
Structure-Based Understanding of ABCA3 Variants

Abstract
:1. Introduction
2. Results
2.1. Survey of the Known Mutations of Human ABCA3
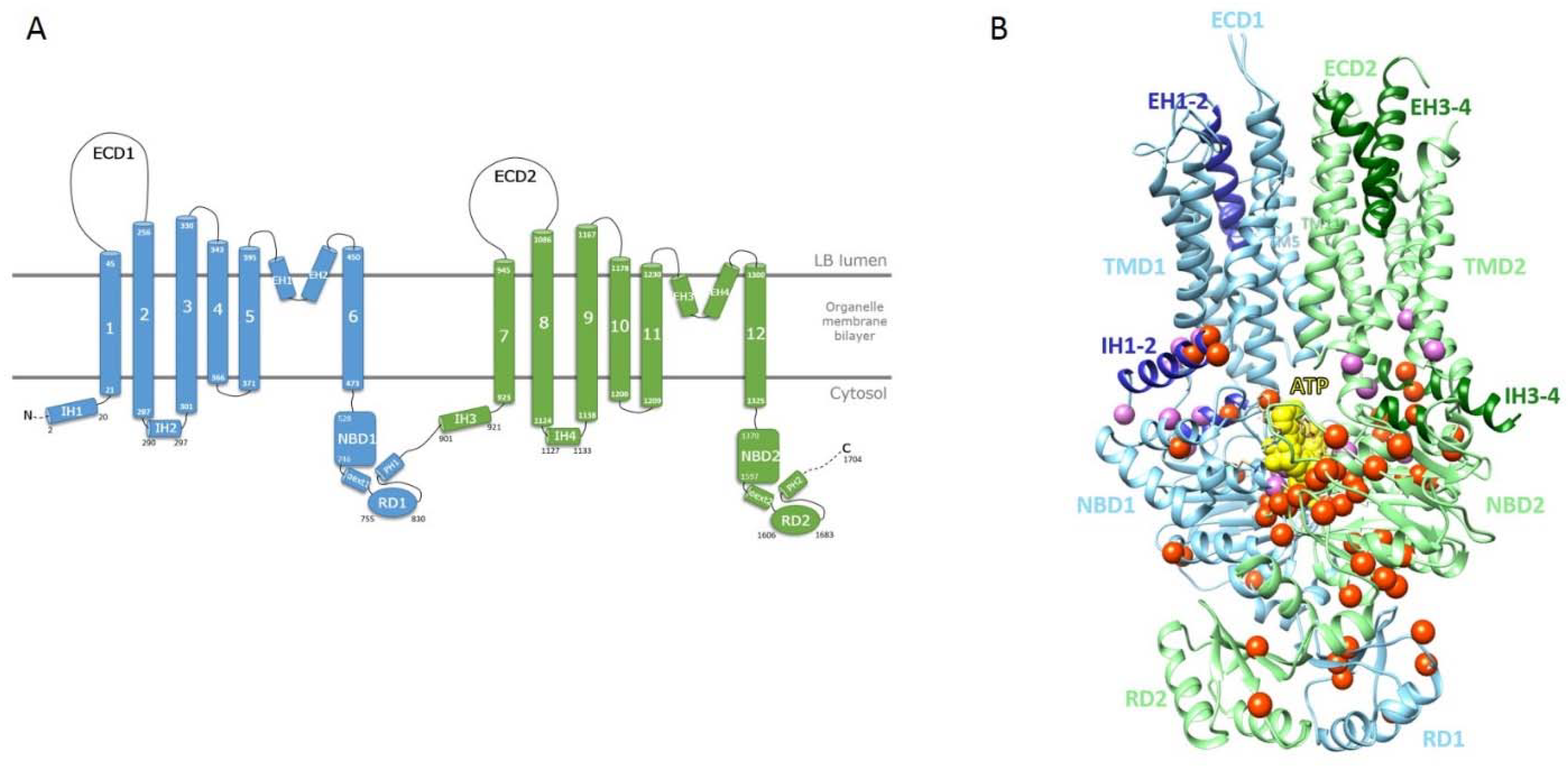
2.2. Mapping of ABCA3 Mutations on a Model of the ATP-Bound 3D Structure of Human ABCA3
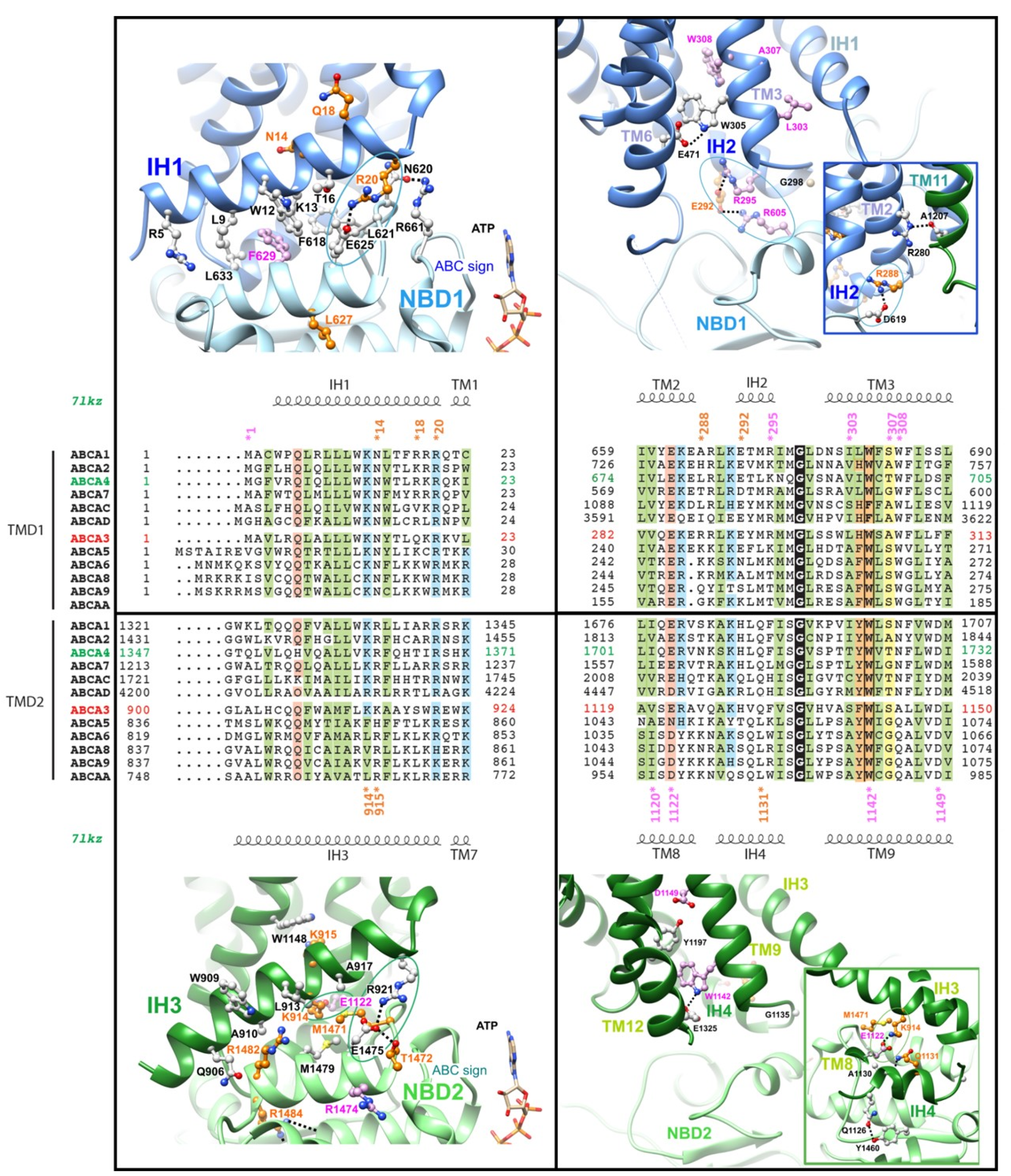
2.3. The Intracellular Helices IH1 to IH4: Connecting TMDs to NBDs
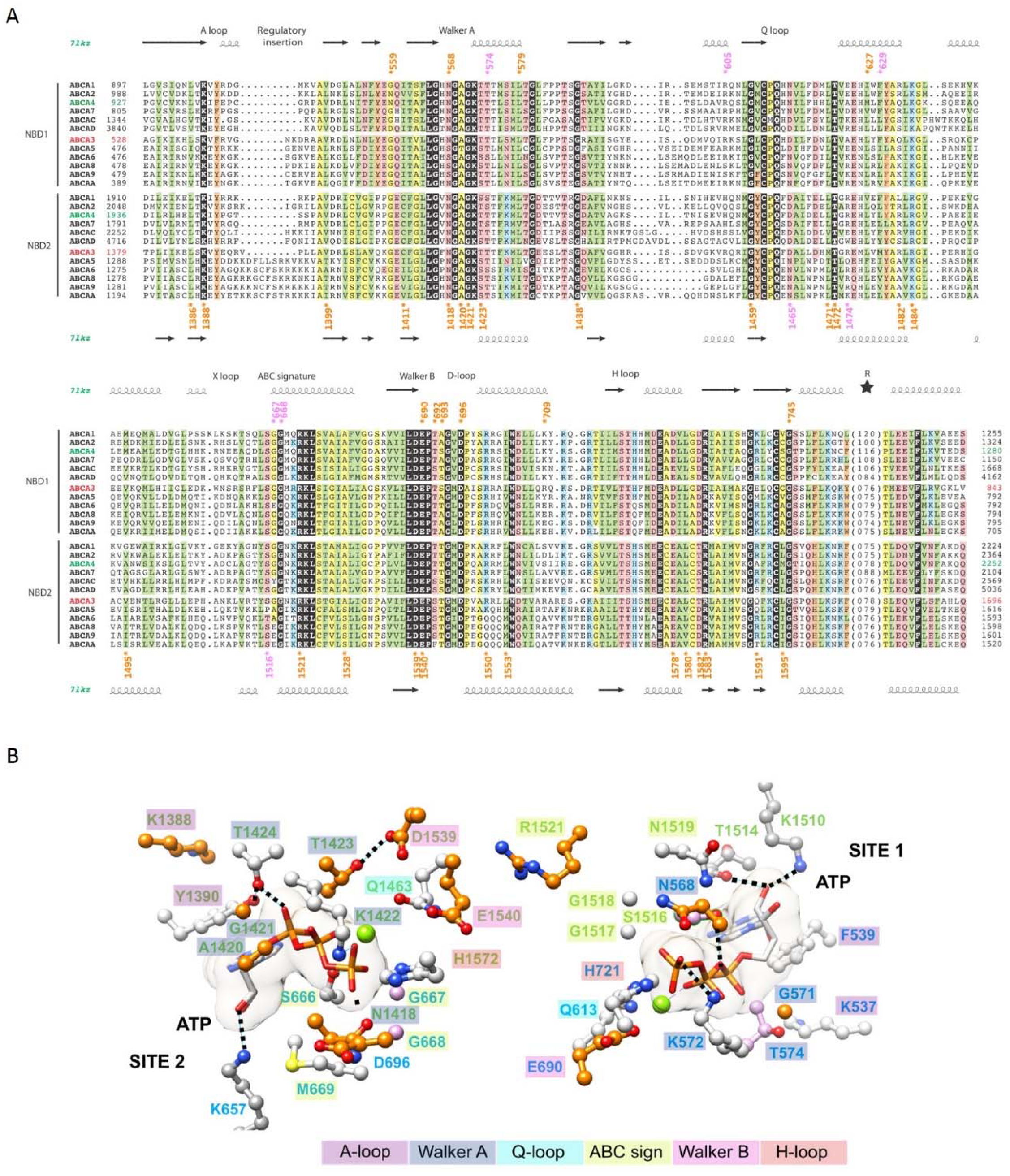
2.4. The ATP-Binding Sites in the NBDs
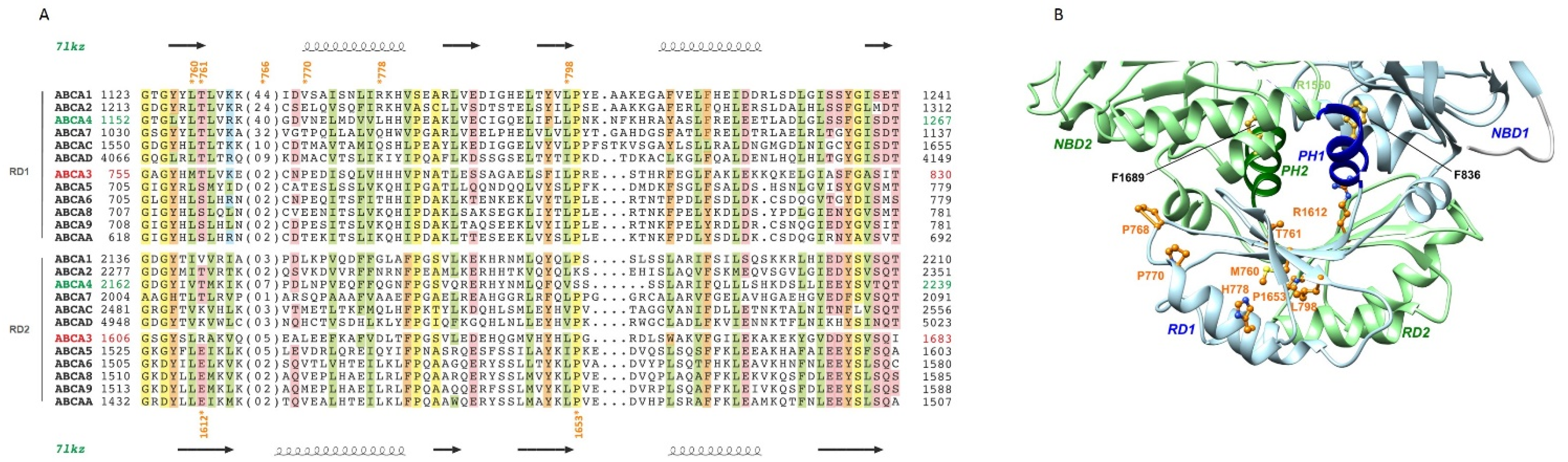
2.5. The Regulatory Domains (RDs)
3. Discussion
4. Materials and Methods
4.1. Variant’s Analysis
4.2. Comparative Modeling and 3D Structure Visualization
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Guagliardo, R.; Pérez-Gil, J.; De Smedt, S.; Raemdonck, K. Pulmonary Surfactant and Drug Delivery: Focusing on the Role of Surfactant Proteins. J. Control. Release 2018, 291, 116–126. [Google Scholar] [CrossRef]
- Beers, M.F.; Mulugeta, S. The Biology of the ABCA3 Lipid Transporter in Lung Health and Disease. Cell Tissue Res. 2017, 367, 481–493. [Google Scholar] [CrossRef]
- Wambach, J.A.; Casey, A.M.; Fishman, M.P.; Wegner, D.J.; Wert, S.E.; Cole, F.S.; Hamvas, A.; Nogee, L.M. Genotype–Phenotype Correlations for Infants and Children with ABCA3 Deficiency. Am. J. Respir. Crit. Care Med. 2014, 189, 1538–1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shulenin, S.; Nogee, L.M.; Annilo, T.; Wert, S.E.; Whitsett, J.A.; Dean, M. ABCA3 Gene Mutations in Newborns with Fatal Surfactant Deficiency. N. Engl. J. Med. 2004, 350, 1296–1303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klay, D.; Platenburg, M.G.J.P.; van Rijswijk, R.H.N.A.J.; Grutters, J.C.; van Moorsel, C.H.M. ABCA3 Mutations in Adult Pulmonary Fibrosis Patients: A Case Series and Review of Literature. Curr. Opin. Pulm. Med. 2020, 26, 293–301. [Google Scholar] [CrossRef] [PubMed]
- Bullard, J.E.; Wert, S.E.; Whitsett, J.A.; Dean, M.; Nogee, L.M. ABCA3 Mutations Associated with Pediatric Interstitial Lung Disease. Am. J. Respir. Crit. Care Med. 2005, 172, 1026–1031. [Google Scholar] [CrossRef] [PubMed]
