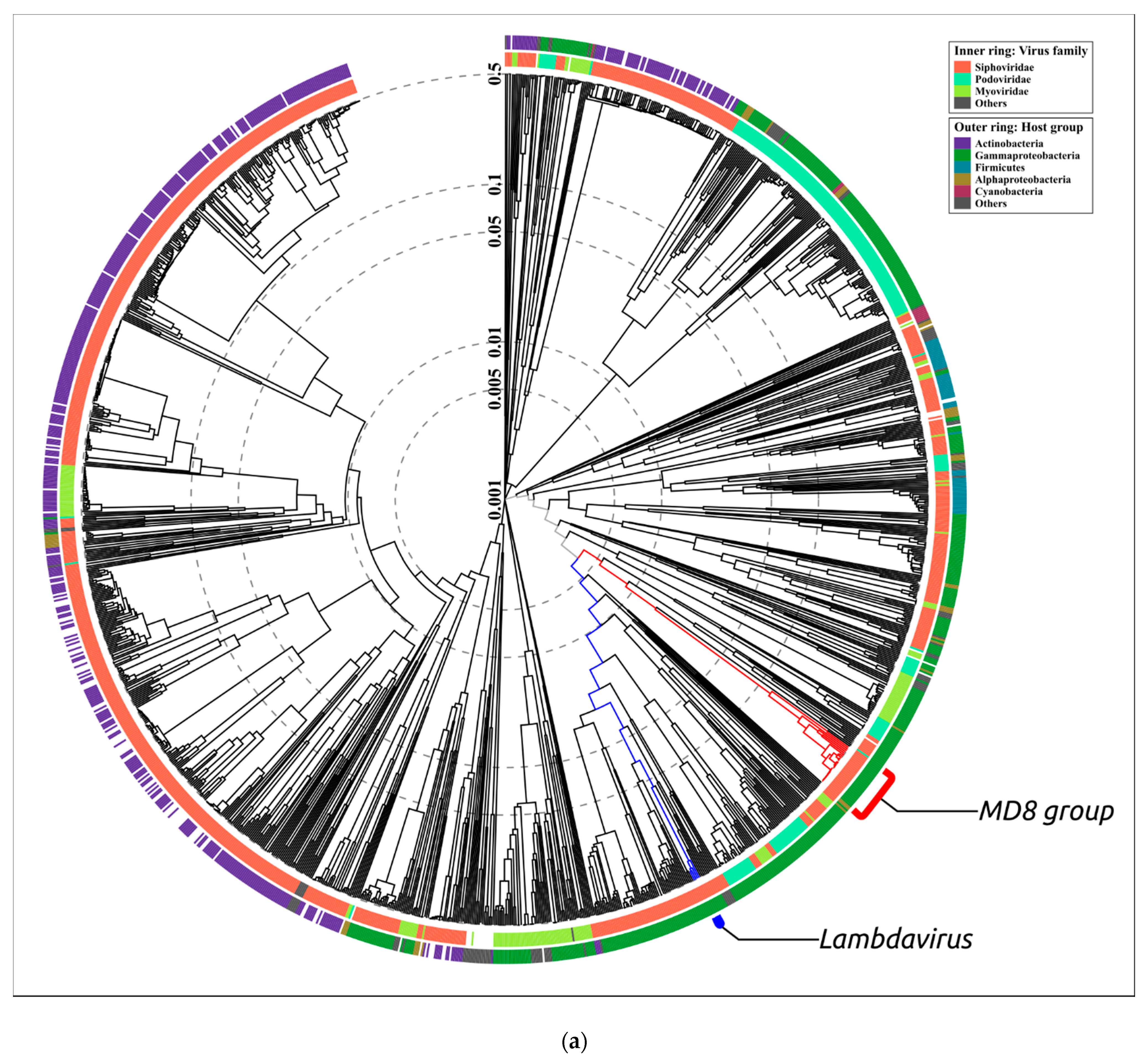
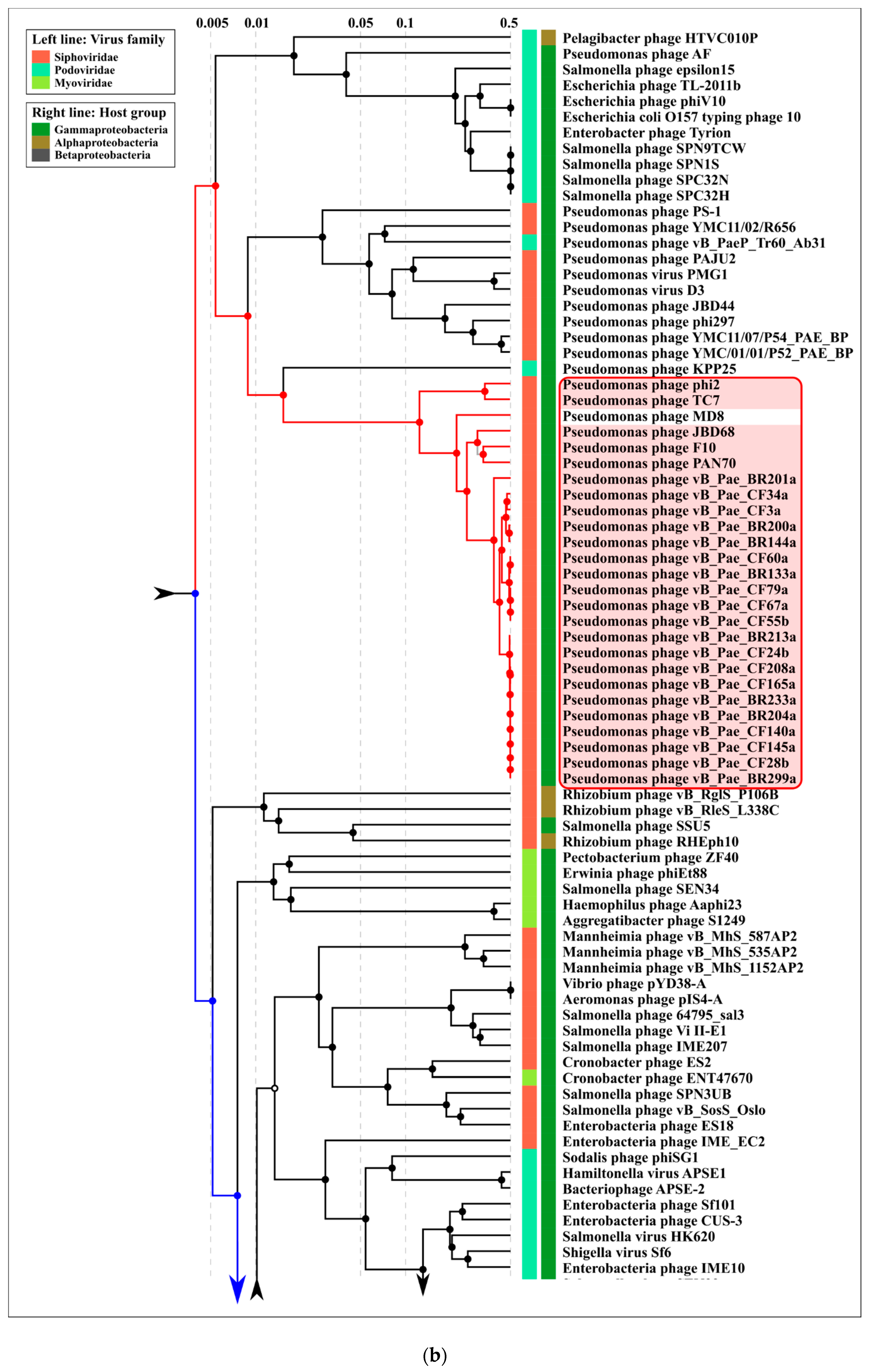
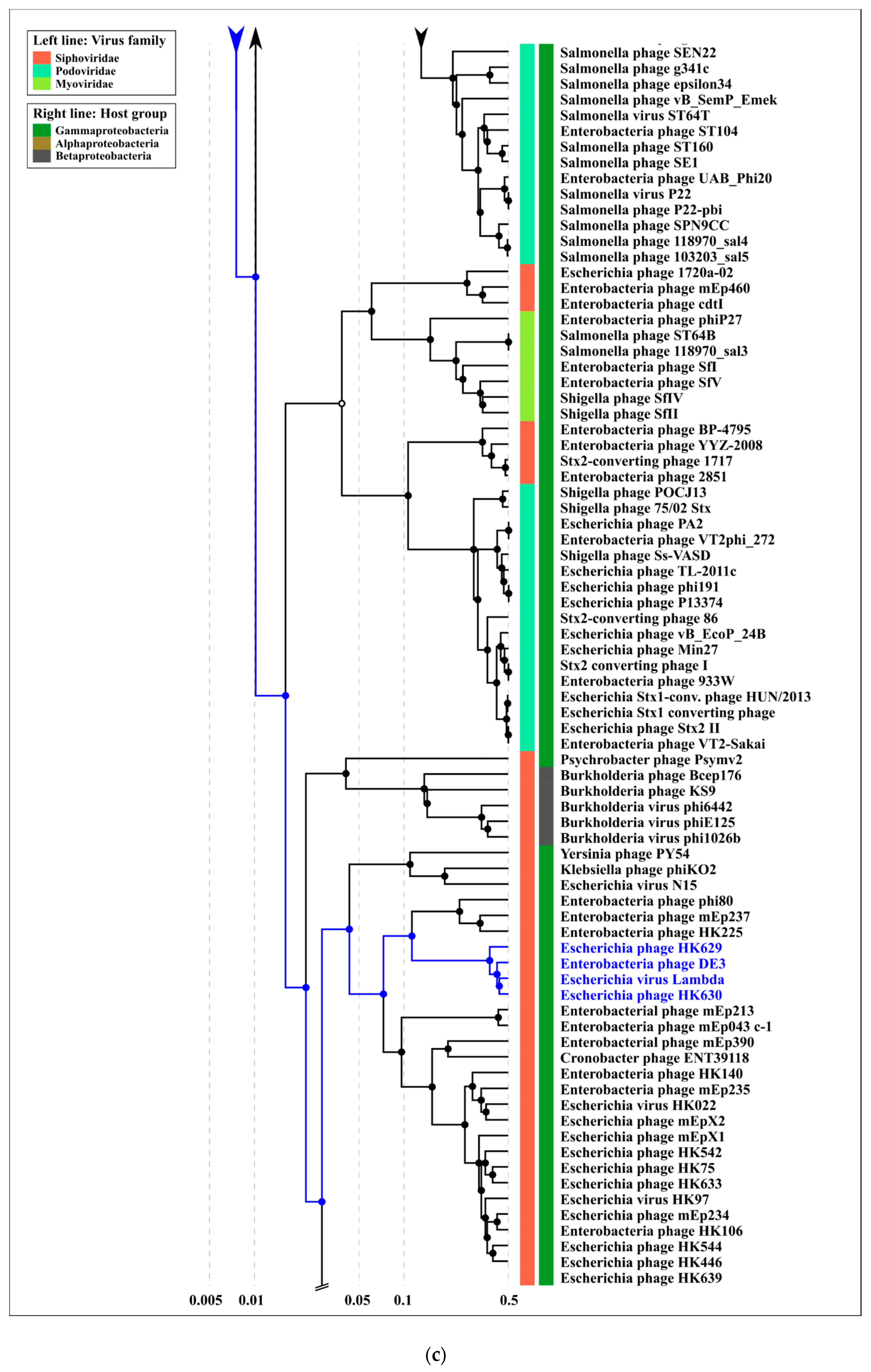
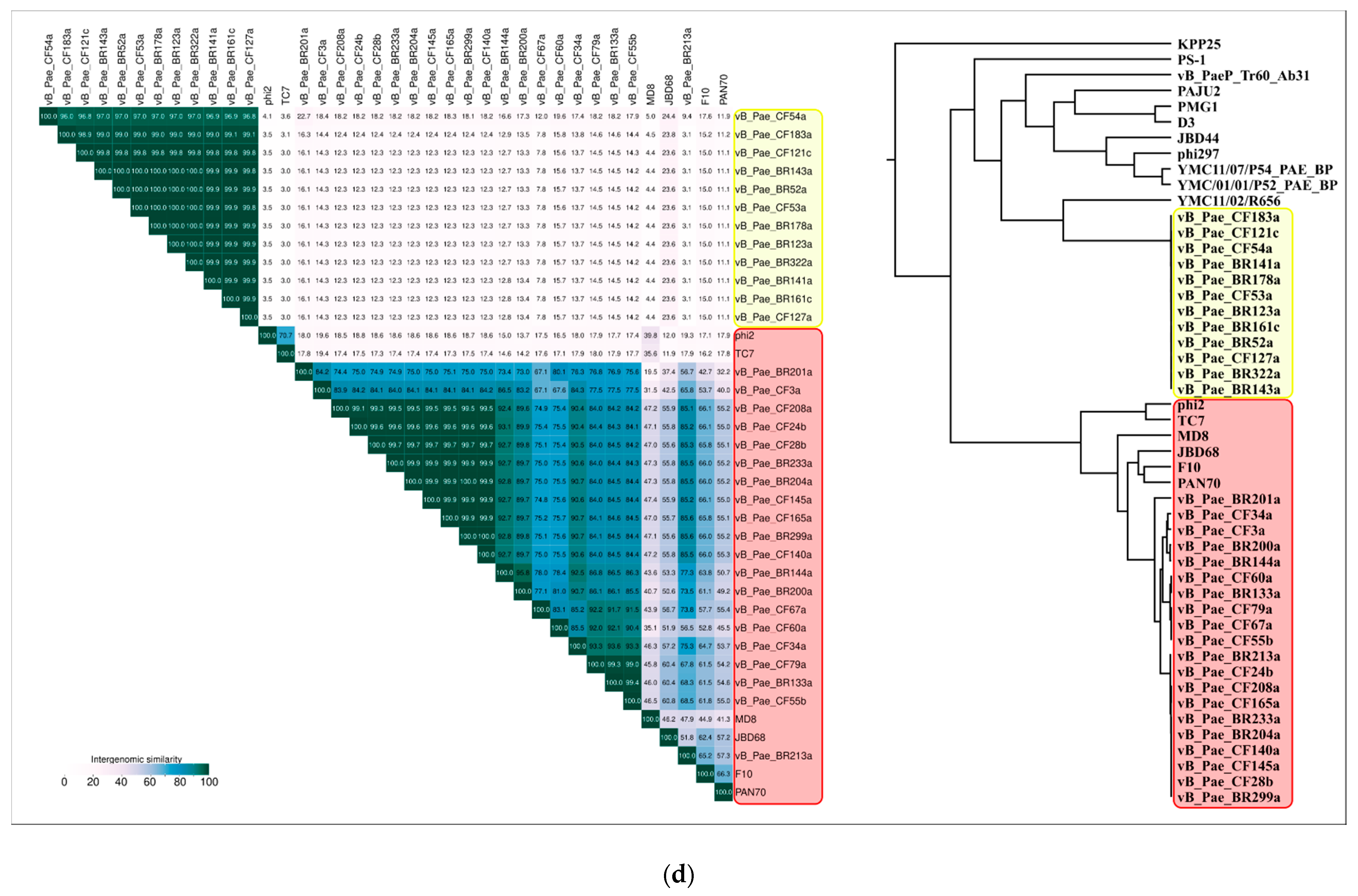
2.3. ANI and VIRIDIC Clustering
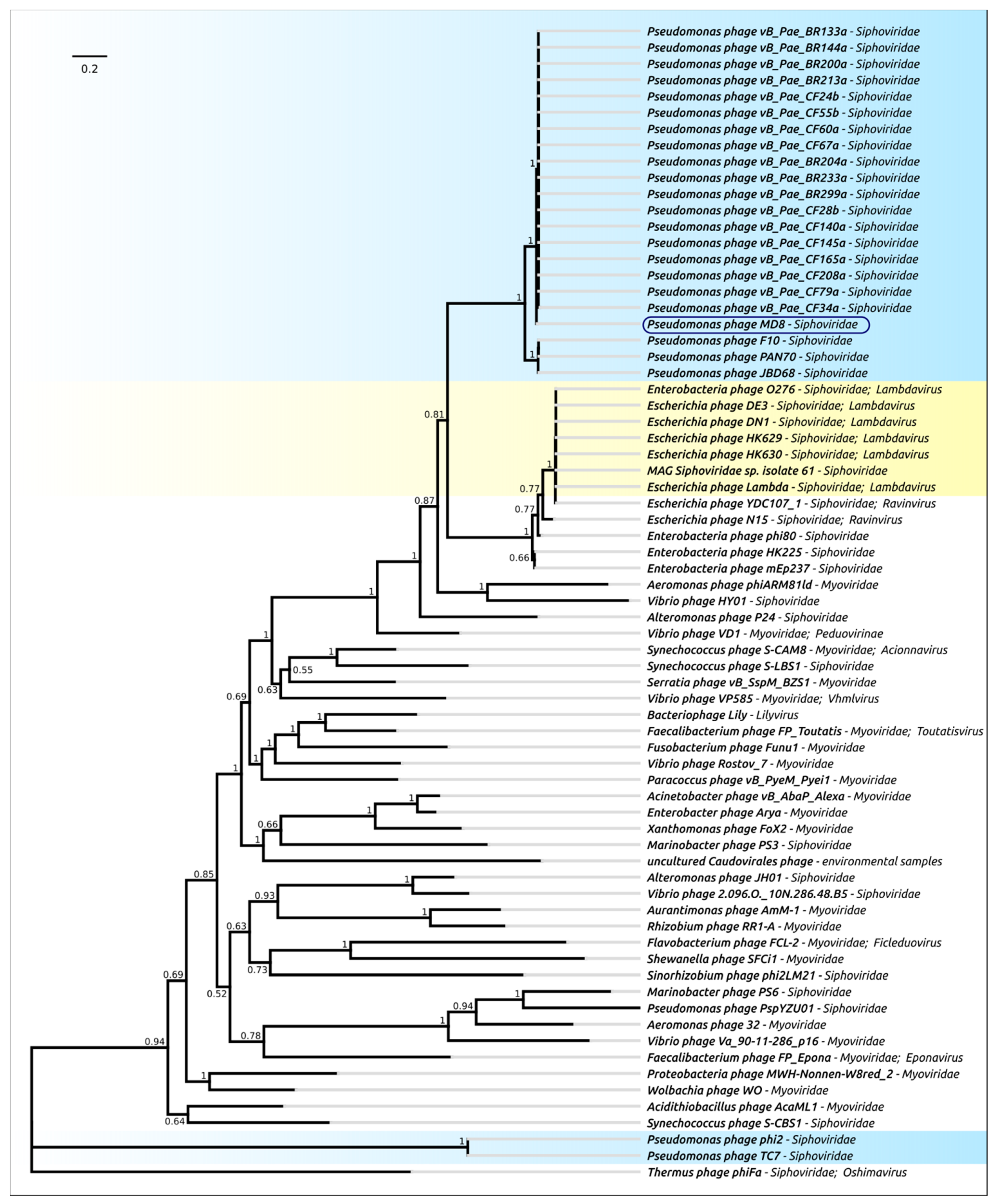
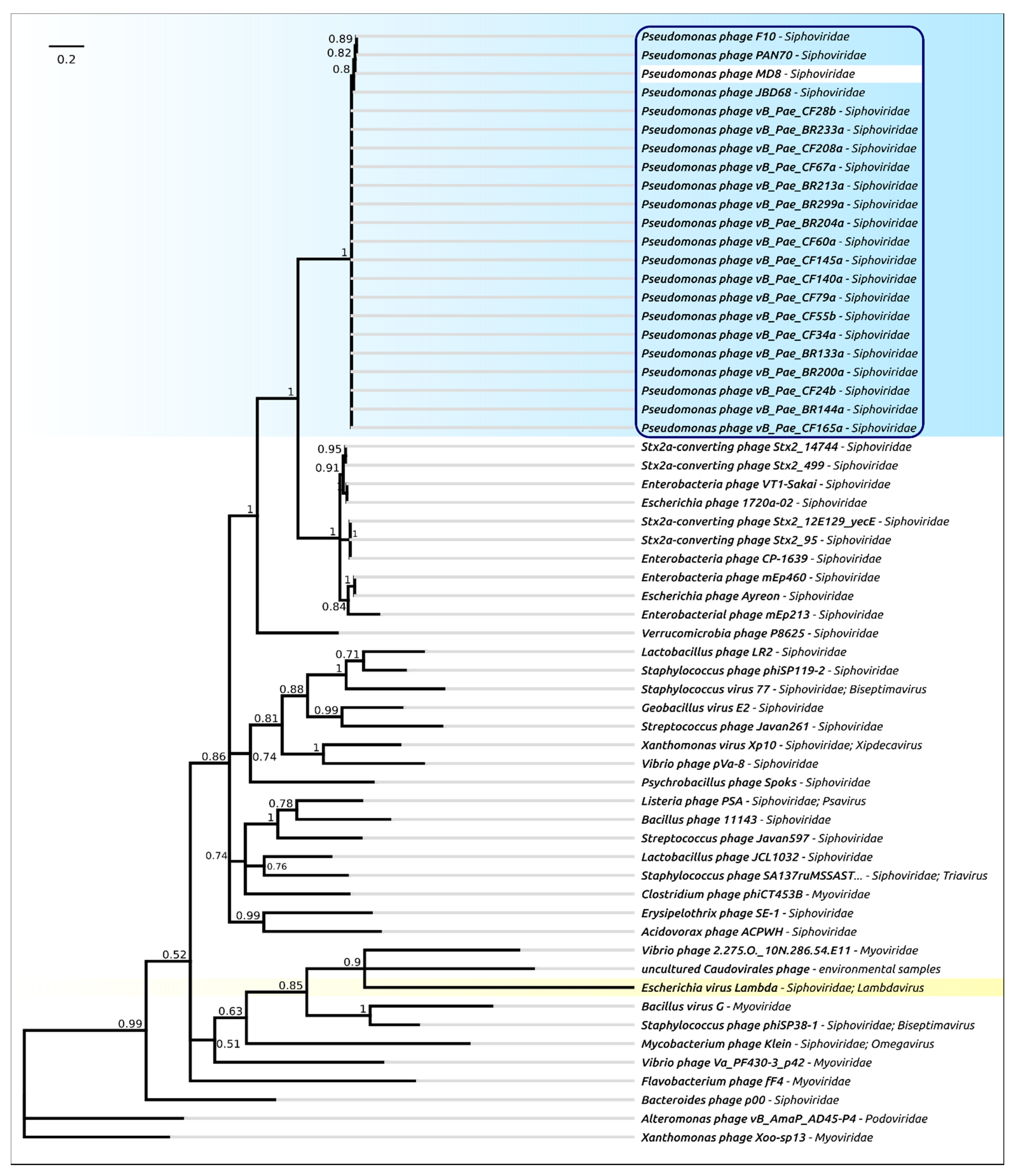
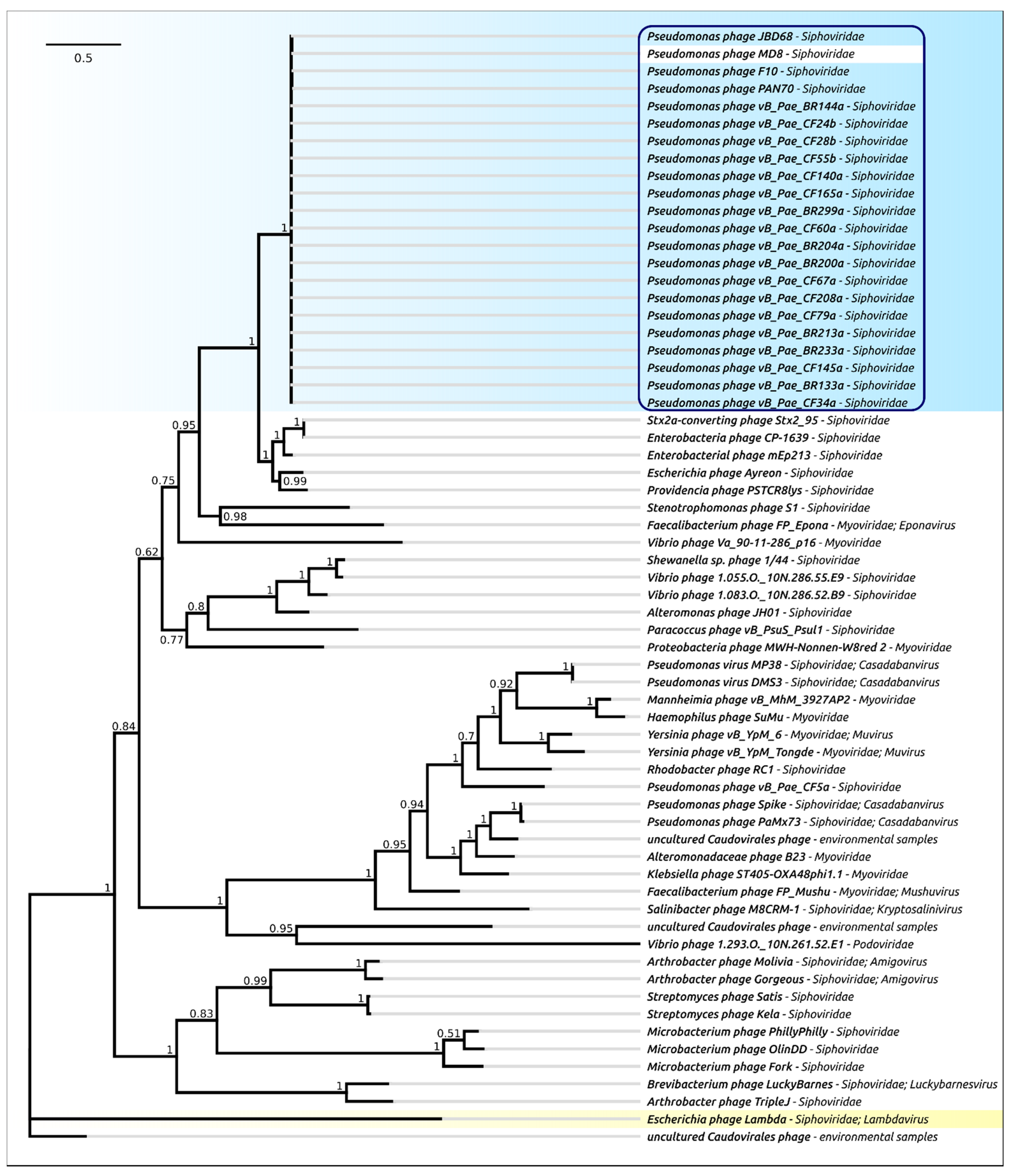
2.4. Terminase Phylogeny
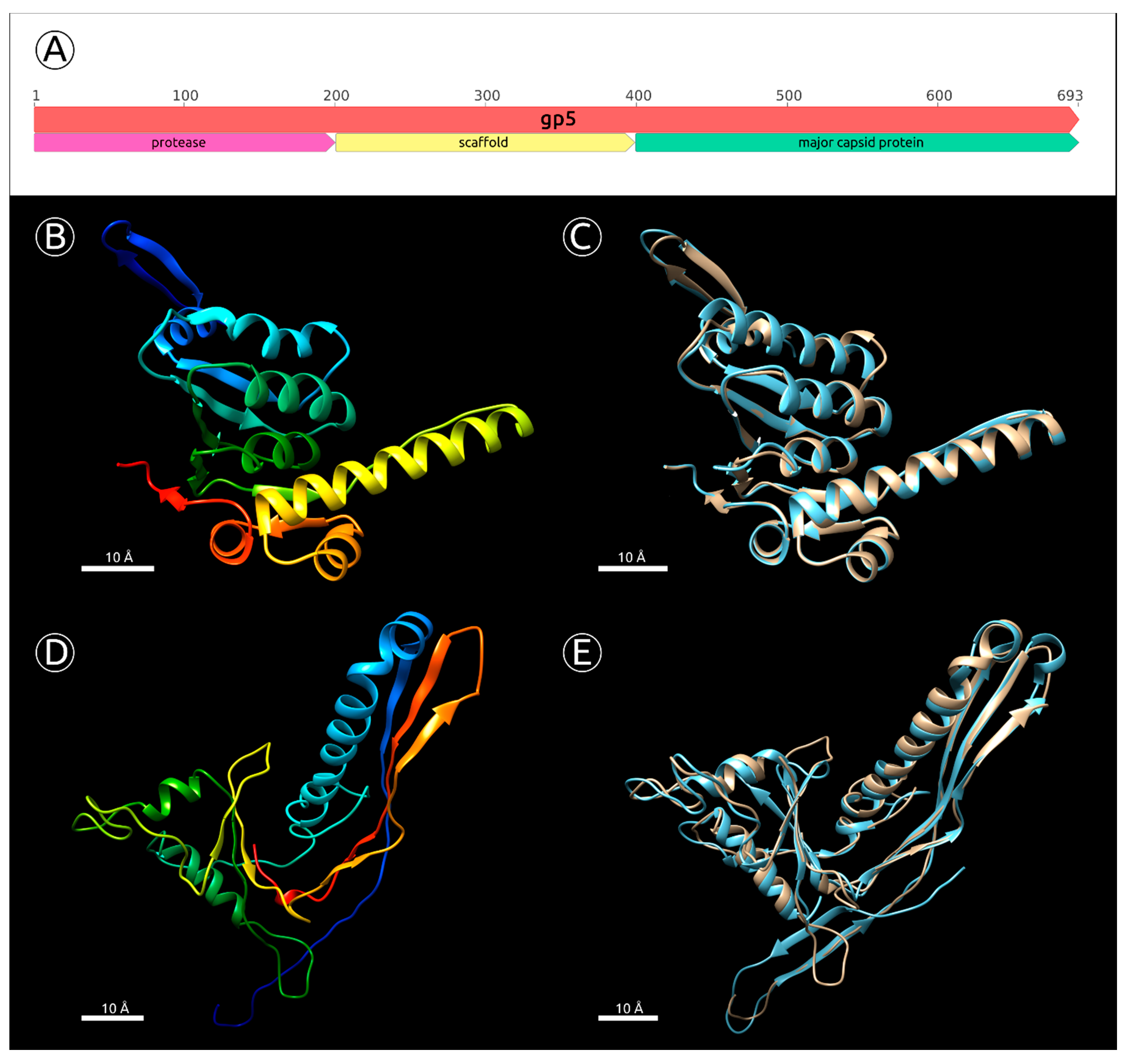
2.5. Major Capsid Protein: Gene, Structure and Phylogeny
- BLAST and HMM-motif searches found significant similarities between the N-terminal part of MD8 gp5 and phage capsid maturation bacterial and other proteases. The length of MD8 gp5 was 693 residues. The Phyre2 modelling server estimated the range of the proteolytic region aligned with PDB structures of known proteases to be about 15–210 amino acid residues of gp5, with 100% confidence. The HHpred HMM search aligned the MD8 gp5 with the protease region of the protease and scaffolding protein of phage λ, with 99.9% probability (Supplementary Figure S5);
- The BLAST and HMM comparison demonstrated significant similarities between the C-terminal part of MD8 gp5 and major capsid proteins of various bacteriophages. Phyre2 modelled the residues 456–690 of MD8 gp5 with bacteriophage HK97 procapsid II, with 98.5% confidence (Figure 6D,E), and the HHpred search had a 98.6% probability. HHpred also pointed to the major capsid protein of Escherichia phage Mu being the closest relative, with a probability of 99.9% and an E-value of 1.3 × 10−28;
