
Analysis of SINE Families B2, Dip, and Ves with Special Reference to Polyadenylation Signals and Transcription Terminators


Abstract
:1. Introduction
2. Results
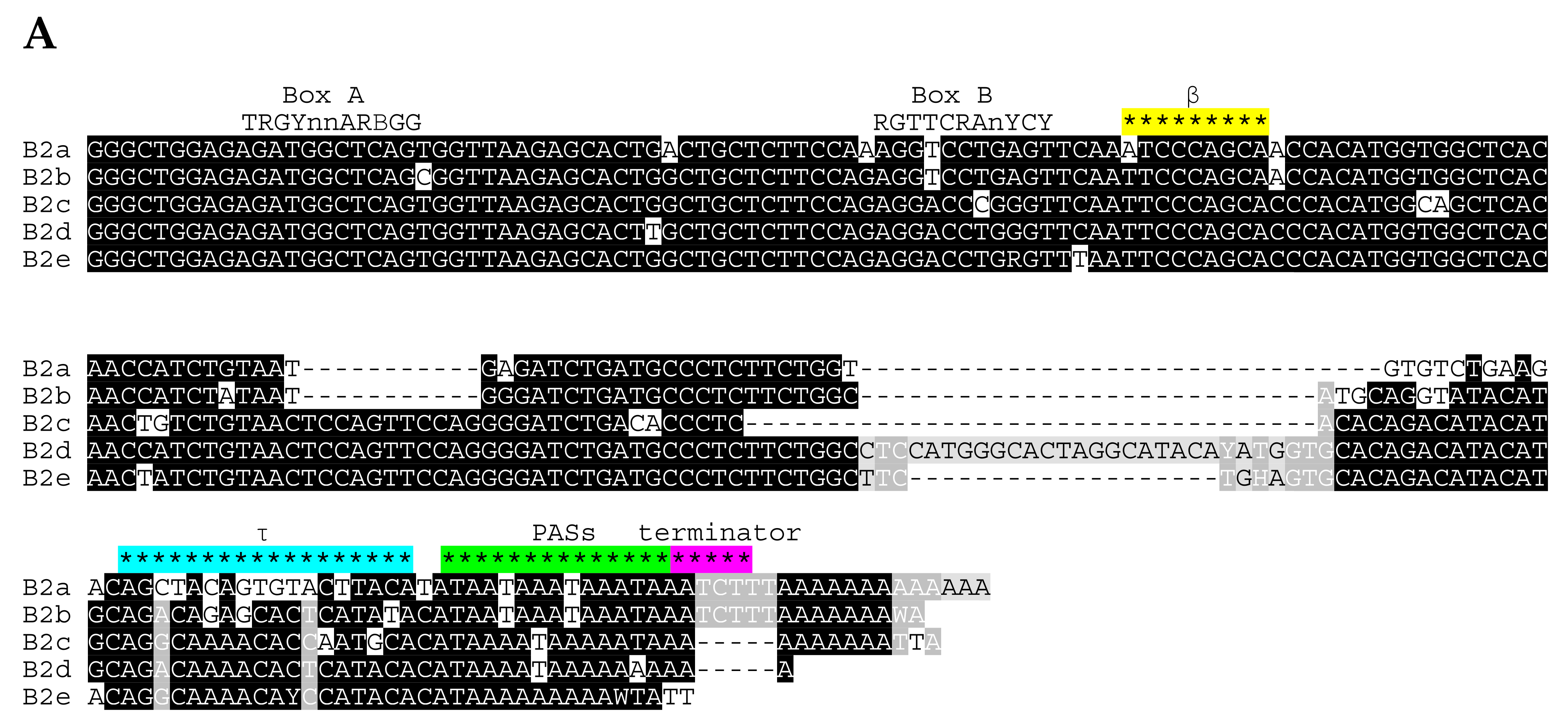
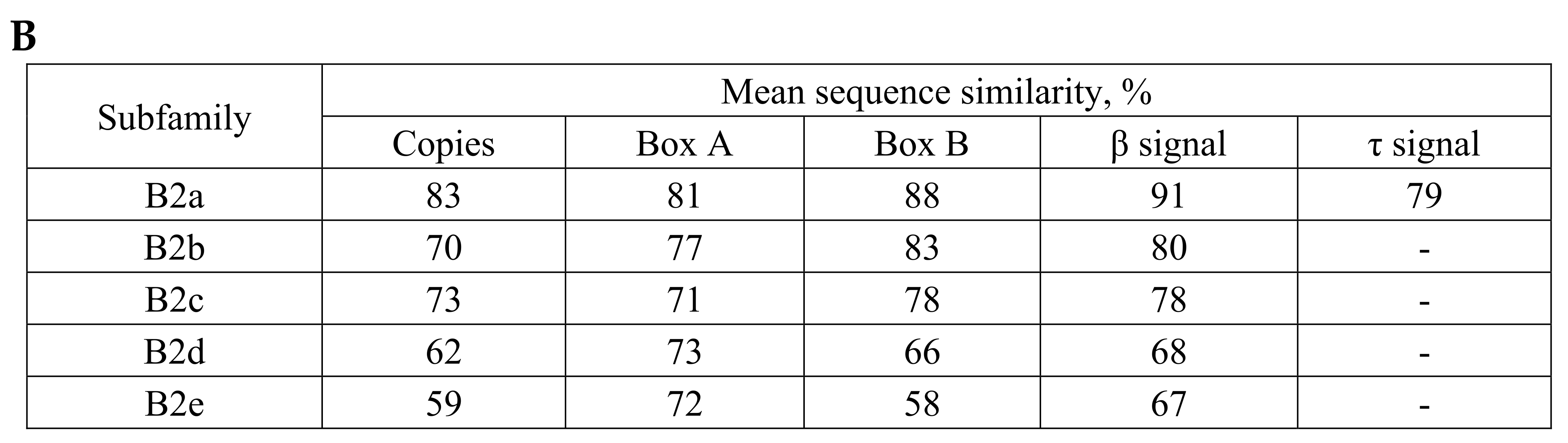
2.1. Overview of B2 SINE Family
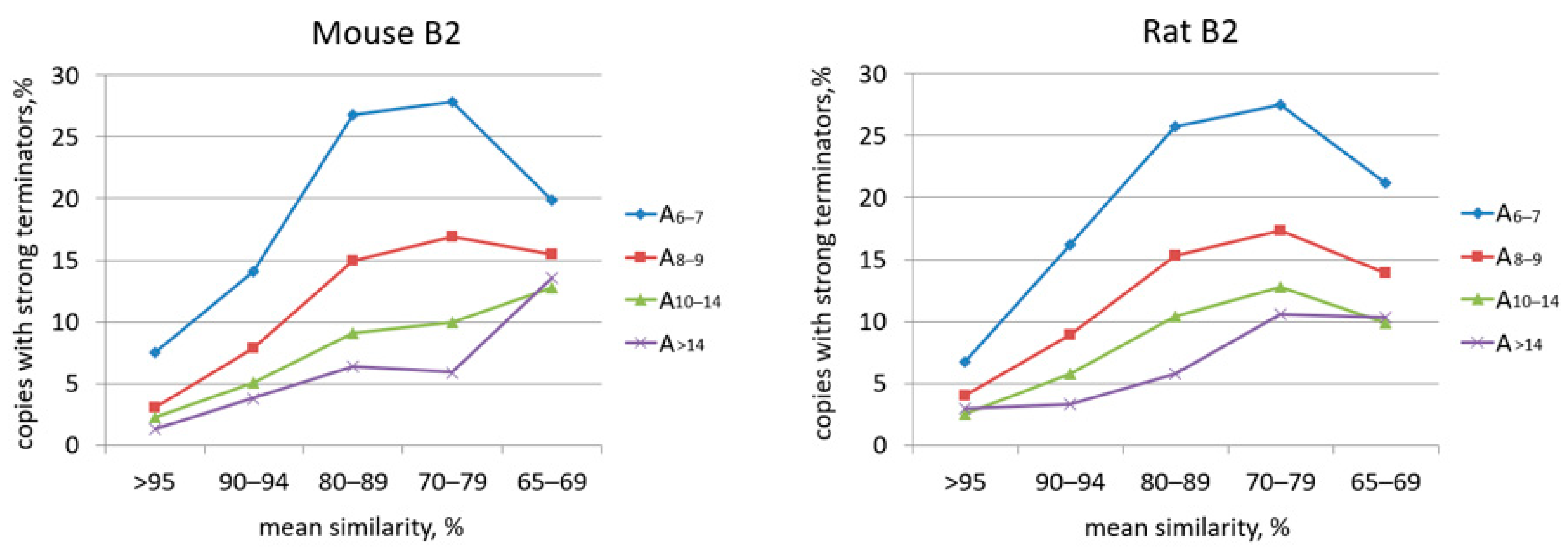
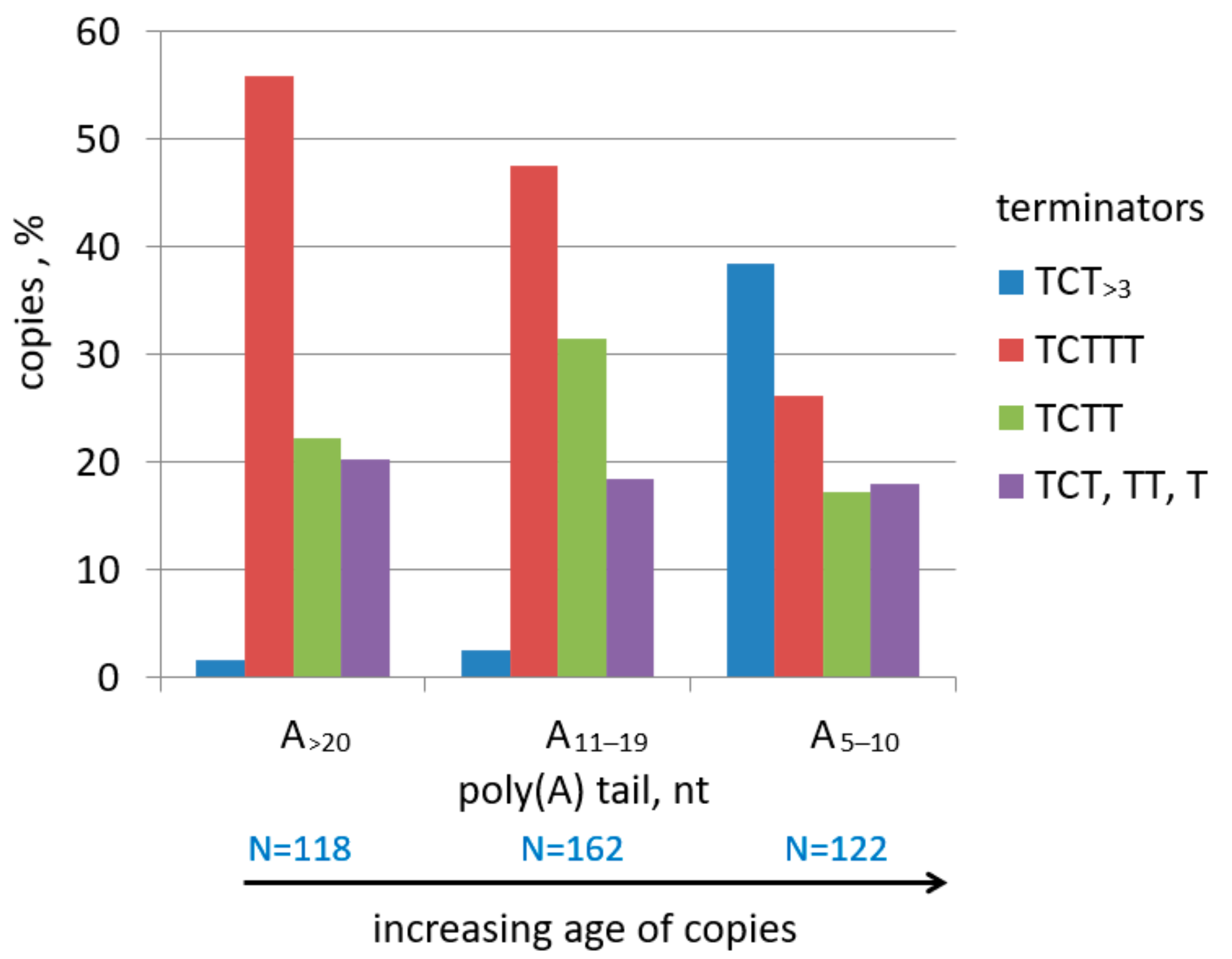
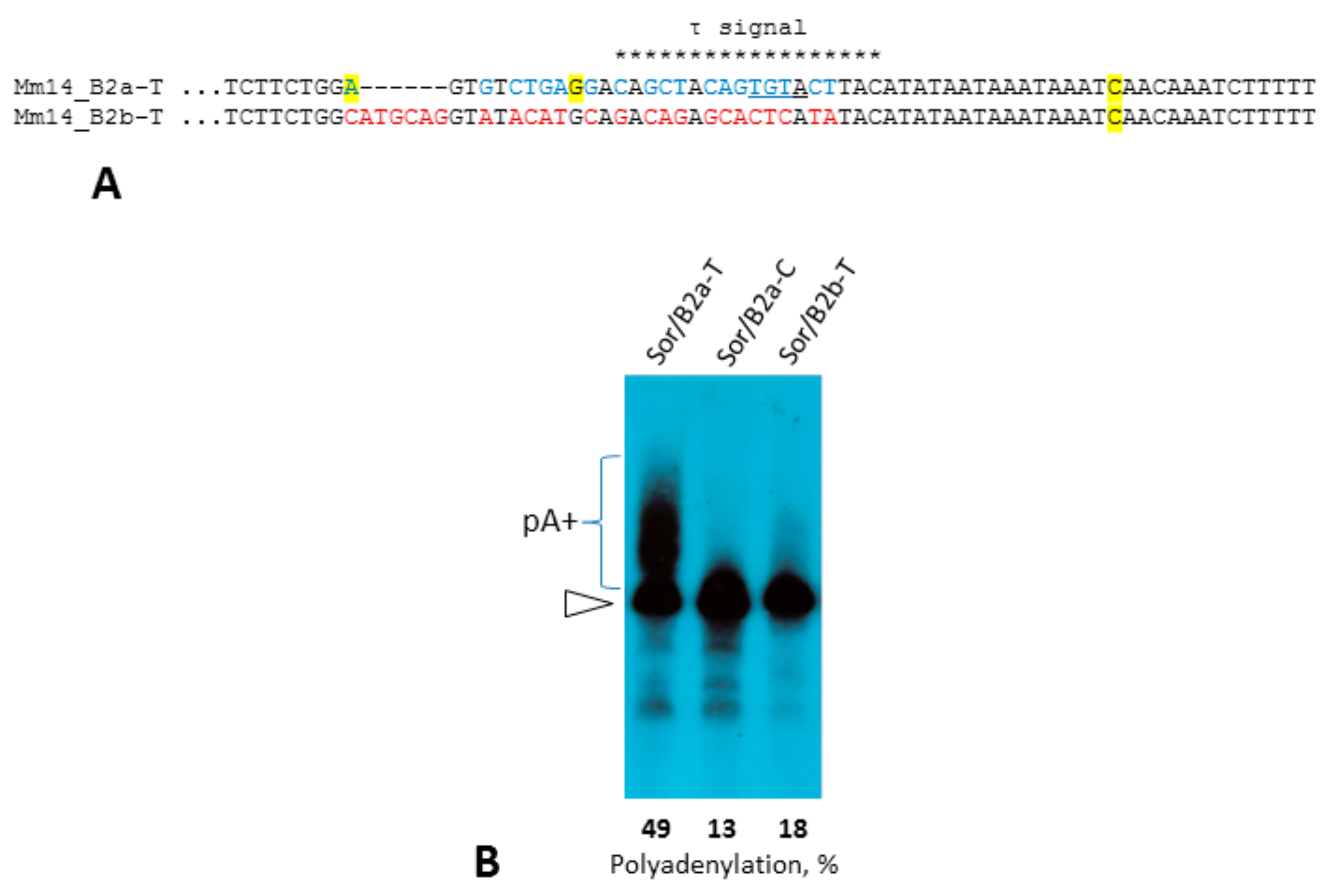
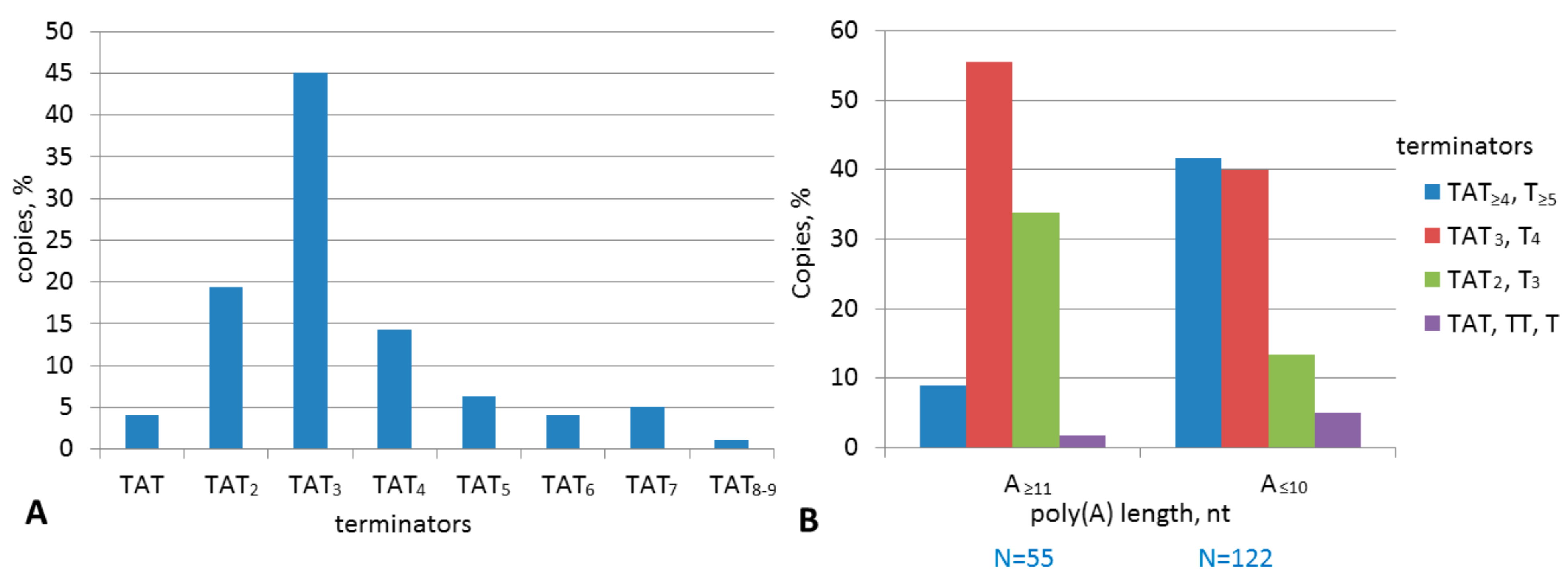
2.2. Analysis of Transcription Terminators and A-Tails in B2a Copies of Different Age
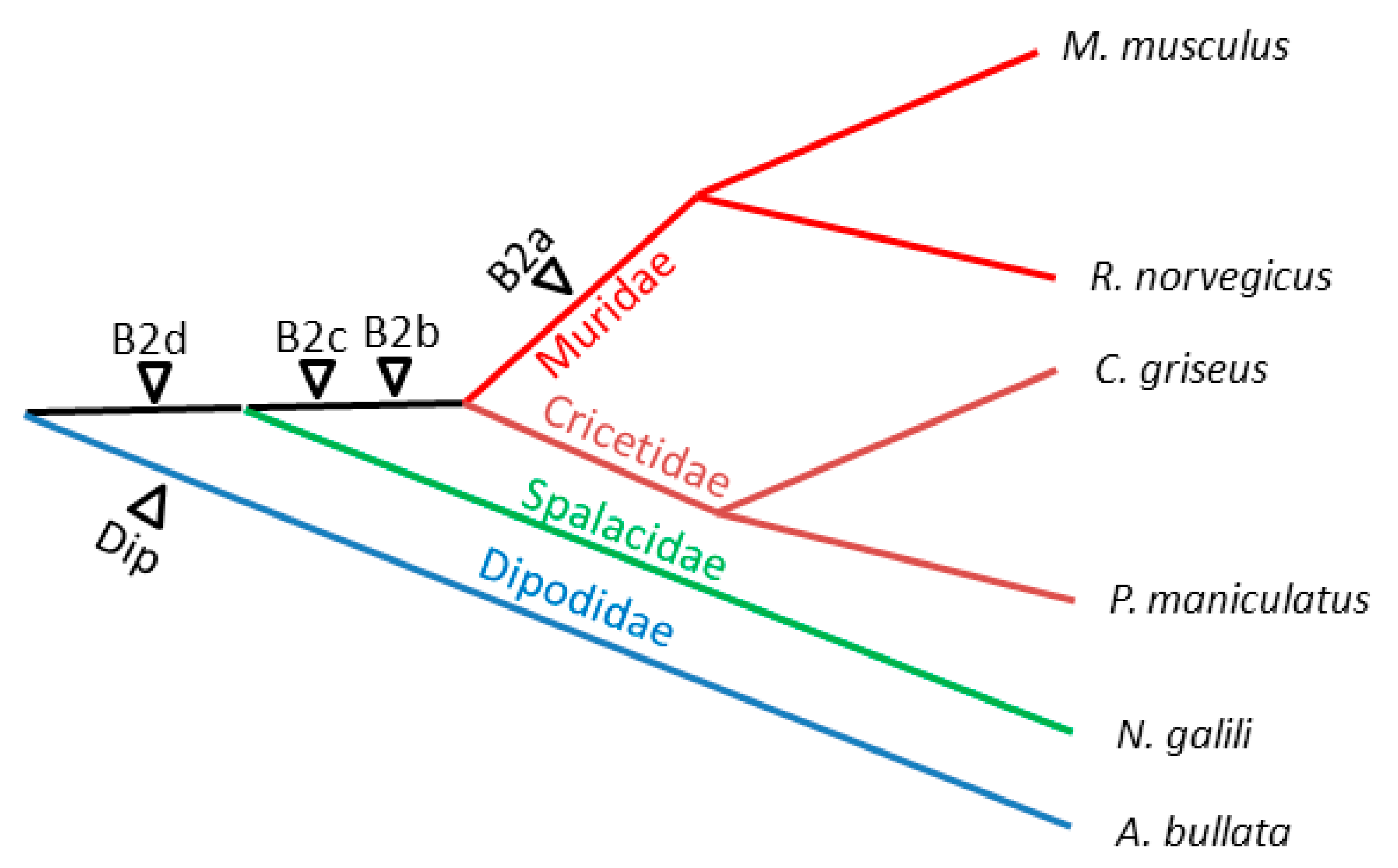
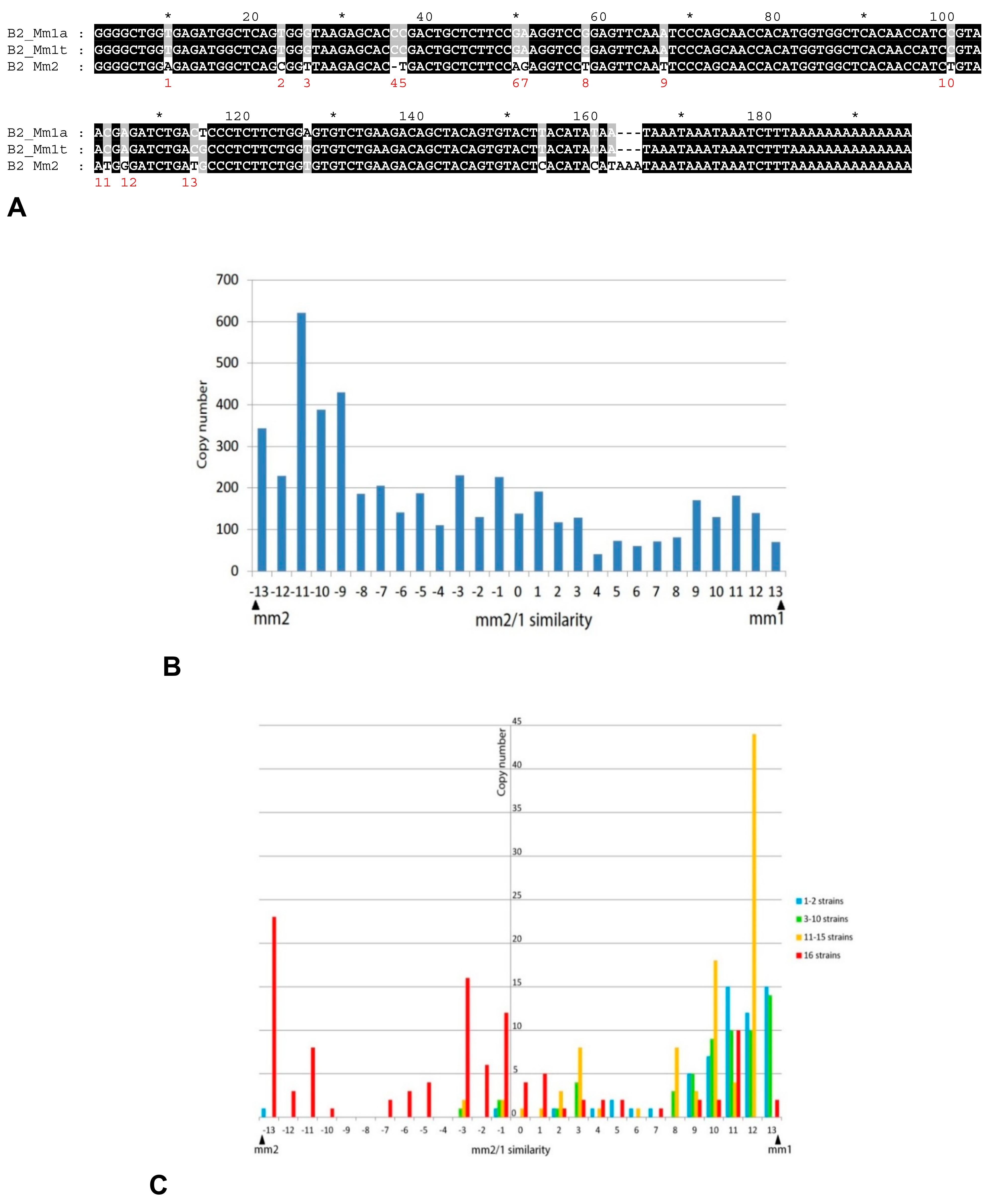
2.3. History of Retrotranspositional Activity of B2a Variants
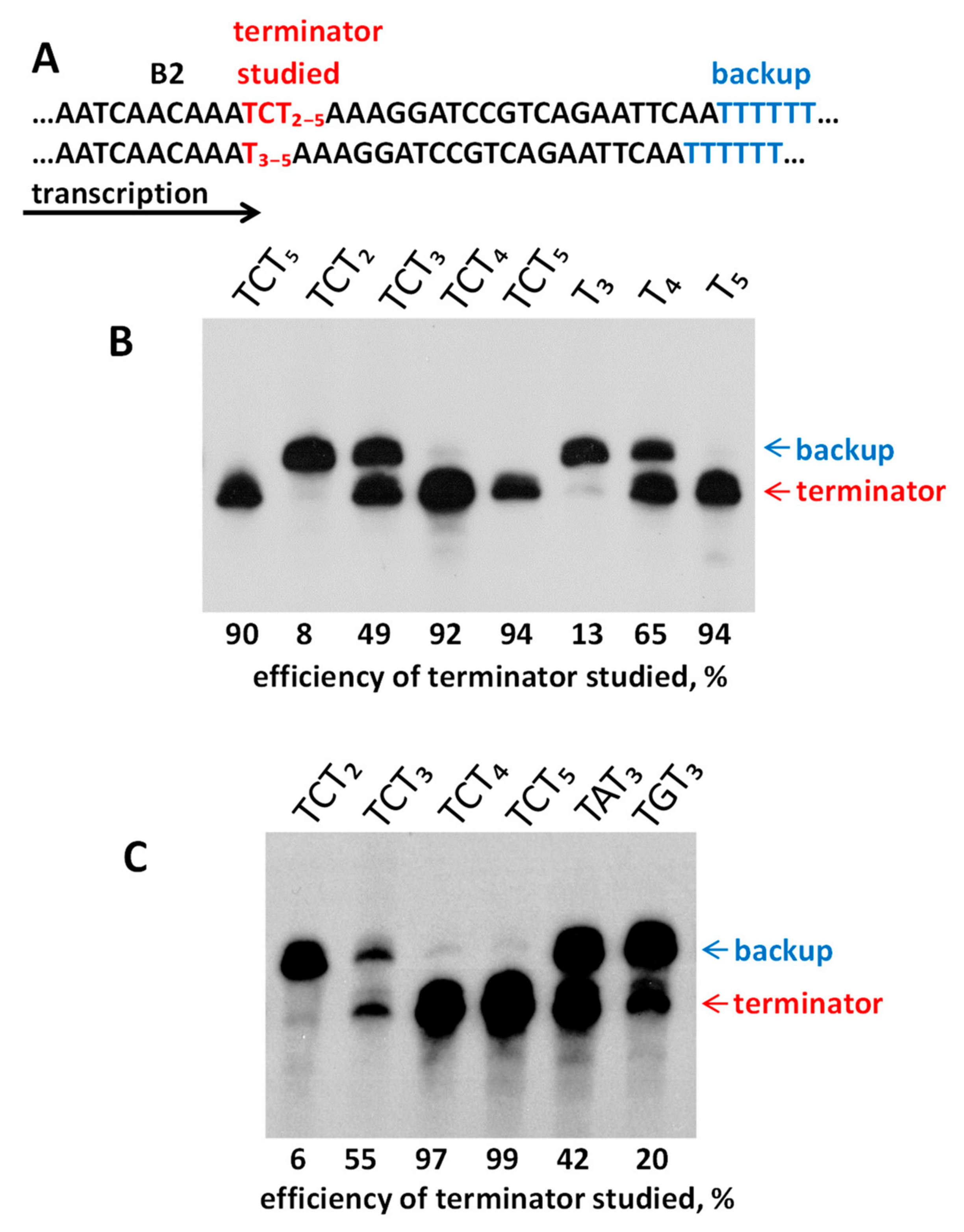
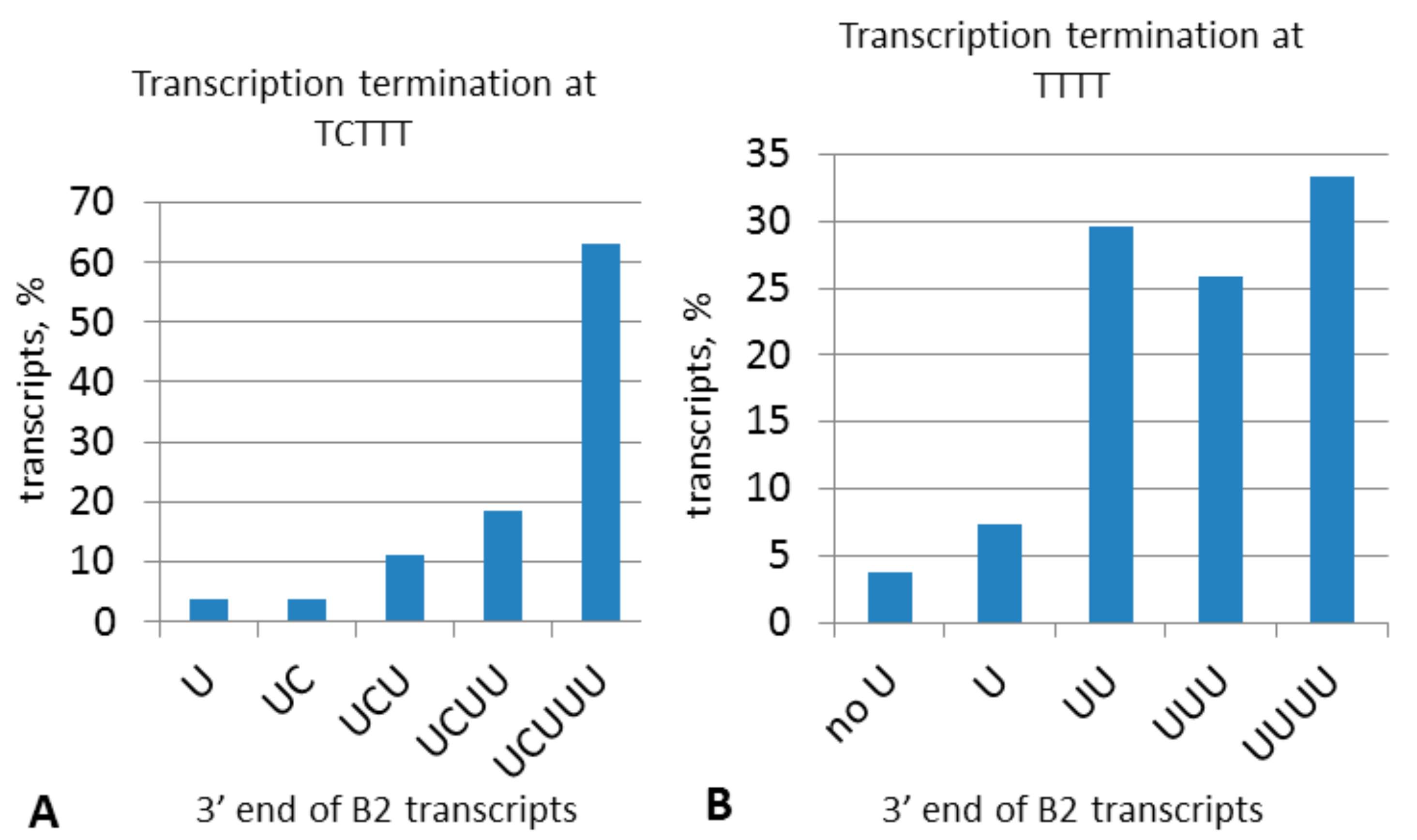
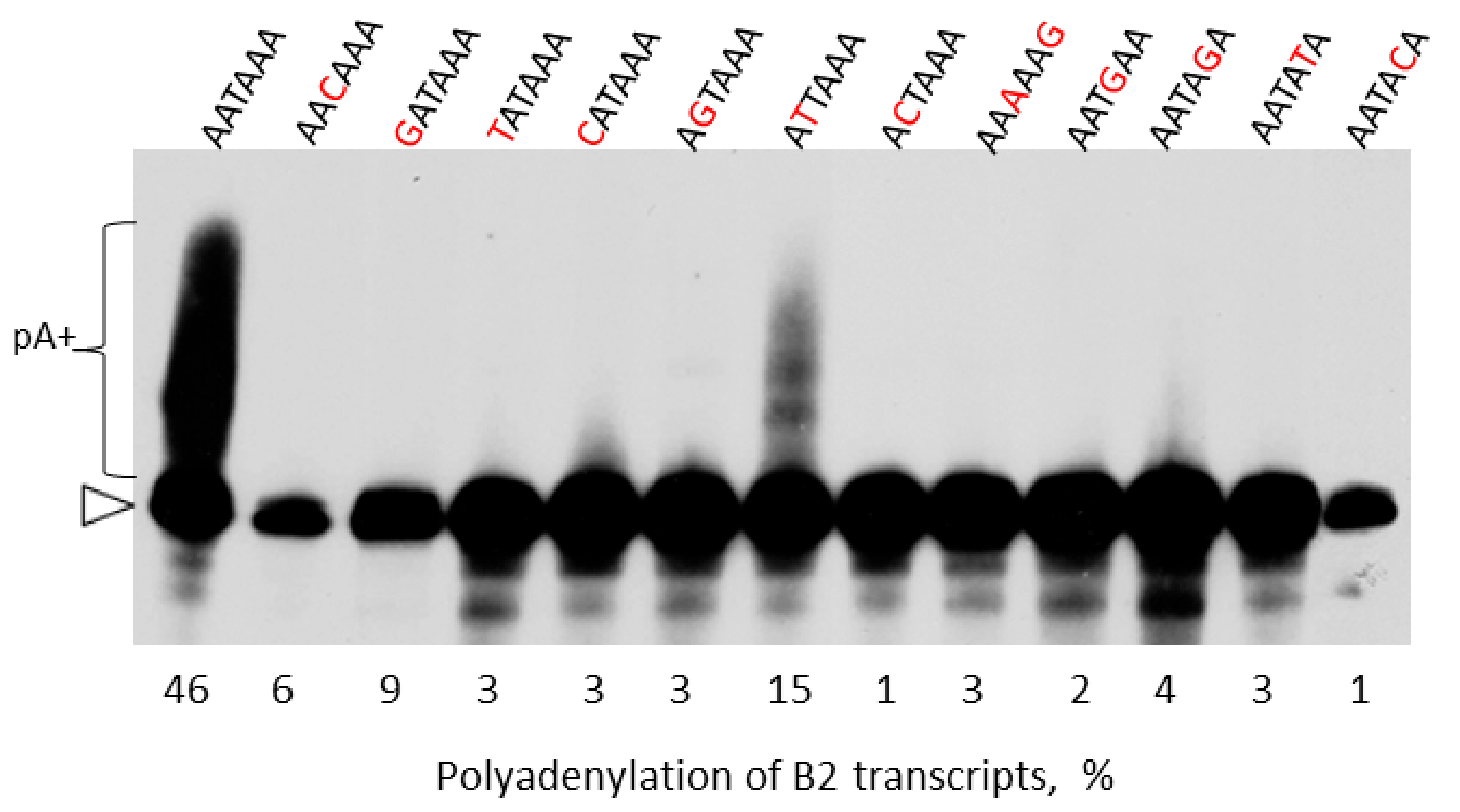
2.4. Experimental Analysis of B2 Transcription Termination and Polyadenylation
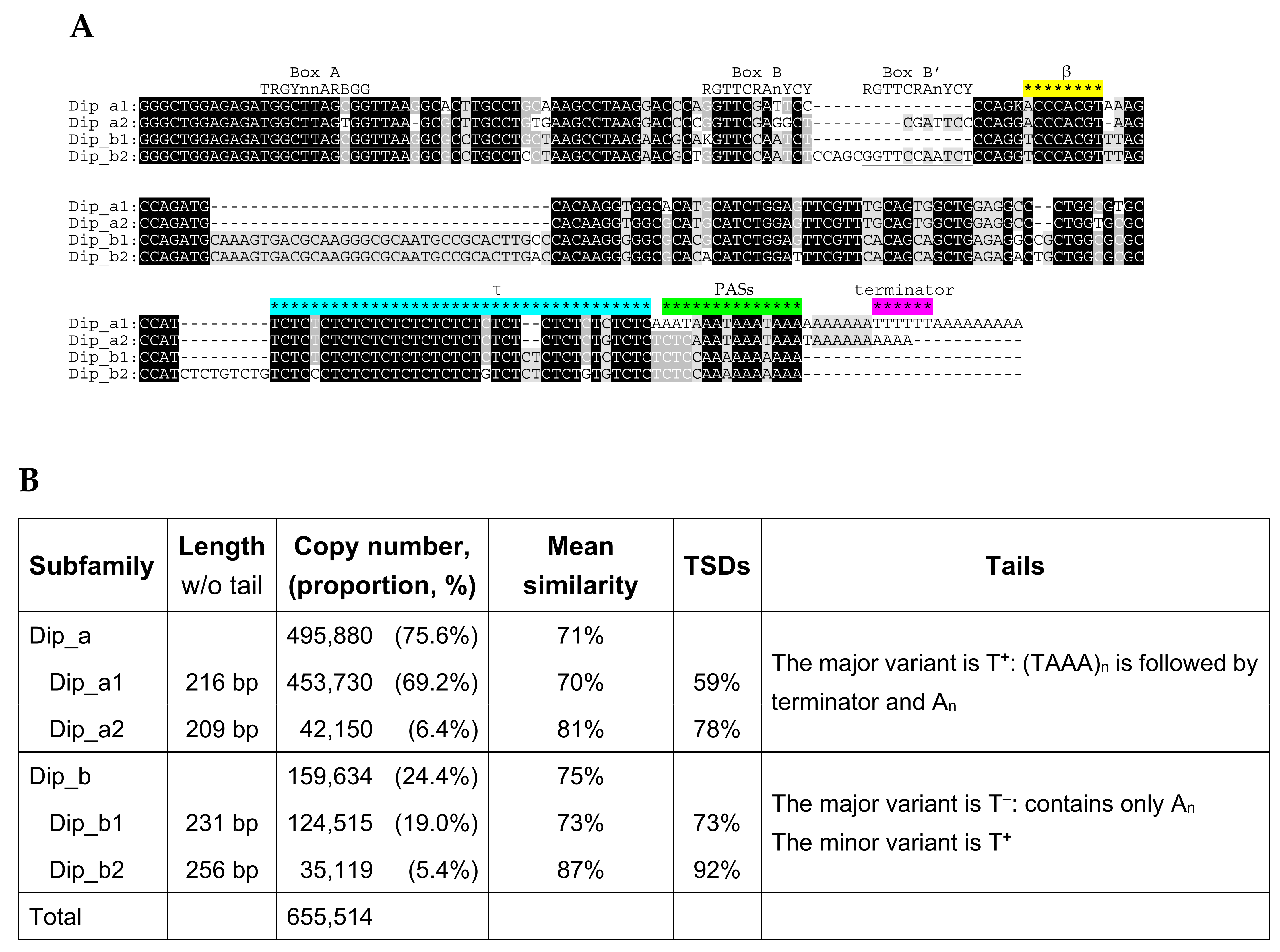
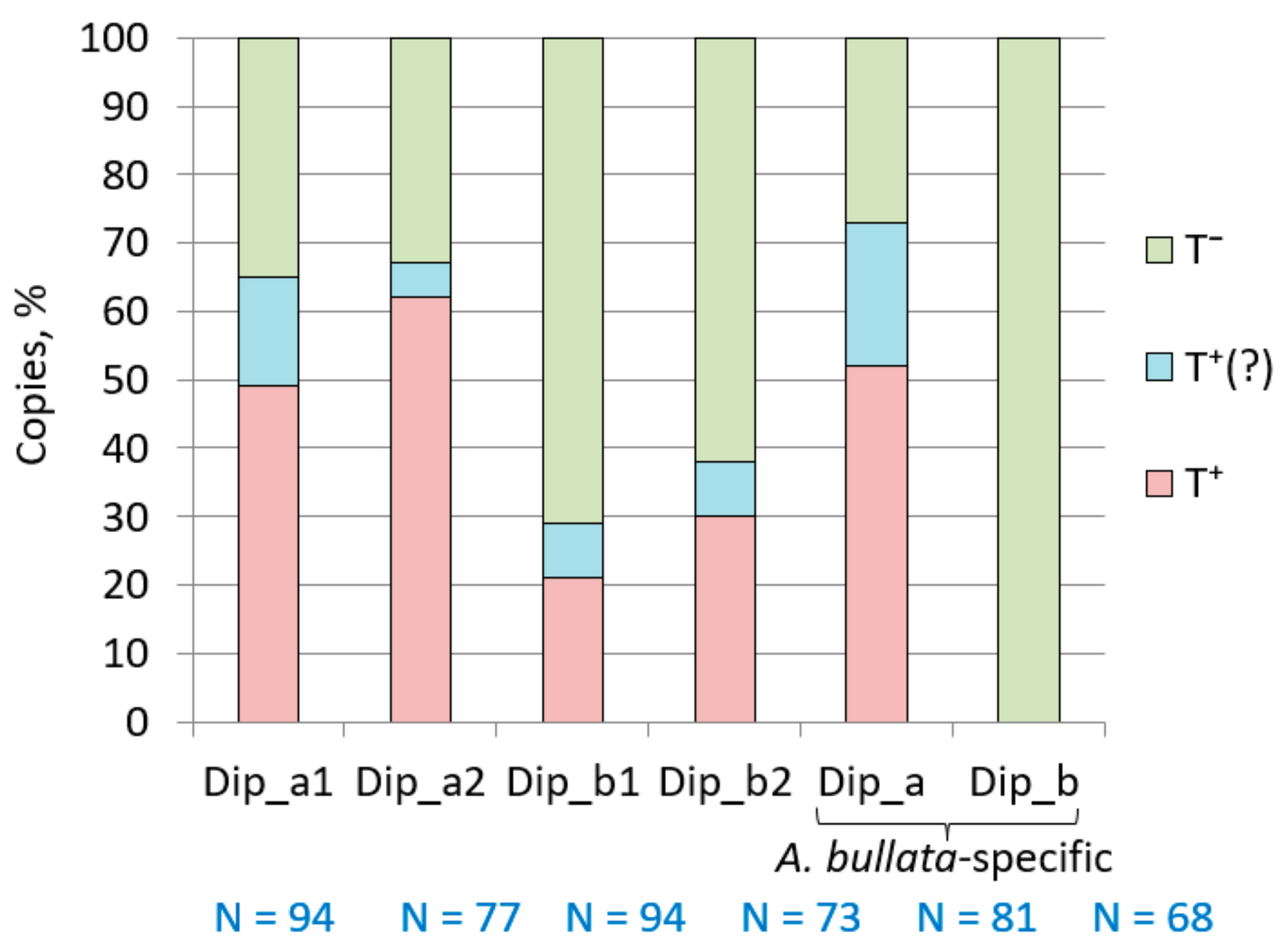
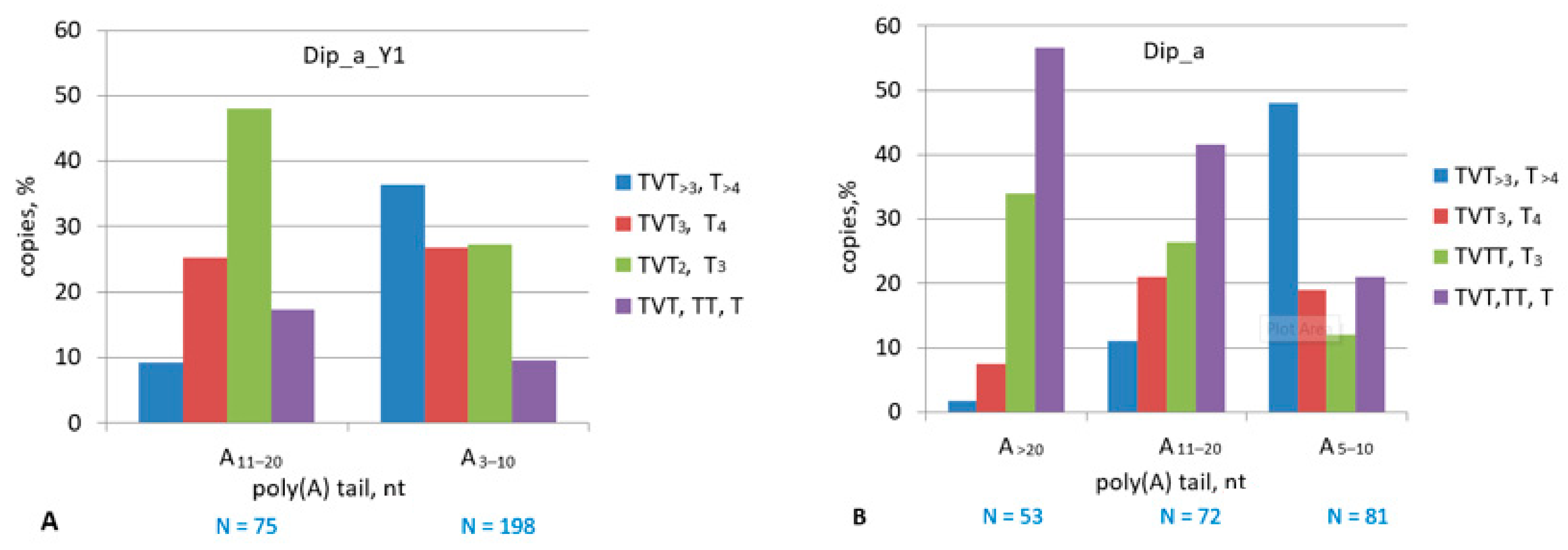
2.5. Dip SINE
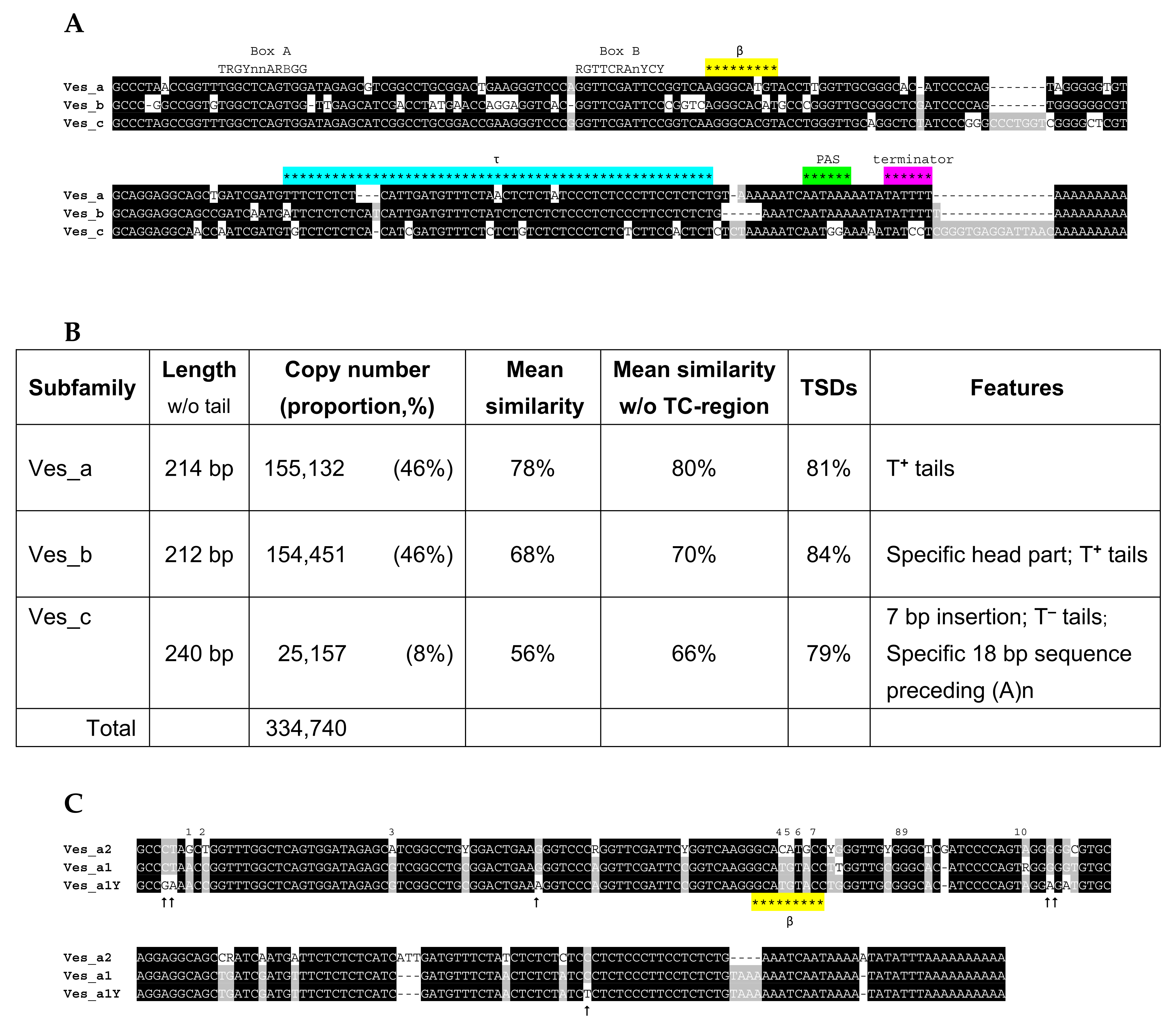
2.6. Ves SINE
3. Discussion
3.1. B2 Family
3.2. Dip Family
3.3. Ves Family
4. Materials and Methods
4.1. Bioinformatics Methods
4.2. Experimental Methods
5. Conclusions
- (i)
- In T+ SINEs, the minimal terminators include TCT3, TAT3, and T4.
- (ii)
- The TCT3 and apparently TAT3 remain intact twice as frequently as T4 at the 3′-ends of pol III-generated transcripts and hence in the daughter SINE copies.
- (iii)
- The minimal and rudimentary terminators tend to elongate and become efficient as SINE copies get older and their poly(A) tails shorten.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| SINE | Short Interspersed Element |
| LINE | Long Interspersed Element |
| PAS | Polyadenylation signal |
| My | Million years |
| Mya | Million years ago |
| RU | Repbase Update |
References
- Kramerov, D.A.; Grigoryan, A.A.; Ryskov, A.P.; Georgiev, G.P. Long double-stranded sequences (dsRNA-B) of nuclear pre-mRNA consist of a few highly abundant classes of sequences: Evidence from DNA cloning experiments. Nucleic Acids Res. 1979, 6, 697–713. [Google Scholar] [CrossRef] [Green Version]
- Houck, C.M.; Rinehart, F.P.; Schmid, C.W. A ubiquitous family of repeated DNA sequences in the human genome. J. Mol. Biol. 1979, 132, 289–306. [Google Scholar] [CrossRef]
- Krayev, A.S.; Kramerov, D.A.; Skryabin, K.G.; Ryskov, A.; Bayev, A.A.; Georgiev, G.P. The nucleotide sequence of the ubiquitous repetitive DNA sequence B1 complementary to the most abundant class of mouse fold-back RNA. Nucleic Acids Res. 1980, 8, 1201–1216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deininger, P.; Jolly, D.J.; Rubin, C.M.; Friedmann, T.; Schmid, C.W. Base sequence studies of 300 nucleotide renatured repeated human DNA clones. J. Mol. Biol. 1981, 151, 17–33. [Google Scholar] [CrossRef]