- Wittmann, T.; Schindlbeck, U.; Höppner, S.; Kinting, S.; Frixel, S.; Kröner, C.; Liebisch, G.; Hegermann, J.; Aslanidis, C.; Brasch, F.; et al. Tools to Explore ABCA3 Mutations Causing Interstitial Lung Disease. Pediatr. Pulmonol. 2016, 51, 1284–1294. [Google Scholar] [CrossRef]
- Peelman, F.; Labeur, C.; Vanloo, B.; Roosbeek, S.; Devaud, C.; Duverger, N.; Denèfle, P.; Rosier, M.; Vandekerckhove, J.; Rosseneu, M. Characterization of the ABCA Transporter Subfamily: Identification of Prokaryotic and Eukaryotic Members, Phylogeny and Topology. J. Mol. Biol. 2003, 325, 259–274. [Google Scholar] [CrossRef]
- Thomas, C.; Aller, S.G.; Beis, K.; Carpenter, E.P.; Chang, G.; Chen, L.; Dassa, E.; Dean, M.; Hoa, F.D.V.; Ekiert, D.; et al. Structural and Functional Diversity Calls for a New Classification of ABC Transporters. FEBS Lett. 2020, 594, 3767–3775. [Google Scholar] [CrossRef]
- Qian, H.; Zhao, X.; Cao, P.; Lei, J.; Yan, N.; Gong, X. Structure of the Human Lipid Exporter ABCA1. Cell 2017, 169, 1228–1239.e10. [Google Scholar] [CrossRef] [Green Version]
- Kaminski, W.E.; Piehler, A.; Wenzel, J.J. ABC A-Subfamily Transporters: Structure, Function and Disease. Biochim. Biophys. Acta Mol. Basis Dis. 2006, 1762, 510–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paolini, A.; Baldassarre, A.; Del Gaudio, I.; Masotti, A. Structural Features of the ATP-Binding Cassette (ABC) Transporter ABCA3. Int. J. Mol. Sci. 2015, 16, 19631–19644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aberuyi, N.; Rahgozar, S.; Dehaghi, Z.K.; Moafi, A.; Masotti, A.; Paolini, A. The Translational Expression of ABCA2 and ABCA3 Is a Strong Prognostic Biomarker for Multidrug Resistance in Pediatric Acute Lymphoblastic Leukemia. OncoTargets Ther. 2017, 10, 3373–3380. [Google Scholar] [CrossRef] [Green Version]
- Kinting, S.; Li, Y.; Forstner, M.; Delhommel, F.; Sattler, M.; Griese, M. Potentiation of ABCA3 Lipid Transport Function by Ivacaftor and Genistein. J. Cell. Mol. Med. 2019, 23, 5225–5234. [Google Scholar] [CrossRef]