- Analysis of the HMM alignment and modelling of the secondary structure demonstrated that the C-terminal part of the phage λ protease and scaffolding protein, containing the scaffolding domain, possessed a similar secondary structure with MD8 gp5 for 80% of the λ scaffolding domain, except for 40 final C-terminal residues. Furthermore, no homologs or similar scaffolding proteins were found by the search of all other predicted proteins of phage MD8.
2.6. Replication Proteins
2.7. Lysogeny and Integration Proteins
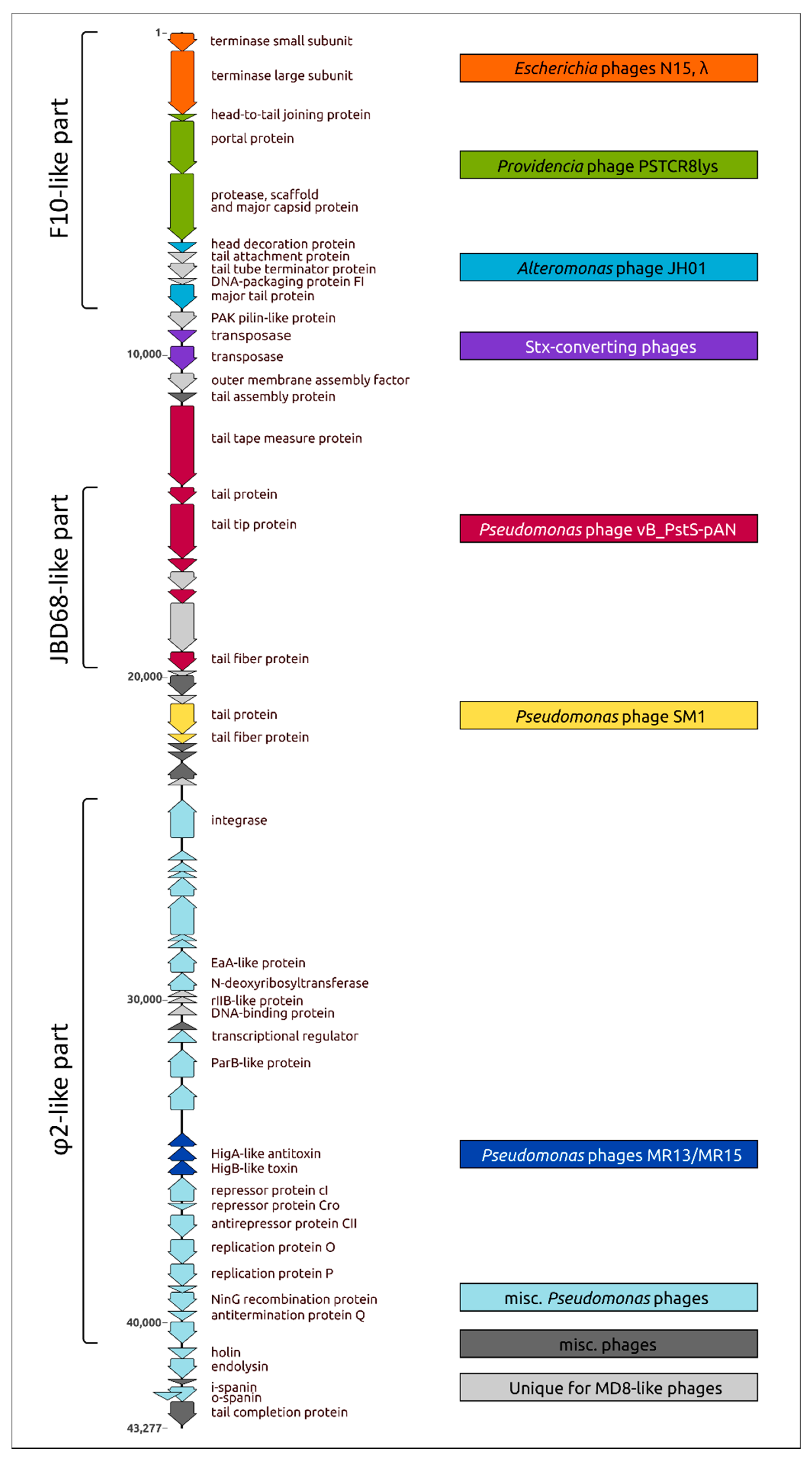
2.8. Adsorption Apparatus
2.9. Lysis Machinery
2.10. Toxin-Antitoxin System, Virulence Factors
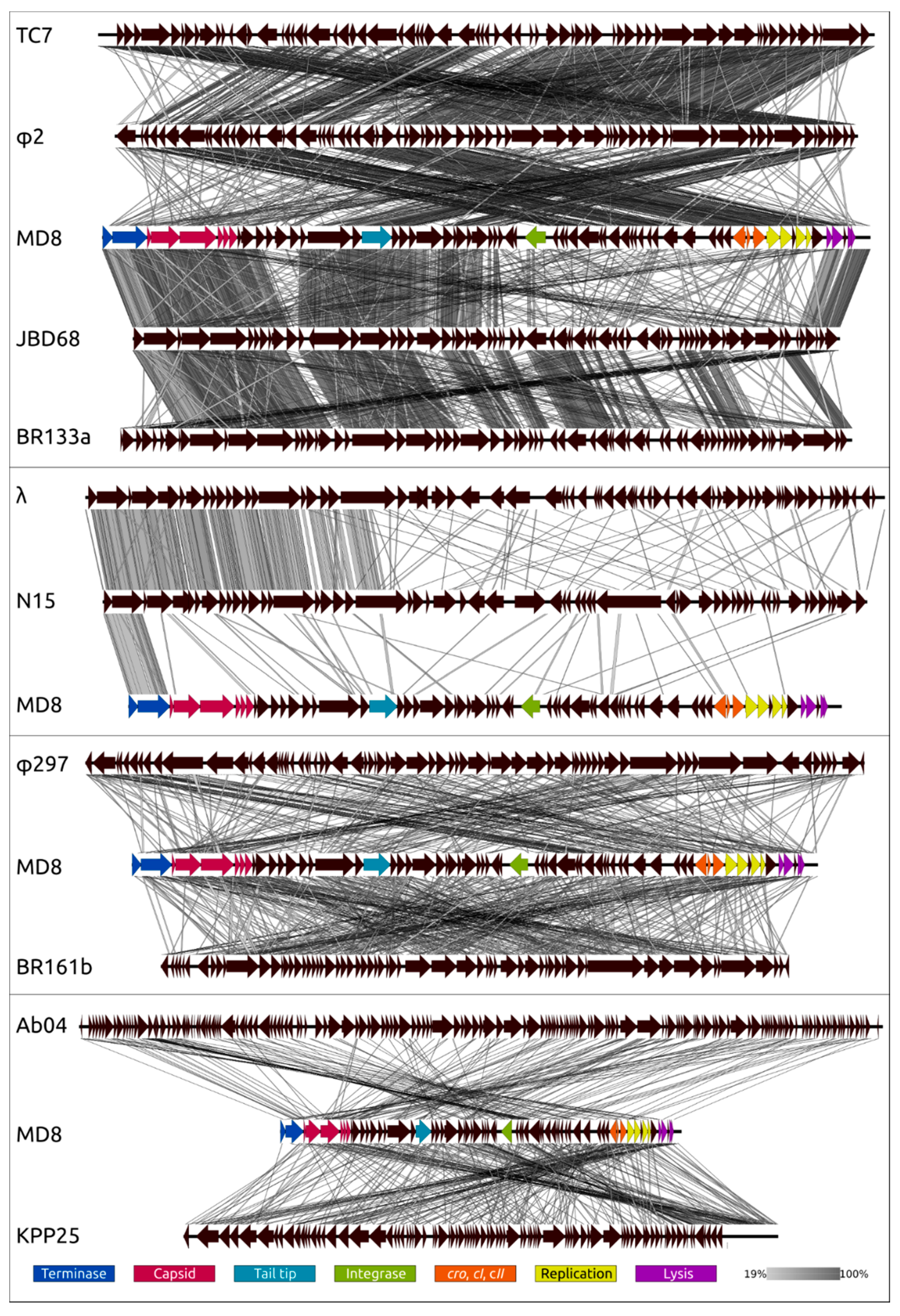
2.11. Proteome Analysis and Comparisons with Related Phages
2.12. Phylogeny of Proteins and Mosaicism of MD8 Genome
3. Discussion
3.1. Phage MD8 and General Problems with the Lambdoids’ Classification
3.2. Biological Properties, Genomic Organisation and Classification at the Level of Family
3.3. Applicability of the Whole-Genome and Proteome Comparisons
3.4. Phylogenetic Analysis of Individual Genes and Genomic Modules
3.5. Delineation of Subfamilies, Genera and Species
4. Materials and Methods
4.1. Phage Propagation and Purification
4.2. Phage Biology Experiments
4.3. Electron Microscopy
4.4. Phage Sequencing and Annotation
4.5. Intergenomic Comparison and Proteome Analysis
4.6. Phylogenetic Analysis
4.7. Analysis and Modelling of Protein Structure
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Esther, M. Zimmer Lederberg Memorial Website. Available online: http://www.estherlederberg.com/home.html (accessed on 14 June 2021).