- Haynes, S.R.; Jelinek, W.R. Low molecular weight RNAs transcribed in vitro by RNA polymerase III from Alu-type dispersed repeats in Chinese hamster DNA are also found in vivo. Proc. Natl. Acad. Sci. USA 1981, 78, 6130–6134. [Google Scholar] [CrossRef] [Green Version]
- Krayev, A.; Markusheva, T.; Kramerov, D.; Ryskov, A.; Skryabin, K.; Bayev, A.; Georgiev, G. Ubiquitous transposon-like repeats B1 and B2 of the mouse genome: B2 sequencing. Nucleic Acids Res. 1982, 10, 7461–7475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vassetzky, N.S.; Kramerov, D.A. SINEBase: A database and tool for SINE analysis. Nucleic Acids Res. 2012, 41, D83–D89. [Google Scholar] [CrossRef] [Green Version]
- Ohshima, K.; Okada, N. SINEs and LINEs: Symbionts of eukaryotic genomes with a common tail. Cytogenet. Genome Res. 2005, 110, 475–490. [Google Scholar] [CrossRef]
- Kramerov, D.A.; Vassetzky, N.S. SINEs. Wiley Interdiscip. Rev. RNA 2011, 2, 772–786. [Google Scholar] [CrossRef] [PubMed]
- Kramerov, D.A.; Vassetzky, N.S. Origin and evolution of SINEs in eukaryotic genomes. Heredity 2011, 107, 487–495. [Google Scholar] [CrossRef] [PubMed]
- Batzer, M.A.; Deininger, P. Alu repeats and human genomic diversity. Nat. Rev. Genet. 2002, 3, 370–379. [Google Scholar] [CrossRef]
- Deininger, P.L. Alu elements: Know the SINEs. Genome Biol. 2011, 12, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Ullu, E.; Tschudi, C. Alu sequences are processed 7SL RNA genes. Nature 1984, 312, 171–172. [Google Scholar] [CrossRef]
- Veniaminova, N.A.; Vassetzky, N.S.; Kramerov, D.A. B1 SINEs in different rodent families. Genomics 2007, 89, 678–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nishihara, H.; Terai, Y.; Okada, N. Characterization of novel Alu- and tRNA-related SINEs from the tree shrew and evolutionary implications of their origins. Mol. Biol. Evol. 2002, 19, 1964–1972. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kriegs, J.O.; Churakov, G.; Jurka, J.; Brosius, J.; Schmitz, J. Evolutionary history of 7SL RNA-derived SINEs in Supraprimates. Trends Genet. 2007, 23, 158–161. [Google Scholar] [CrossRef]
- Kojima, K.K. Hagfish genome reveals parallel evolution of 7SL RNA-derived SINEs. Mob. DNA 2020, 11, 1–9. [Google Scholar] [CrossRef]
- Kapitonov, V.V.; Jurka, J. A novel class of SINE elements derived from 5S rRNA. Mol. Biol. Evol. 2003, 20, 694–702. [Google Scholar] [CrossRef] [Green Version]
- Gogolevsky, K.P.; Vassetzky, N.S.; Kramerov, D.A. 5S rRNA-derived and tRNA-derived SINEs in fruit bats. Genomics 2009, 93, 494–500. [Google Scholar] [CrossRef]