- Liu, F.; Lee, J.; Chen, J. Molecular Structures of the Eukaryotic Retinal Importer ABCA4. eLife 2021, 10, e63524. [Google Scholar] [CrossRef]
- Xie, T.; Zhang, Z.; Fang, Q.; Du, B.; Gong, X. Structural Basis of Substrate Recognition and Translocation by Human ABCA4. Nat. Commun. 2021, 12, 3853. [Google Scholar] [CrossRef] [PubMed]
- Grant, G.A. The ACT Domain: A Small Molecule Binding Domain and Its Role as a Common Regulatory Element. J. Biol. Chem. 2006, 281, 33825–33829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chipman, D.M.; Shaanan, B. The ACT Domain Family. Curr. Opin. Struct. Biol. 2001, 11, 694–700. [Google Scholar] [CrossRef]
- Ban, N.; Matsumura, Y.; Sakai, H.; Takanezawa, Y.; Sasaki, M.; Arai, H.; Inagaki, N. ABCA3 as a Lipid Transporter in Pulmonary Surfactant Biogenesis. J. Biol. Chem. 2007, 282, 9628–9634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagata, K.; Yamamoto, A.; Ban, N.; Tanaka, A.R.; Matsuo, M.; Kioka, N.; Inagaki, N.; Ueda, K. Human ABCA3, a Product of a Responsible Gene for Abca3 for Fatal Surfactant Deficiency in Newborns, Exhibits Unique ATP Hydrolysis Activity and Generates Intracellular Multilamellar Vesicles. Biochem. Biophys. Res. Commun. 2004, 324, 262–268. [Google Scholar] [CrossRef] [PubMed]
- Le, L.T.M.; Thompson, J.R.; Aikawa, T.; Kanikeyo, T.; Alam, A. Cryo-EM Structure of Lipid Embedded Human ABCA7 at 3.6 Å Resolution. bioRxiv 2021. [Google Scholar] [CrossRef]
- Bækvad-Hansen, M.; Nordestgaard, B.G.; Dahl, M. Heterozygosity for E292V in ABCA3, Lung Function and COPD in 64,000 Individuals. Respir. Res. 2012, 13, 67. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.Y.; Yang, P.; Wegner, D.J.; Heins, H.B.; Luke, C.J.; Li, F.; White, F.V.; Silverman, G.A.; Cole, F.S.; Wambach, J.A. Functional Characterization of Four ATP-Binding Cassette Transporter A3 Gene (ABCA3) Variants. Hum. Mutat. 2020, 41, 1298–1307. [Google Scholar] [CrossRef]