- Lederberg, E.M.; Lederberg, J. Genetic studies of lysogenicity in Escherichia coli. Genetics 1953, 38, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Weigle, J.J. Induction of Mutations in a Bacterial Virus. Proc. Natl. Acad. Sci. USA 1953, 39, 628–636. [Google Scholar] [CrossRef] [Green Version]
- Jacob, F.; Wollman, E.L. Induction of Phage Development in Lysogenic Bacteria. Cold Spring Harb. Symp. Quant. Biol. 1953, 18, 101–121. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, A.D. A genetic study of the temperate coliphage λ. Virology 1955, 1, 424–443. [Google Scholar] [CrossRef]
- Campbell, A. Sensitive mutants of bacteriophage λ. Virology 1961, 14, 22–32. [Google Scholar] [CrossRef]
- Casjens, S.R.; Hendrix, R.W. Bacteriophage lambda: Early pioneer and still relevant. Virology 2015, 479–480, 310–330. [Google Scholar] [CrossRef] [Green Version]
- Dove, W.F. The Genetics of the Lambdoid Phages. Annu. Rev. Genet. 1968, 2, 305–340. [Google Scholar] [CrossRef]
- Campbell, A. Comparative Molecular Biology of Lambdoid Phages. Annu. Rev. Microbiol. 1994, 48, 193–222. [Google Scholar] [CrossRef]
- Kameyama, L.; Fernandez, L.; Calderon, J.; Ortiz-Rojas, A.; Patterson, T.A. Characterization of Wild Lambdoid Bacteriophages: Detection of a Wide Distribution of Phage Immunity Groups and Identification of a Nus-Dependent, Nonlambdoid Phage Group. Virology 1999, 263, 100–111. [Google Scholar] [CrossRef] [Green Version]