- Wang, J.; Wang, A.; Han, Z.; Zhang, Z.; Li, F.; Li, X. Characterization of Three Novel SINE Families with Unusual Features in Helicoverpa armigera. PLoS ONE 2012, 7, e31355. [Google Scholar] [CrossRef] [PubMed]
- Nishihara, H.; Smit, A.F.; Okada, N. Functional noncoding sequences derived from SINEs in the mammalian genome. Genome Res. 2006, 16, 864–874. [Google Scholar] [CrossRef] [Green Version]
- Okada, N. SINEs: Short interspersed repeated elements of the eukaryotic genome. Trends Ecol. Evol. 1991, 6, 358–361. [Google Scholar] [CrossRef]
- Moran, J.V.; Holmes, S.E.; Naas, T.; DeBerardinis, R.J.; Boeke, J.D.; Kazazian, H.H. High Frequency Retrotransposition in Cultured Mammalian Cells. Cell 1996, 87, 917–927. [Google Scholar] [CrossRef] [Green Version]
- Dewannieux, M.; Esnault, C.; Heidmann, T. LINE-mediated retrotransposition of marked Alu sequences. Nat. Genet. 2003, 35, 41–48. [Google Scholar] [CrossRef]
- Chesnokov, I.; Schmid, C.W. Flanking sequences of an Alu source stimulate transcription in vitro by interacting with sequence-specific transcription factors. J. Mol. Evol. 1996, 42, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Tatosyan, K.A.; Stasenko, D.V.; Koval, A.P.; Gogolevskaya, I.K.; Kramerov, D.A. TATA-Like Boxes in RNA Polymerase III Promoters: Requirements for Nucleotide Sequences. Int. J. Mol. Sci. 2020, 21, 3706. [Google Scholar] [CrossRef] [PubMed]
- Paolella, G.; Lucero, M.A.; Murphy, M.H.; Baralle, F.E. The Alu family repeat promoter has a tRNA-like bipartite structure. EMBO J. 1983, 2, 691–696. [Google Scholar] [CrossRef]
- Ohshima, K.; Hattori, M.; Yada, T.; Gojobori, T.; Sakaki, Y.; Okada, N. Whole-genome screening indicates a possible burst of formation of processed pseudogenes and Alu repeats by particular L1 subfamilies in ancestral primates. Genome Biol. 2003, 4, R74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borodulina, O.R.; Kramerov, D. Short interspersed elements (SINEs) from insectivores. Two classes of mammalian SINEs distinguished by A-rich tail structure. Mamm. Genome 2001, 12, 779–786. [Google Scholar] [CrossRef]
- Borodulina, O.R.; Kramerov, D.A. Transcripts synthesized by RNA polymerase III can be polyadenylated in an AAUAAA-dependent manner. RNA 2008, 14, 1865–1873. [Google Scholar] [CrossRef] [Green Version]
- Borodulina, O.R.; Golubchikova, J.S.; Ustyantsev, I.G.; Kramerov, D.A. Polyadenylation of RNA transcribed from mammalian SINEs by RNA polymerase III: Complex requirements for nucleotide sequences. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2016, 1859, 355–365. [Google Scholar] [CrossRef] [PubMed]