- Schindlbeck, U.; Wittmann, T.; Höppner, S.; Kinting, S.; Liebisch, G.; Hegermann, J.; Griese, M. ABCA3 Missense Mutations Causing Surfactant Dysfunction Disorders Have Distinct Cellular Phenotypes. Hum. Mutat. 2018, 39, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Coghlan, M.A.; Shifren, A.; Huang, H.J.; Russell, T.D.; Mitra, R.D.; Zhang, Q.; Wegner, D.J.; Cole, F.S.; Hamvas, A. Sequencing of Idiopathic Pulmonary Fibrosis-Related Genes Reveals Independent Single Gene Associations. BMJ Open Respir. Res. 2014, 1, e000057. [Google Scholar] [CrossRef] [Green Version]
- Brasch, F.; Schimanski, S.; Mühlfeld, C.; Barlage, S.; Langmann, T.; Aslanidis, C.; Boettcher, A.; Dada, A.; Schroten, H.; Mildenberger, E.; et al. Alteration of the Pulmonary Surfactant System in Full-Term Infants with Hereditary ABCA3 Deficiency. Am. J. Respir. Crit. Care Med. 2006, 174, 571–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wittmann, T.; Frixel, S.; Höppner, S.; Schindlbeck, U.; Schams, A.; Kappler, M.; Hegermann, J.; Wrede, C.; Liebisch, G.; Vierzig, A.; et al. Increased Risk of Interstitial Lung Disease in Children with a Single R288K Variant of ABCA3. Mol. Med. 2016, 22, 183. [Google Scholar] [CrossRef]
- Wambach, J.A.; Yang, P.; Wegner, D.J.; Heins, H.B.; Kaliberova, L.N.; Kaliberov, S.A.; Curiel, D.T.; White, F.V.; Hamvas, A.; Hackett, B.P.; et al. Functional Characterization of ATP-Binding Cassette Transporter A3 Mutations from Infants with Respiratory Distress Syndrome. Am. J. Respir. Cell Mol. Biol. 2016, 55, 716–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumura, Y.; Ban, N.; Ueda, K.; Inagaki, N. Characterization and Classification of ATP-Binding Cassette Transporter ABCA3 Mutants in Fatal Surfactant Deficiency. J. Biol. Chem. 2006, 281, 34503–34514. [Google Scholar] [CrossRef] [Green Version]
- Höppner, S.; Kinting, S.; Torrano, A.A.; Schindlbeck, U.; Bräuchle, C.; Zarbock, R.; Wittmann, T.; Griese, M. Quantification of Volume and Lipid Filling of Intracellular Vesicles Carrying the ABCA3 Transporter. Biochim. Biophys. Acta BBA Mol. Cell Res. 2017, 1864, 2330–2335. [Google Scholar] [CrossRef]