- Campbell, A.; Botstein, D. Evolution of the Lambdoid Phages. Cold Spring Harb. Monogr. Arch. 1983, 13, 365–380. [Google Scholar] [CrossRef]
- Juhala, R.J.; E Ford, M.; Duda, R.; Youlton, A.; Hatfull, G.F.; Hendrix, R.W. Genomic sequences of bacteriophages HK97 and HK022: Pervasive genetic mosaicism in the lambdoid bacteriophages. J. Mol. Biol. 2000, 299, 27–51. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brüssow, H.; Hendrix, R.W. Phage Genomics: Small Is Beautiful. Cell 2002, 108, 13–16. [Google Scholar] [CrossRef] [Green Version]
- Clark, A.J.; Inwood, W.; Cloutier, T.; Dhillon, T. Nucleotide sequence of coliphage HK620 and the evolution of lambdoid phages. J. Mol. Biol. 2001, 311, 657–679. [Google Scholar] [CrossRef] [PubMed]
- Campbell, A.; Schneider, S.J.; Song, B. Lambdoid phages as elements of bacterial genomes (integrase/phage21/Escherichia coli K-12/icd gene). Genetica 1992, 86, 259–267. [Google Scholar] [CrossRef] [PubMed]
- Brüssow, H.; Canchaya, C.; Hardt, W.-D. Phages and the Evolution of Bacterial Pathogens: From Genomic Rearrangements to Lysogenic Conversion. Microbiol. Mol. Biol. Rev. 2004, 68, 560–602. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofer, U. Stop that plasmid. Nat. Rev. Genet. 2020, 18, 264–265. [Google Scholar] [CrossRef] [PubMed]
- Drucker, V.V.; Dutova, N.V. Study of the morphological diversity of bacteriophages in Lake Baikal. Dokl. Biol. Sci. 2006, 410, 421–423. [Google Scholar] [CrossRef] [PubMed]