- Ustyantsev, I.G.; Borodulina, O.R.; Kramerov, D.A. Identification of nucleotide sequences and some proteins involved in polyadenylation of RNA transcribed by Pol III from SINEs. RNA Biol. 2020, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Wang, X.; Forouzmand, E.; Jeong, J.; Qiao, F.; Sowd, G.A.; Engelman, A.N.; Xie, X.; Hertel, K.; Shi, Y. Molecular Mechanisms for CFIm-Mediated Regulation of mRNA Alternative Polyadenylation. Mol. Cell 2017, 69, 62–74. [Google Scholar] [CrossRef] [Green Version]
- Ustyantsev, I.G.; Tatosyan, K.A.; Stasenko, D.V.; Kochanova, N.Y.; Borodulina, O.R.; Kramerov, D.A. Polyadenylation of Sine Transcripts Generated by RNA Polymerase III Dramatically Prolongs Their Lifetime in Cells. Mol. Biol. 2020, 54, 78–86. [Google Scholar] [CrossRef]
- Goss, D.J.; Kleiman, F.E. Poly(A) binding proteins: Are they all created equal? Wiley Interdiscip. Rev. RNA 2013, 4, 167–179. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, A.; Pasquinelli, A.E. Tales of Detailed Poly(A) Tails. Trends Cell Biol. 2018, 29, 191–200. [Google Scholar] [CrossRef]
- Dewannieux, M.; Heidmann, T. Role of poly(A) tail length in Alu retrotransposition. Genomics 2005, 86, 378–381. [Google Scholar] [CrossRef]
- Engel, A.; Salem, A.H.; Oyeniran, O.O.; Deininger, L.; Hedges, D.J.; Kilroy, G.E.; Batzer, M.A.; Deininger, P. Active Alu Element “A-Tails”: Size Does Matter. Genome Res. 2002, 12, 1333–1344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odom, G.L.; Robichaux, J.L.; Deininger, P.L. Predicting Mammalian SINE Subfamily Activity from A-tail Length. Mol. Biol. Evol. 2004, 21, 2140–2148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagstaff, B.J.; Hedges, D.J.; Derbes, R.S.; Sanchez, R.C.; Chiaromonte, F.; Makova, K.D.; Roy-Engel, A.M. Rescuing Alu: Recovery of New Inserts Shows LINE-1 Preserves Alu Activity through A-Tail Expansion. PLoS Genet. 2012, 8, e1002842. [Google Scholar] [CrossRef] [Green Version]
- Maraia, R.J.; Lamichhane, T.N. 3′ processing of eukaryotic precursor tRNAs. Wiley Interdiscip. Rev. RNA 2010, 2, 362–375. [Google Scholar] [CrossRef] [Green Version]
- Arimbasseri, G.A.; Rijal, K.; Maraia, R.J. Transcription termination by the eukaryotic RNA polymerase III. Biochim. Biophys. Acta (BBA)-Gene Regul. Mech. 2013, 1829, 318–330. [Google Scholar] [CrossRef] [Green Version]
- Gogolevskaya, I.K.; Kramerov, D.A. Evolutionary History of 4.5SI RNA and Indication That It Is Functional. J. Mol. Evol. 2002, 54, 354–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gogolevskaya, I.K.; Koval, A.P.; Kramerov, D.A. Evolutionary history of 4.5SH RNA. Mol. Biol. Evol. 2005, 22, 1546–1554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gogolevskaya, I.K.; Kramerov, D. 4.5SI RNA genes and the role of their 5′-flanking sequences in the gene transcription. Gene 2010, 451, 32–37. [Google Scholar] [CrossRef]