- Kinting, S.; Höppner, S.; Schindlbeck, U.; Forstner, M.E.; Harfst, J.; Wittmann, T.; Griese, M. Functional Rescue of Misfolding ABCA3 Mutations by Small Molecular Correctors. Hum. Mol. Genet. 2018, 27, 943–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tordai, H.; Jakab, K.; Gyimesi, G.; András, K.; Brózik, A.; Sarkadi, B.; Hegedűs, T. ABCMdb Reloaded: Updates on Mutations in ATP Binding Cassette Proteins. Database 2017, 2017, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Steinhaus, R.; Proft, S.; Schuelke, M.; Cooper, D.N.; Schwarz, J.M.; Seelow, D. MutationTaster2021. Nucleic Acids Res. 2021, 2, 34. [Google Scholar] [CrossRef]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the Deleteriousness of Variants throughout the Human Genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef] [PubMed]
- Kopanos, C.; Tsiolkas, V.; Kouris, A.; Chapple, C.E.; Albarca Aguilera, M.; Meyer, R.; Massouras, A. VarSome: The Human Genomic Variant Search Engine. Bioinformatics 2019, 35, 1978–1980. [Google Scholar] [CrossRef]
- Li, Q.; Wang, K. InterVar: Clinical Interpretation of Genetic Variants by the 2015 ACMG-AMP Guidelines. Am. J. Hum. Genet. 2017, 100, 267–280. [Google Scholar] [CrossRef] [Green Version]
- Robert, X.; Gouet, P. Deciphering Key Features in Protein Structures with the New ENDscript Server. Nucleic Acids Res. 2014, 42, W320–W324. [Google Scholar] [CrossRef] [Green Version]
- Kelley, L.A.; Mezulis, S.; Yates, C.M.; Wass, M.N.; Sternberg, M.J.E. The Phyre2 Web Portal for Protein Modeling, Prediction and Analysis. Nat. Protoc. 2015, 10, 845–858. [Google Scholar] [CrossRef] [Green Version]
- Webb, B.; Sali, A. Protein Structure Modeling with MODELLER. In Structural Genomics: General Applications; Chen, Y.W., Yiu, C.-P.B., Eds.; Methods in Molecular Biology; Springer: New York, NY, USA, 2021; pp. 239–255. ISBN 978-1-07-160892-0. [Google Scholar]