- Drucker, V.V.; Potapov, S.A.; Gorshkova, A.S.; Belykh, O.I. Bacteriophages of Lake Baikal; SO RAS: Novosibirsk, Russia, 2020; ISBN 978-5-7692-1678-7. [Google Scholar]
- Matsumoto, T.; Fujita, M. Chronic Pseudomonas aeruginosa Infection as the Pathogenesis of Chronic Obstructive Pulmonary Disease. In Bacterial Pathogenesis and Antibacterial Control; InTech: London, UK, 2018. [Google Scholar]
- Crull, M.R.; Somayaji, R.; Ramos, K.J.; Caldwell, E.; Mayer-Hamblett, N.; Aitken, M.L.; Nichols, D.P.; Rowhani-Rahbar, A.; Goss, C.H. Changing Rates of Chronic Pseudomonas aeruginosa Infections in Cystic Fibrosis: A Population-Based Cohort Study. Clin. Infect. Dis. 2018, 67, 1089–1095. [Google Scholar] [CrossRef]
- Serra, R.; Grande, R.; Butrico, L.; Rossi, A.; Settimio, U.F.; Caroleo, B.; Amato, B.; Gallelli, L.; de Franciscis, S. Chronic wound infections: The role of Pseudomonas aeruginosa and Staphylococcus aureus. Expert Rev. Anti-Infect. Ther. 2015, 13, 605–613. [Google Scholar] [CrossRef]
- Kwan, T.; Liu, J.; DuBow, M.; Gros, P.; Pelletier, J. Comparative Genomic Analysis of 18 Pseudomonas aeruginosa Bacteriophages. J. Bacteriol. 2006, 188, 1184–1187. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.; Xu, J.; Gu, Y.; Zhang, R.; Zhu, Y.; Liu, X. Molecular Characterization and Comparative Genomic Analysis of vB_PaeP_YA3, a Novel Temperate Bacteriophage of Pseudomonas aeruginosa. Front. Microbiol. 2020, 11, 947. [Google Scholar] [CrossRef]
- International Committee on Taxonomy of Viruses (ICTV). Available online: https://talk.ictvonline.org/?Redirected=true (accessed on 14 June 2021).