- Kwak, J.E.; Wickens, M. A family of poly(U) polymerases. RNA 2007, 13, 860–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scott, D.D.; Norbury, C.J. RNA decay via 3′ uridylation. Biochim. Biophys. Acta 2013, 1829, 654–665. [Google Scholar] [CrossRef] [PubMed]
- Menezes, M.R.; Balzeau, J.; Hagan, J.P. 3′ RNA Uridylation in Epitranscriptomics, Gene Regulation, and Disease. Front Mol. Biosci. 2018, 5, 61. [Google Scholar] [CrossRef] [Green Version]
- Serdobova, I.M.; Kramerov, D. Short Retroposons of the B2 Superfamily: Evolution and Application for the Study of Rodent Phylogeny. J. Mol. Evol. 1998, 46, 202–214. [Google Scholar] [CrossRef] [PubMed]
- Borodulina, O.R.; Kramerov, D.A. Wide distribution of short interspersed elements among eukaryotic genomes. FEBS Lett. 1999, 457, 409–413. [Google Scholar] [CrossRef]
- Kawai, K.; Nikaido, M.; Harada, M.; Matsumura, S.; Lin, L.-K.; Wu, Y.; Hasegawa, M.; Okada, N. Intra- and Interfamily Relationships of Vespertilionidae Inferred by Various Molecular Markers Including SINE Insertion Data. J. Mol. Evol. 2002, 55, 284–301. [Google Scholar] [CrossRef] [PubMed]
- Ray, D.A.; Pagan, H.J.; Platt, R.N., 2nd; Kroll, A.R.; Schaack, S.; Stevens, R.D. Differential SINE evolution in vesper and non-vesper bats. Mob. DNA 2015, 6, 10. [Google Scholar] [CrossRef] [Green Version]
- Jurka, J.; Kapitonov, V.; Pavlicek, A.; Klonowski, P.; Kohany, O.; Walichiewicz, J. Repbase Update, a database of eukaryotic repetitive elements. Cytogenet. Genome Res. 2005, 110, 462–467. [Google Scholar] [CrossRef] [PubMed]
- Dunnen, J.T.D.; Schoenmakers, J.G.G. Consensus sequences of theRattus norvegicusB1- and B2 repeats. Nucleic Acids Res. 1987, 15, 2772. [Google Scholar] [CrossRef] [Green Version]
- Keane, T.; Goodstadt, L.; Danecek, P.; White, M.A.; Wong, K.; Yalcin, B.; Heger, A.; Agam, A.; Slater, G.; Goodson, M.; et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature 2011, 477, 289–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kember, R.L.; Fernandes, C.; Tunbridge, E.M.; Liu, L.; Paya-Cano, J.L.; Parsons, M.J.; Schalkwyk, L.C. A B2 SINE insertion in the Comt1 gene (Comt1(B2i)) results in an overexpressing, behavior modifying allele present in classical inbred mouse strains. Genes Brain Behav. 2010, 9, 925–932. [Google Scholar] [CrossRef] [Green Version]
- Leong, D.W.; Komen, J.C.; Hewitt, C.A.; Arnaud, E.; McKenzie, M.; Phipson, B.; Bahlo, M.; Laskowski, A.; Kinkel, S.A.; Davey, G.; et al. Proteomic and Metabolomic Analyses of Mitochondrial Complex I-deficient Mouse Model Generated by Spontaneous B2 Short Interspersed Nuclear Element (SINE) Insertion into NADH Dehydrogenase (Ubiquinone) Fe-S Protein 4 (Ndufs4) Gene. J. Biol. Chem. 2012, 287, 20652–20663. [Google Scholar] [CrossRef] [Green Version]