- Shen, M.; Sali, A. Statistical Potential for Assessment and Prediction of Protein Structures. Protein Sci. 2006, 15, 2507–2524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Meng, E.C.; Couch, G.S.; Croll, T.I.; Morris, J.H.; Ferrin, T.E. UCSF ChimeraX: Structure Visualization for Researchers, Educators, and Developers. Protein Sci. 2021, 30, 70–82. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nomenclature | Epidemiologic Data (gnomAD) | In Silico Prediction and Aggregators | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| cDNA Name | Protein Name | rsID | Allelic Freq | Homozygous Count | ClinVar | SIFT | Poly-Phen 2 | MutationTaster a | CADD a | Varsome a | InterVar a | |
| IH1 | c.42C>G | p.(Asn14Lys) N14K | - | - | 0 | - | D | D | D | 24.0 | VUS | VUS |
| c.53A>G | p.(Gln18Arg) Q18R | - | - | 0 | - | D | B | D | 27.4 | Lik. P | VUS | |
| c.59G>T | p.(Arg20Leu) R20L | rs201777730 | 0.00% | 0 | - | D | D | D | 28.1 | VUS | VUS | |
| ICL1 and IH2 | c.863G>A | p.(Arg288Lys) R288K † | rs117603931 | 0.61% ‡ | 12 | B, Lik. B, VUS | B | B | B | 0.15 | B | Lik. B |
| c.875A>T | p.(Glu292Val) E292V | rs149989682 | 0.23% | 3 | P, Lik. P | D | D | D | 32 | P | VUS | |
| NBD1 | c.1675G>A | p.(Gly559Arg) G559R | rs976333358 | - | 0 | - | D | D | D | 23.9 | Lik. P | Lik. P |
| c.1702A>G | p.(Asn568Asp) N568D | rs121909184 | - | 0 | P | D | D | D | 24.4 | Lik. P | Lik. P | |
| c.1736T>C | p.(Leu579Pro) L579P | - | - | 0 | - | D | D | D | 25.1 | VUS | VUS | |
| c.1880T>A | p.(Leu627His) L627H | - | - | 0 | - | D | D | D | 25.1 | VUS | VUS | |
| c.2068G>A | p.(Glu690Lys) E690K | - | - | 0 | - | D | D | D | 28.2 | VUS | VUS | |
| c.2069A>G | p.(Glu690Gly) E690G | - | - | 0 | - | D | D | D | 27.6 | VUS | VUS | |
| c.2074A>C | p.(Thr692Pro) T692P | - | - | 0 | - | D | D | D | 25 | VUS | VUS | |
| c.2078C>T | p.(Ser693Leu) S693L † | rs200835546 | 0.03% | 0 | - | D | D | D | 23.6 | VUS | VUS | |
| c.2086G>A | p.(Asp696Asn) D696N | rs193920904 | 0.00% | 0 | VUS | D | D | D | 26.6 | VUS | VUS | |
| c.2125C>T | p.(Arg709Trp) R709W | rs148671332 | 0.14% | 0 | VUS | B | B | B | 24.5 | VUS | VUS | |
| c.2233G>A | p.(Gly745Arg) G745R | - | - | 0 | - | D | D | D | 26.7 | VUS | VUS | |
| RD1 | c.2279T>G | p.(Met760Arg) M760R | - | - | 0 | - | D | D | D | 24.2 | VUS | VUS |
| c.2282C>T | p.(Thr761Met) T761M | rs369081312 | - | 0 | - | D | D | D | 22.4 | VUS | VUS | |