- Shapiro, J.W.; Putonti, C. Gene Co-occurrence Networks Reflect Bacteriophage Ecology and Evolution. mBio 2018, 9, e01870-17. [Google Scholar] [CrossRef] [Green Version]
- Chibani, C.M.; Farr, A.; Klama, S.; Dietrich, S.; Liesegang, H. Classifying the Unclassified: A Phage Classification Method. Viruses 2019, 11, 195. [Google Scholar] [CrossRef] [Green Version]
- Holguín, A.V.; Rangel, G.; Clavijo, V.; Prada, C.; Mantilla, M.; Gomez, M.C.; Kutter, E.; Taylor, C.; Fineran, P.C.; Barrios, A.F.G.; et al. Phage ΦPan70, a Putative Temperate Phage, Controls Pseudomonas aeruginosa in Planktonic, Biofilm and Burn Mouse Model Assays. Viruses 2015, 7, 4602–4623. [Google Scholar] [CrossRef]
- Tariq, M.A.; Everest, F.L.C.; Cowley, L.; Wright, R.; Holt, G.S.; Ingram, H.; Duignan, L.A.; Nelson, A.; Lanyon, C.V.; Perry, A.; et al. Temperate Bacteriophages from Chronic Pseudomonas aeruginosa Lung Infections Show Disease-Specific Changes in Host Range and Modulate Antimicrobial Susceptibility. mSystems 2019, 4, e00191-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Belkum, A.; Soriaga, L.B.; LaFave, M.C.; Akella, S.; Veyrieras, J.-B.; Barbu, E.M.; Shortridge, D.; Blanc, B.; Hannum, G.; Zambardi, G.; et al. Phylogenetic Distribution of CRISPR-Cas Systems in Antibiotic-Resistant Pseudomonas aeruginosa. mBio 2015, 6, e01796-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moraru, C.; Varsani, A.; Kropinski, A. VIRIDIC—A Novel Tool to Calculate the Intergenomic Similarities of Prokaryote-Infecting Viruses. Viruses 2020, 12, 1268. [Google Scholar] [CrossRef] [PubMed]
- Smith, K.C.; Castro-Nallar, E.; Fisher, J.N.; Breakwell, D.P.; Grose, J.H.; Burnett, S.H. Phage cluster relationships identified through single gene analysis. BMC Genom. 2013, 14, 1–16. [Google Scholar] [CrossRef] [Green Version]
- King, J.; Casjens, S. Catalytic head assembling protein in virus morphogenesis. Nat. Cell Biol. 1974, 251, 112–119. [Google Scholar] [CrossRef]
- Duda, R.L.; Oh, B.; Hendrix, R.W. Functional Domains of the HK97 Capsid Maturation Protease and the Mechanisms of Protein Encapsidation. J. Mol. Biol. 2013, 425, 2765–2781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fokine, A.; Rossmann, M.G. Common Evolutionary Origin of Procapsid Proteases, Phage Tail Tubes, and Tubes of Bacterial Type VI Secretion Systems. Structure 2016, 24, 1928–1935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duda, R.L.; Hempel, J.; Michel, H.; Shabanowitz, J.; Hunt, D.; Hendrix, R.W. Structural transitions during bacteriophage HK97 head assembly. J. Mol. Biol. 1995, 247, 618–635. [Google Scholar] [CrossRef]
- Medina, E.; Wieczorek, D.; Medina, E.M.; Yang, Q.; Feiss, M.; Catalano, C.E. Assembly and Maturation of the Bacteriophage Lambda Procapsid: GpC Is the Viral Protease. J. Mol. Biol. 2010, 401, 813–830. [Google Scholar] [CrossRef]
- Chang, J.R.; Spilman, M.S.; Rodenburg, C.M.; Dokland, T. Functional domains of the bacteriophage P2 scaffolding protein: Identification of residues involved in assembly and protease activity. Virology 2009, 384, 144–150. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Sun, L.; Zhang, Z.; Fokine, A.; Padilla-Sanchez, V.; Hanein, D.; Jiang, W.; Rossmann, M.G.; Rao, V.B. Cryo-EM structure of the bacteriophage T4 isometric head at 3.3-Å resolution and its relevance to the assembly of icosahedral viruses. Proc. Natl. Acad. Sci. USA 2017, 114, E8184–E8193. [Google Scholar] [CrossRef] [Green Version]
- Guo, F.; Liu, Z.; Fang, P.-A.; Zhang, Q.; Wright, E.T.; Wu, W.; Zhang, C.; Vago, F.; Ren, Y.; Jakana, J.; et al. Capsid expansion mechanism of bacteriophage T7 revealed by multistate atomic models derived from cryo-EM reconstructions. Proc. Natl. Acad. Sci. USA 2014, 111, E4606–E4614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casjens, S.; King, J. P22 morphogenesis I: Catalytic scaffolding protein in capsid assembly. J. Supramol. Struct. 1974, 2, 202–224. [Google Scholar] [CrossRef] [PubMed]
- Nelson, R.A.; E Reilly, B.; Anderson, D.L. Morphogenesis of bacteriophage phi 29 of Bacillus subtilis: Preliminary isolation and characterization of intermediate particles of the assembly pathway. J. Virol. 1976, 19, 518–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shaw, J.E.; Murialdo, H. Morphogenetic genes C and Nu3 overlap in bacteriophage λ. Nature 1980, 283, 30–35. [Google Scholar] [CrossRef]
- Zylicz, M.; Liberek, K.; Wawrzynow, A.; Georgopoulos, C. Formation of the Preprimosome Protects λ O from RNA Transcription-Dependent Proteolysis by ClpP/ClpX. Proc. Natl. Acad. Sci. USA 1998, 95, 15259–15263. [Google Scholar] [CrossRef] [Green Version]
- Roberts, R.; McMacken, R. The bacteriopbage λ O replication protein: Isolation and characterization of the amplified initiator. Nucleic Acids Res. 1983, 11, 7435–7452. [Google Scholar] [CrossRef]
- Hayes, S.; Erker, C.; Horbay, M.A.; Marciniuk, K.; Wang, W.; Hayes, C. Phage Lambda p Protein: Trans-Activation, Inhibition Phenotypes and Their Suppression. Viruses 2013, 5, 619–653. [Google Scholar] [CrossRef]
- Van der Wilk, F.; Dullemans, A.; Verbeek, M.; Heuvel, J.F.V.D. Isolation and Characterization of APSE-1, a Bacteriophage Infecting the Secondary Endosymbiont of Acyrthosiphon pisum. Virology 1999, 262, 104–113. [Google Scholar] [CrossRef] [Green Version]
- Ravin, V.; Ravin, N.; Casjens, S.; Ford, M.E.; Hatfull, G.F.; Hendrix, R.W. Genomic sequence and analysis of the atypical temperate bacteriophage N15. J. Mol. Biol. 2000, 299, 53–73. [Google Scholar] [CrossRef]
- Tarkowski, T.A.; Mooney, D.; Thomason, L.C.; Stahl, F.W. Gene Products Encoded in the NinR Region of Phage λ Participate in Red-Mediated Recombination. Genes Cells 2002, 7, 351–363. [Google Scholar] [CrossRef]
- Hollifield, W.C.; Kaplan, E.N.; Huang, H.V. Efficient recABC-dependent, homologous recombination between coliphage lambda and plasmids requires a phage ninR region gene. Mol. Genet. Genom. 1987, 210, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Kaiser, A.D. Mutations in a temperate bacteriophage affecting its ability to lysogenize Escherichia coli. Virology 1957, 3, 42–61. [Google Scholar] [CrossRef]
- Johnson, A.; Meyer, B.J.; Ptashne, M. Mechanism of action of the cro protein of bacteriophage lambda. Proc. Natl. Acad. Sci. USA 1978, 75, 1783–1787. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- García, P.; Ladero, V.; Alonso, J.C.; Suárez, J.E. Cooperative Interaction of CI Protein Regulates Lysogeny of Lactobacillus casei by Bacteriophage A2. J. Virol. 1999, 73, 3920–3929. [Google Scholar] [CrossRef] [Green Version]
- Kornitzer, D.; Altuvia, S.; Oppenheim, A.B. The activity of the CIII regulator of lambdoid bacteriophages resides within a 24-amino acid protein domain. Proc. Natl. Acad. Sci. USA 1991, 88, 5217–5221. [Google Scholar] [CrossRef] [Green Version]
- Kobiler, O.; Rokney, A.; Oppenheim, A.B. Phage Lambda CIII: A Protease Inhibitor Regulating the Lysis-Lysogeny Decision. PLoS ONE 2007, 2, e363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayes, F.; Austin, S.J. Specificity determinants of the P1 and P7 plasmid centromere analogs. Proc. Natl. Acad. Sci. USA 1993, 90, 9228–9232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Letarov, A.V.; Kulikov, E. Adsorption of bacteriophages on bacterial cells. Biochemistry 2017, 82, 1632–1658. [Google Scholar] [CrossRef]