- Lacey, D.; Hickey, P.; Arhatari, B.D.; O’Reilly, L.A.; Rohrbeck, L.; Kiriazis, H.; Du, X.-J.; Bouillet, P. Spontaneous retrotransposon insertion into TNF 3′UTR causes heart valve disease and chronic polyarthritis. Proc. Natl. Acad. Sci. USA 2015, 112, 9698–9703. [Google Scholar] [CrossRef] [Green Version]
- Moulson, C.L.; Martin, D.R.; Lugus, J.J.; Schaffer, J.E.; Lind, A.C.; Miner, J.H. Cloning of wrinkle-free, a previously uncharacterized mouse mutation, reveals crucial roles for fatty acid transport protein 4 in skin and hair development. Proc. Natl. Acad. Sci. USA 2003, 100, 5274–5279. [Google Scholar] [CrossRef] [Green Version]
- Nesterovitch, A.B.; Szanto, S.; Gonda, A.; Bardos, T.; Kis-Toth, K.; Adarichev, V.A.; Olasz, K.; Ghassemi-Najad, S.; Hoffman, M.D.; Tharp, M.D.; et al. Spontaneous Insertion of a B2 Element in the Ptpn6 Gene Drives a Systemic Autoinflammatory Disease in Mice Resembling Neutrophilic Dermatosis in Humans. Am. J. Pathol. 2011, 178, 1701–1714. [Google Scholar] [CrossRef]
- Morelli, K.H.; Seburn, K.L.; Schroeder, D.G.; Spaulding, E.L.; Dionne, L.A.; Cox, G.A.; Burgess, R.W. Severity of Demyelinating and Axonal Neuropathy Mouse Models Is Modified by Genes Affecting Structure and Function of Peripheral Nodes. Cell Rep. 2017, 18, 3178–3191. [Google Scholar] [CrossRef]
- Kass, D.H.; Batzer, M.A.; Deininger, P.L. Gene conversion as a secondary mechanism of short interspersed element (SINE) evolution. Mol. Cell. Biol. 1995, 15, 19–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, A.M.; Carroll, M.L.; Nguyen, S.V.; Salem, A.-H.; Oldridge, M.; Wilkie, A.O.M.; Batzer, M.A.; Deininger, P.L. Potential Gene Conversion and Source Genes for Recently Integrated Alu Elements. Genome Res. 2000, 10, 1485–1495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orioli, A.; Pascali, C.; Quartararo, J.; Diebel, K.W.; Praz, V.; Romascano, D.; Percudani, R.; van Dyk, L.; Hernandez, N.; Teichmann, M.; et al. Widespread occurrence of non-canonical transcription termination by human RNA polymerase III. Nucleic Acids Res. 2011, 39, 5499–5512. [Google Scholar] [CrossRef] [Green Version]
- Beaudoing, E.; Freier, S.; Wyatt, J.R.; Claverie, J.-M.; Gautheret, D. Patterns of Variant Polyadenylation Signal Usage in Human Genes. Genome Res. 2000, 10, 1001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Xia, L.; Kimura, Y.; Shenbrot, G.; Zhang, Z.; Ge, D.; Yang, Q. Tracing the Origin and Diversification of Dipodoidea (Order: Rodentia): Evidence from Fossil Record and Molecular Phylogeny. Evol. Biol. 2012, 40, 32–44. [Google Scholar] [CrossRef]