| c.2296C>T | p.(Pro766Ser) P766S † | rs45592239 | 0.17% | 0 | - | B | B | D | 22.7 | Lik. B | VUS | |
| c.2309C>T | p.(Pro770Leu) P770L † | rs143929832 | 0.15% | 0 | VUS, Lik. B | B | B | B | 14.4 | VUS | VUS | |
| c.2333A>G | p.(His778Arg) H778R † | rs34912779 | 0.12% ‡ | 2 | Lik. B, B | B | B | B | 1.2 | Lik. B | Lik. B | |
| c.2393T>C | p.(Leu798Pro) L798P | - | - | 0 | - | D | D | D | 25.3 | VUS | VUS | |
| IH3 | c.2741A>G | p.(Lys914Arg) K914R | rs763862811 | - | 0 | - | D | D | D | 28.1 | VUS | VUS |
| c.2745G>C | p.(Lys915Asn) K915N | rs1459105468 | 0.00% | 0 | - | D | D | D | 24.0 | VUS | VUS | |
| IH4 | c.3392A>G | p.(Gln1131Arg) Q1131R | - | - | 0 | - | D | D | D | 23.9 | VUS | VUS |
| NBD2 | c.4157T>C | p.(Leu1386Pro) L1386P | - | - | 0 | - | D | D | D | 26.8 | VUS | VUS |
| c.4164G>C | p.(Lys1388Asn) K1388N | - | - | 0 | - | D | D | D | 35 | Lik. P | VUS | |
| c.4195G>A | p.(Val1399Met) V1399M | rs763166660 | 0.00% | 0 | - | D | D | D | 25.6 | VUS | VUS | |
| c.4231T>C | p.(Cys1411Arg) C1411R | - | - | 0 | - | D | D | D | 28 | VUS | VUS | |
| c.4253A>G | p.(Asn1418Ser) N1418S | rs147036502 | 0.01% | 0 | - | D | D | D | 25.1 | VUS | VUS | |
| c.4258G>C | p.(Ala1420Pro) A1420P | rs1167324185 | 0.00% | 0 | - | D | D | D | 28.2 | VUS | VUS | |
| c.4261G>A | p.(Gly1421Arg) G1421R | rs776453529 | 0.12% | 0 | - | D | D | D | 29.2 | VUS | VUS | |
| c.4268C>T | p.(Thr1423Ile) T1423I | rs764069673 | - | 0 | - | D | D | D | 25.7 | VUS | VUS | |
| c.4313G>T | p.(Gly1438Val) G1438V | - | - | 0 | - | D | D | D | 28.5 | VUS | VUS | |
| c.4376G>A | p.(Gly1459Asp) G1459D | - | - | 0 | - | D | D | D | 26.4 | VUS | VUS | |
| c.4411A>G | p.(Met1471Val) M1471V | rs754896003 | 0.00% | 0 | - | D | D | D | 23.8 | VUS | VUS | |
| c.4415C>G | p.(Thr1472Arg) T1472R | - | - | 0 | - | D | D | D | 25 | VUS | VUS | |
| c.4444C>T | p.(Arg1482Trp) R1482W | rs892042868 | - | 0 | - | D | D | D | 32 | VUS | VUS | |
| c.4451G>C | p.(Arg1484Pro) R1484P | - | - | 0 | - | D | D | D | 26.8 | Lik. P | VUS | |