- Dupont, K.; Vogensen, F.K.; Neve, H.; Bresciani, J.; Josephsen, J. Identification of the Receptor-Binding Protein in 936-Species Lactococcal Bacteriophages. Appl. Environ. Microbiol. 2004, 70, 5818–5824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fokine, A.; Islam, M.Z.; Zhang, Z.; Bowman, V.D.; Rao, V.B.; Rossmann, M.G. Structure of the Three N-Terminal Immunoglobulin Domains of the Highly Immunogenic Outer Capsid Protein from a T4-Like Bacteriophage. J. Virol. 2011, 85, 8141–8148. [Google Scholar] [CrossRef] [Green Version]
- Werts, C.; Michel, V.; Hofnung, M.; Charbit, A. Adsorption of bacteriophage lambda on the LamB protein of Escherichia coli K-12: Point mutations in gene J of lambda responsible for extended host range. J. Bacteriol. 1994, 176, 941–947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Randall-Hazelbauer, L.; Schwartz, M. Isolation of the Bacteriophage Lambda Receptor from Escherichia coli. J. Bacteriol. 1973, 116, 1436–1446. [Google Scholar] [CrossRef] [Green Version]
- Hendrix, R.; Duda, R. Bacteriophage lambda PaPa: Not the mother of all lambda phages. Science 1992, 258, 1145–1148. [Google Scholar] [CrossRef]
- Berry, J.; Rajaure, M.; Pang, T.; Young, R. The Spanin Complex Is Essential for Lambda Lysis. J. Bacteriol. 2012, 194, 5667–5674. [Google Scholar] [CrossRef] [Green Version]
- Young, R. Phage lysis: Three steps, three choices, one outcome. J. Microbiol. 2014, 52, 243–258. [Google Scholar] [CrossRef]
- Evseev, P.; Lukianova, A.; Shneider, M.; Korzhenkov, A.; Bugaeva, E.; Kabanova, A.; Miroshnikov, K.; Kulikov, E.; Toshchakov, S.; Ignatov, A.; et al. Origin and Evolution of Studiervirinae Bacteriophages Infecting Pectobacterium: Horizontal Transfer Assists Adaptation to New Niches. Microorganisms 2020, 8, 1707. [Google Scholar] [CrossRef]
- Shneider, M.M.; Lukianova, A.A.; Evseev, P.V.; Shpirt, A.M.; Kabilov, M.R.; Tokmakova, A.D.; Miroshnikov, K.K.; Obraztsova, E.A.; Baturina, O.A.; Shashkov, A.S.; et al. Autographivirinae Bacteriophage Arno 160 Infects Pectobacterium carotovorum via Depolymerization of the Bacterial O-Polysaccharide. Int. J. Mol. Sci. 2020, 21, 3170. [Google Scholar] [CrossRef] [PubMed]
- Kędzierska, B.; Hayes, F. Emerging Roles of Toxin-Antitoxin Modules in Bacterial Pathogenesis. Molecules 2016, 21, 790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Gao, Z.; Liu, G.; Geng, Z.; Dong, Y.; Zhang, H. Structural Insights Into the Transcriptional Regulation of HigBA Toxin–Antitoxin System by Antitoxin HigA in Pseudomonas Aeruginosa. Front. Microbiol. 2020, 10, 3158. [Google Scholar] [CrossRef]
- Craig, L.; Taylor, R.K.; E Pique, M.; Adair, B.D.; Arvai, A.S.; Singh, M.; Lloyd, S.J.; Shin, D.S.; Getzoff, E.D.; Yeager, M.; et al. Type IV Pilin Structure and Assembly. Mol. Cell 2003, 11, 1139–1150. [Google Scholar] [CrossRef]
- Nishimura, Y.; Yoshida, T.; Kuronishi, M.; Uehara, H.; Ogata, H.; Goto, S. ViPTree: The viral proteomic tree server. Bioinformatics 2017, 33, 2379–2380. [Google Scholar] [CrossRef]
- Latino, L.; Essoh, C.; Blouin, Y.; Thien, H.V.; Pourcel, C. A novel Pseudomonas aeruginosa Bacteriophage, Ab31, a Chimera Formed from Temperate Phage PAJU2 and P. putida Lytic Phage AF: Characteristics and Mechanism of Bacterial Resistance. PLoS ONE 2014, 9, e93777. [Google Scholar] [CrossRef] [Green Version]
- Botstein, D. A Theory of Modular Evolution for bacteriophages. Ann. N. Y. Acad. Sci. 1980, 354, 484–491. [Google Scholar] [CrossRef]
- Labrie, S.; Frois-Moniz, K.; Osburne, M.; Kelly, L.; Roggensack, S.E.; Sullivan, M.B.; Gearin, G.; Zeng, Q.; Fitzgerald, M.; Henn, M.R.; et al. Genomes of marine cyanopodoviruses reveal multiple origins of diversity. Environ. Microbiol. 2013, 15, 1356–1376. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Zhang, S.; Jiao, N.; Chen, F. Comparative Genomic and Phylogenomic Analyses Reveal a Conserved Core Genome Shared by Estuarine and Oceanic Cyanopodoviruses. PLoS ONE 2015, 10, e0142962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mizuno, C.M.; Rodriguez-Valera, F.; Kimes, N.E.; Ghai, R. Expanding the Marine Virosphere Using Metagenomics. PLoS Genet. 2013, 9, e1003987. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Qin, F.; Zhang, R.; Giovannoni, S.J.; Zhang, Z.; Sun, J.; Du, S.; Rensing, C. Pelagiphages in thePodoviridaefamily integrate into host genomes. Environ. Microbiol. 2019, 21, 1989–2001. [Google Scholar] [CrossRef] [PubMed]
- Glusman, G.; Smit, A.F.A. Genome Organization. In Encyclopedia of Complexity and Systems Science; Springer: New York, NY, USA, 2009; pp. 4160–4178. [Google Scholar]
- Zheng, H.; Xie, W. The role of 3D genome organization in development and cell differentiation. Nat. Rev. Mol. Cell Biol. 2019, 20, 535–550. [Google Scholar] [CrossRef]
- Misteli, T. Beyond the Sequence: Cellular Organization of Genome Function. Cell 2007, 128, 787–800. [Google Scholar] [CrossRef] [Green Version]
- International Committee on Taxonomy of Viruses Executive Committee. The new scope of virus taxonomy: Partitioning the virosphere into 15 hierarchical ranks. Nat. Microbiol. 2020, 5, 668–674. [Google Scholar] [CrossRef] [PubMed]
- Kang, H.S.; McNair, K.; Cuevas, D.; Bailey, B.; Segall, A.; Edwards, R. Prophage Genomics Reveals Patterns in Phage Genome Organization and Replication. bioRxiv 2017, 114819. [Google Scholar] [CrossRef] [Green Version]
- Low, S.J.; Džunková, M.; Chaumeil, P.-A.; Parks, D.H.; Hugenholtz, P. Evaluation of a concatenated protein phylogeny for classification of tailed double-stranded DNA viruses belonging to the order Caudovirales. Nat. Microbiol. 2019, 4, 1306–1315. [Google Scholar] [CrossRef]
- Gontcharov, A.A.; Marin, B.; Melkonian, M. Are Combined Analyses Better Than Single Gene Phylogenies? A Case Study Using SSU rDNA and rbcL Sequence Comparisons in the Zygnematophyceae (Streptophyta). Mol. Biol. Evol. 2003, 21, 612–624. [Google Scholar] [CrossRef]
- Glazko, G.; Makarenkov, V.; Liu, J.; Mushegian, A. Evolutionary history of bacteriophages with double-stranded DNA genomes. Biol. Direct 2007, 2, 36. [Google Scholar] [CrossRef] [Green Version]
- Serwer, P.; Hayes, S.J.; A Thomas, J.; Hardies, S.C. Propagating the missing bacteriophages: A large bacteriophage in a new class. Virol. J. 2007, 4, 21. [Google Scholar] [CrossRef] [Green Version]
- Colombet, J.; Robin, A.; Lavie, L.; Bettarel, Y.; Cauchie, H.; Sime-Ngando, T. Virioplankton ‘pegylation’: Use of PEG (polyethylene glycol) to concentrate and purify viruses in pelagic ecosystems. J. Microbiol. Methods 2007, 71, 212–219. [Google Scholar] [CrossRef]
- Czajkowski, R.; Ozymko, Z.; Lojkowska, E. Isolation and characterization of novel soilborne lytic bacteriophages infectingDickeyaspp. biovar 3 (‘D. solani’). Plant Pathol. 2014, 63, 758–772. [Google Scholar] [CrossRef]
- Brenner, S.; Horne, R. A negative staining method for high resolution electron microscopy of viruses. Biochim. Biophys. Acta (BBA)-Bioenerg. 1959, 34, 103–110. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Seemann, T. Prokka: Rapid Prokaryotic Genome Annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Goodacre, N.; Aljanahi, A.; Nandakumar, S.; Mikailov, M.; Khan, A.S. A Reference Viral Database (RVDB) To Enhance Bioinformatics Analysis of High-Throughput Sequencing for Novel Virus Detection. mSphere 2018, 3, e00069-18. [Google Scholar] [CrossRef] [Green Version]
- Hyatt, D.; Chen, G.-L.; Locascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delcher, A.L. Improved microbial gene identification with GLIMMER. Nucleic Acids Res. 1999, 27, 4636–4641. [Google Scholar] [CrossRef] [PubMed]
- Geneious Prime. Available online: http://www.geneious.com/ (accessed on 7 April 2020).