- Lebedev, V.S.; Bannikova, A.A.; Pagès, M.; Pisano, J.; Michaux, J.R.; Shenbrot, G.I. Molecular phylogeny and systematics of Dipodoidea: A test of morphology-based hypotheses. Zool. Scr. 2012, 42, 231–249. [Google Scholar] [CrossRef]
- Jebb, D.; Huang, Z.; Pippel, M.; Hughes, G.M.; Lavrichenko, K.; Devanna, P.; Winkler, S.; Jermiin, L.S.; Skirmuntt, E.C.; Katzourakis, A.; et al. Six reference-quality genomes reveal evolution of bat adaptations. Nature 2020, 583, 578–584. [Google Scholar] [CrossRef]
- Bhak, Y.; Jeon, Y.; Jeon, S.; Chung, O.; Jho, S.; Jun, J.; Kim, H.M.; Cho, Y.; Yoon, C.; Lee, S.; et al. Myotis rufoniger genome sequence and analyses: M. rufoniger’s genomic feature and the decreasing effective population size of Myotis bats. PLoS ONE 2017, 12, e0180418. [Google Scholar] [CrossRef]
- Ichiyanagi, T.; Katoh, H.; Mori, Y.; Hirafuku, K.; Boyboy, B.A.; Kawase, M.; Ichiyanagi, K. B2 SINE Copies Serve as a Transposable Boundary of DNA Methylation and Histone Modifications in the Mouse. Mol. Biol. Evol. 2021, 38, 2380–2395. [Google Scholar] [CrossRef]
- Koval, A.P.; Veniaminova, N.A.; Kramerov, D.A. Additional box B of RNA polymerase III promoter in SINE B1 can be functional. Gene 2011, 487, 113–117. [Google Scholar] [CrossRef]
- Yamada, K.D.; Tomii, K.; Katoh, K. Application of the MAFFT sequence alignment program to large data—reexamination of the usefulness of chained guide trees. Bioinformatics 2016, 32, 3246–3251. [Google Scholar] [CrossRef] [Green Version]
- Yan, T.; Yoo, D.; Berardini, T.; Mueller, L.A.; Weems, D.C.; Weng, S.; Cherry, J.M.; Rhee, S.Y. PatMatch: A program for finding patterns in peptide and nucleotide sequences. Nucleic Acids Res. 2005, 33, W262–W266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- James, B.; Luczak, B.B.; Girgis, H.Z. MeShClust: An intelligent tool for clustering DNA sequences. Nucleic Acids Res. 2018, 46, e83. [Google Scholar] [CrossRef] [Green Version]
- Li, H. Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv 2013, arXiv:1303.3997. [Google Scholar]
- Gibbs, R.A.; Weinstock, G.M.; Metzker, M.L.; Muzny, D.M.; Sodergren, E.J.; Scherer, S.; Scott, G.; Steffen, D.; Worley, K.C.; Burch, P.E.; et al. Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature 2004, 428, 493–521. [Google Scholar] [PubMed] [Green Version]
- Chomczynski, P.; Sacchi, N. The single-step method of RNA isolation by acid guanidinium thiocyanate–phenol–chloroform extraction: Twenty-something years on. Nat. Protoc. 2006, 1, 581–585. [Google Scholar] [CrossRef]


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| B2a | B2b | B2c | B2d | B2e | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| B2b1 | B2b2 | B2b3 | B2c1 | B2c2 | B2c3 | B2c4 | B2d1 | B2d2 | ||||||||