| c.4483G>A | p.(Val1495Met) V1495M † | rs141058709 | 0.08% | 0 | - | D | D | D | 29 | VUS | VUS | |
| c.4561C>T | p.(Arg1521Trp) R1521W | rs760872079 | 0.0004 | 0 | - | D | D | D | 27.8 | VUS | VUS | |
| c.4583C>T | p.(Ala1528Val) A1528V | - | - | 0 | - | D | D | D | 25.9 | VUS | VUS | |
| c.4615G>C | p.(Asp1539His) D1539H | - | - | 0 | - | D | D | D | 26.6 | VUS | VUS | |
| c.4618G>A | p.(Glu1540Lys) E1540K | rs968080956 | - | 0 | - | D | D | D | 25.4 | VUS | VUS | |
| c.4648C>T | p.(Arg1550Trp) R1550W | rs781422468 | 0.0004 | 0 | - | D | D | D | 25 | VUS | VUS | |
| c.4658T>C | p.(Leu1553Pro) L1553P | rs121909183 | - | 0 | P | D | D | D | 23.7 | Lik. P | Lik. P | |
| c.4732G>A | p.(Glu1578Lys) E1578K | rs1034626421 | 0.0064 | 0 | - | D | D | D | 27.2 | VUS | VUS | |
| c.4739T>C | p.(Leu1580Pro) L1580P | - | - | 0 | - | D | D | D | 25.8 | VUS | VUS | |
| c.4745C>G | p.(Thr1582Ser) T1582S | rs574182515 | 0.00% | 0 | - | B | D | D | 24.4 | Lik. P | VUS | |
| c.4747C>T | p.(Arg1583Trp) R1583W | - | - | 0 | - | D | D | D | 26 | VUS | VUS | |
| c.4772A>C | p.(Gln1591Pro) Q1591P | rs28936691 | - | 0 | P | D | D | D | 25 | Lik. P | Lik. P | |
| c.4784T>C | p.(Leu1595Pro) L1595P | - | - | 0 | - | D | D | D | 29.5 | VUS | VUS | |
| RD2 | c.4835G>C | p.(Arg1612Pro) R1612P | - | - | 0 | - | B | B | B | 19.9 | VUS | VUS |
| c.4958C>T | p.(Pro1653Leu) P1653L † | rs774227126 | 0.00% | 0 | - | D | D | D | 26.3 | VUS | VUS | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Onnée, M.; Fanen, P.; Callebaut, I.; de Becdelièvre, A. Structure-Based Understanding of ABCA3 Variants. Int. J. Mol. Sci. 2021, 22, 10282. https://doi.org/10.3390/ijms221910282
Onnée M, Fanen P, Callebaut I, de Becdelièvre A. Structure-Based Understanding of ABCA3 Variants. International Journal of Molecular Sciences. 2021; 22(19):10282. https://doi.org/10.3390/ijms221910282
Chicago/Turabian StyleOnnée, Marion, Pascale Fanen, Isabelle Callebaut, and Alix de Becdelièvre. 2021. "Structure-Based Understanding of ABCA3 Variants" International Journal of Molecular Sciences 22, no. 19: 10282. https://doi.org/10.3390/ijms221910282