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 web portal for protein modeling, prediction and analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [Green Version]
- Zimmermann, L.; Stephens, A.; Nam, S.-Z.; Rau, D.; Kübler, J.; Lozajic, M.; Gabler, F.; Söding, J.; Lupas, A.N.; Alva, V. A Completely Reimplemented MPI Bioinformatics Toolkit with a New HHpred Server at its Core. J. Mol. Biol. 2018, 430, 2237–2243. [Google Scholar] [CrossRef] [PubMed]
- Söding, J.; Biegert, A.; Lupas, A.N. The HHpred interactive server for protein homology detection and structure prediction. Nucleic Acids Res. 2005, 33, W244–W248. [Google Scholar] [CrossRef] [Green Version]
- Schattner, P.; Brooks, A.N.; Lowe, T.M. The tRNAscan-SE, snoscan and snoGPS web servers for the detection of tRNAs and snoRNAs. Nucleic Acids Res. 2005, 33, W686–W689. [Google Scholar] [CrossRef]
- Laslett, D.; Canback, B. ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res. 2004, 32, 11–16. [Google Scholar] [CrossRef]
- Edgar, R.C. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 2010, 26, 2460–2461. [Google Scholar] [CrossRef] [Green Version]
- Lee, I.; Kim, Y.O.; Park, S.-C.; Chun, J. OrthoANI: An improved algorithm and software for calculating average nucleotide identity. Int. J. Syst. Evol. Microbiol. 2016, 66, 1100–1103. [Google Scholar] [CrossRef] [PubMed]
- Yoon, S.-H.; Ha, S.-M.; Lim, J.; Kwon, S.; Chun, J. A large-scale evaluation of algorithms to calculate average nucleotide identity. Antonie Van Leeuwenhoek 2017, 110, 1281–1286. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.J.; Petty, N.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Capella-Gutierrez, S.; Silla-Martinez, J.M.; Gabaldon, T. trimAl: A tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 2009, 25, 1972–1973. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Posada, D.; Kozlov, A.M.; Stamatakis, A.; Morel, B.; Flouri, T. ModelTest-NG: A New and Scalable Tool for the Selection of DNA and Protein Evolutionary Models. Mol. Biol. Evol. 2020, 37, 291–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flouri, T.; Izquierdo-Carrasco, F.; Darriba, D.; Aberer, A.; Nguyen, L.-T.; Minh, B.Q.; Von Haeseler, A.; Stamatakis, A. The Phylogenetic Likelihood Library. Syst. Biol. 2015, 64, 356–362. [Google Scholar] [CrossRef] [Green Version]
- Ronquist, F.; Huelsenbeck, J.P. MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 2003, 19, 1572–1574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing Large Minimum Evolution Trees with Profiles instead of a Distance Matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree 2–Approximately Maximum-Likelihood Trees for Large Alignments. PLoS ONE 2010, 5, e9490. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 2006, 22, 2688–2690. [Google Scholar] [CrossRef]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Le, S.Q.; Gascuel, O. An Improved General Amino Acid Replacement Matrix. Mol. Biol. Evol. 2008, 25, 1307–1320. [Google Scholar] [CrossRef] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera-A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NCBI Accession | ANI | Average Aligned Length | Genome Length | Phage |
|---|---|---|---|---|
| KX198612 | 100.00 | 42,840 | 42,840 | Pseudomonas phage MD8 |
| MK510975 | 96.19 | 10,023 | 39,780 | Pseudomonas phage vB_Pae_CF145a |
| MK510976 | 95.86 | 11,062 | 39,780 | Pseudomonas phage vB_Pae_CF165a |
| MK510964 | 95.08 | 10,158 | 39,780 | Pseudomonas phage vB_Pae_CF28b |
| MK510968 | 94.95 | 13,160 | 41,820 | Pseudomonas phage vB_Pae_CF55b |
| MK510969 | 94.51 | 9288 | 34,680 | Pseudomonas phage vB_Pae_CF60a |
| MK510986 | 94.08 | 15,886 | 30,600 | Pseudomonas phage vB_Pae_BR213a |
| MK510988 | 93.96 | 14,820 | 39,780 | Pseudomonas phage vB_Pae_BR299a |
| MK510962 | 93.92 | 7927 | 33,660 | Pseudomonas phage vB_Pae_CF3a |
| MK510990 | 93.91 | 11,921 | 40,800 | Pseudomonas phage vB_Pae_BR133a |
| MK510963 | 93.88 | 16,786 | 39,780 | Pseudomonas phage vB_Pae_CF24b |
| MK510970 | 93.68 | 14,779 | 34,680 | Pseudomonas phage vB_Pae_CF67a |
| MK510974 | 93.68 | 17,260 | 39,780 | Pseudomonas phage vB_Pae_CF140a |
| MK510987 | 93.58 | 17,309 | 39,780 | Pseudomonas phage vB_Pae_BR233a |
| MK510971 | 93.57 | 14,897 | 40,800 | Pseudomonas phage vB_Pae_CF79a |
| MK510985 | 93.56 | 17,306 | 39,780 | Pseudomonas phage vB_Pae_BR204a |
| MK510978 | 93.25 | 15,465 | 39,780 | Pseudomonas phage vB_Pae_CF208a |
| KT887558 | 93.11 | 11,613 | 41,820 | Pseudomonas phage φ2 |
| MK510965 | 93.07 | 15,680 | 40,800 | Pseudomonas phage vB_Pae_CF34a |
| MK510984 | 92.96 | 10,307 | 37,740 | Pseudomonas phage vB_Pae_BR200a |
| MK510992 | 92.93 | 12,157 | 39,780 | Pseudomonas phage vB_Pae_BR144a |
| MG707188 | 92.39 | 10,879 | 42,840 | Pseudomonas phage TC7 |
| KY707339 | 92.35 | 17,040 | 39,780 | Pseudomonas phage JBD68 |
| KJ959591 | 91.60 | 15,111 | 38,760 | Pseudomonas phage PAN70 |
| DQ163912 | 91.59 | 16,514 | 38,760 | Pseudomonas phage F10 |
| MK510993 | 89.11 | 5095 | 37,740 | Pseudomonas phage vB_Pae_BR201a |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Evseev, P.; Lukianova, A.; Sykilinda, N.; Gorshkova, A.; Bondar, A.; Shneider, M.; Kabilov, M.; Drucker, V.; Miroshnikov, K. Pseudomonas Phage MD8: Genetic Mosaicism and Challenges of Taxonomic Classification of Lambdoid Bacteriophages. Int. J. Mol. Sci. 2021, 22, 10350. https://doi.org/10.3390/ijms221910350
Evseev P, Lukianova A, Sykilinda N, Gorshkova A, Bondar A, Shneider M, Kabilov M, Drucker V, Miroshnikov K. Pseudomonas Phage MD8: Genetic Mosaicism and Challenges of Taxonomic Classification of Lambdoid Bacteriophages. International Journal of Molecular Sciences. 2021; 22(19):10350. https://doi.org/10.3390/ijms221910350
Chicago/Turabian StyleEvseev, Peter, Anna Lukianova, Nina Sykilinda, Anna Gorshkova, Alexander Bondar, Mikhail Shneider, Marsel Kabilov, Valentin Drucker, and Konstantin Miroshnikov. 2021. "Pseudomonas Phage MD8: Genetic Mosaicism and Challenges of Taxonomic Classification of Lambdoid Bacteriophages" International Journal of Molecular Sciences 22, no. 19: 10350. https://doi.org/10.3390/ijms221910350