| Mus musculus | proportion | 63.9% | 7.8% | 5.4% | 20.2% | 1.6% | ||||||||||
| total number | ~59,013 | ~7203 | ~4987 | ~18,655 | ~1478 | |||||||||||
| mean similarity | 83% | 70% | 73% | 62% | 59% | |||||||||||
| TSDs | 73% | 64% | 62% | 54% | 23% | |||||||||||
| TCT3 | **** | *** | * | * | * | |||||||||||
| Rattus norvegicus | proportion | 71.0% | 6.2% | 5.5% | 15.3% | 2.0% | ||||||||||
| total number | ~85,872 | ~7499 | ~6652 | ~18,505 | ~2419 | |||||||||||
| mean similarity | 81% | 76% | 66% | 59% | 62% | |||||||||||
| TSDs | 76.9% | 64.7% | 43.3% | 44.6% | 60.0% | |||||||||||
| TCT3 | *** | ** | * | |||||||||||||
| Cricetulus griseus | proportion | 46.3% | 19.4% | 11.7% | 3.3% | 2.2% | 2.2% | 34.3% | ||||||||
| total number | ~33,945 | ~14,223 | ~8578 | ~2419 | ~1613 | ~1613 | ~25,147 | |||||||||
| mean similarity | 80% | 70% | 76% | 68% | 77% | 63% | ||||||||||
| TSDs | 76.9% | 67.7% | 68.4% | 54.5% | 69.2% | 58.9% | ||||||||||
| TCT3 | *** | * | * | * | ||||||||||||
| Peromyscus maniculatus | proportion | 39.5% | 24.7% | 9.3% | 5.5% | 20.4% | 15.8% | 1.8% | 2.8% | 40% | ||||||
| total number | ~32,914 | ~20,582 | ~7749 | ~4583 | ~16,999 | ~13,166 | ~1500 | ~2333 | ~33,330 | |||||||
| mean similarity | 79% | 81% | 92% | 74% | 80% | 80% | 64% | |||||||||
| TSDs | 68.8% | 80.8% | 97.0% | 76.6% | 81.8% | 64.7% | 61.1% | |||||||||
| TCT3 | ** | *** | **** | |||||||||||||
| Nannospalax galili | proportion | 100% | 93.5% | 6.5% | ||||||||||||
| total number | ~245,653 | ~229,686 | ~15,967 | |||||||||||||
| mean similarity | 73% | 75% | 64% | |||||||||||||
| TSDs | 58.5% | 59.0% | 57.6% | |||||||||||||
| TCT3 | *** | |||||||||||||||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Vassetzky, N.S.; Borodulina, O.R.; Ustyantsev, I.G.; Kosushkin, S.A.; Kramerov, D.A. Analysis of SINE Families B2, Dip, and Ves with Special Reference to Polyadenylation Signals and Transcription Terminators. Int. J. Mol. Sci. 2021, 22, 9897. https://doi.org/10.3390/ijms22189897
Vassetzky NS, Borodulina OR, Ustyantsev IG, Kosushkin SA, Kramerov DA. Analysis of SINE Families B2, Dip, and Ves with Special Reference to Polyadenylation Signals and Transcription Terminators. International Journal of Molecular Sciences. 2021; 22(18):9897. https://doi.org/10.3390/ijms22189897
Chicago/Turabian StyleVassetzky, Nikita S., Olga R. Borodulina, Ilia G. Ustyantsev, Sergei A. Kosushkin, and Dmitri A. Kramerov. 2021. "Analysis of SINE Families B2, Dip, and Ves with Special Reference to Polyadenylation Signals and Transcription Terminators" International Journal of Molecular Sciences 22, no. 18: 9897. https://doi.org/10.3390/ijms22189897