
Aggregation of Cystatin C Changes Its Inhibitory Functions on Protease Activities and Amyloid β Fibril Formation

 , and
, and {kind=link}
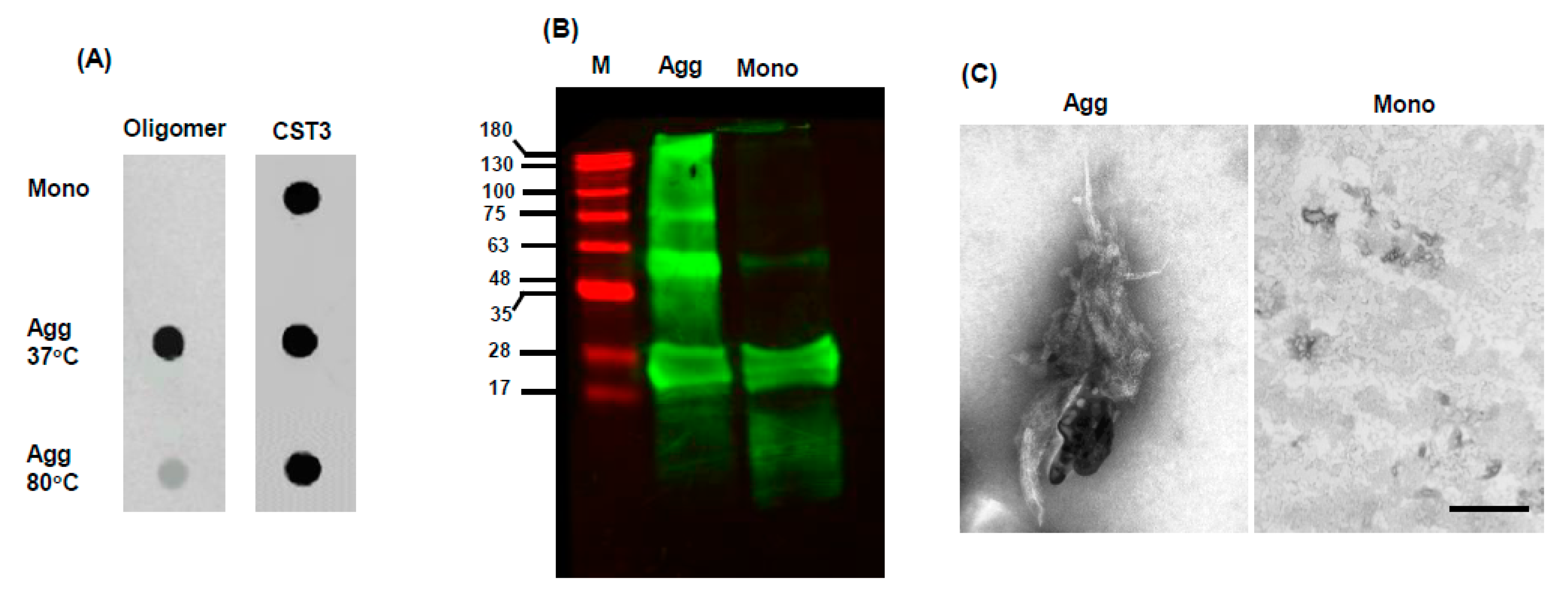{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Preparation of Recombinant CST3
5.2. Aggregation of CST3
5.3. Aggregation Assay
5.4. Dot-Blot Oligomer Assay
5.5. Electron Microscopy
5.6. Western Blotting
5.7. Cathepsin B Assay
5.8. Aβ1-40 Peptide Fibril Formation
5.9. Assessment of Fibril Levels Using ThT Fluorescence
5.10. Cell Culture
5.10.1. CCF-STTG1 Culture
5.10.2. Culture of a Mouse Neuronal Stem Cell Line and Differentiation to Mature Neurons
5.11. (3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium Bromide (MTT) Assay
5.12. Immunocytochemistry
5.13. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mussap, M.; Plebani, M. Biochemistry and clinical role of human cystatin C. Crit. Rev. Clin. Lab. Sci. 2004, 41, 467–550. [Google Scholar] [CrossRef]
- Yamaze, T.; Mino, S.; Atsuta, I.; Danjo, A.; Kagia, T.; Nishijima, K.; Jang, J.; Kido, M.A.; Tanaka, T. Localization of the endogenous cysteine proteinase Inhibitor, cystatin C, and the cysteine proteinase, cathepsin B, to the junctional epithelium in rat gingiva. Acta Histochem. Cytochem. 2005, 38, 121–129. [Google Scholar] [CrossRef] [Green Version]
- Deng, A.; Irizarry, M.C.; Nitsch, R.M.; Growdon, J.H.; Rebeck, G.W. Elevation of cystatin C in susceptible neurons in Alzheimer’s disease. Am. J. Pathol. 2001, 159, 1061–1068. [Google Scholar] [CrossRef]
- Watanabe, S.; Hayakawa, T.; Wakasugi, K.; Yamanaka, K. Cystatin C protects neuronal cells against mutant copper-zinc superoxide dismutase-mediated toxicity. Cell Death Dis. 2014, 5, e1497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abrahamson, M.; Barrett, A.; Salvesen, G.; Grubb, A. Isolation of six cysteine proteinase inhibitors from human urine. Their physicochemical and enzyme kinetic properties and concentrations in biological fluids. J. Biol. Chem. 1986, 261, 11282–11289. [Google Scholar] [CrossRef]
- Zavasnik-Bergant, T.; Bergant, M.; Jaras, M.; Griffiths, G. Different localisation of cystatin C in immature and mature dendritic cells. Radiol. Oncol. 2006, 40, 183–188. [Google Scholar]
- Lutgens, S.P.M.; Cleutjens, K.B.J.M.; Daemen, M.; Heeneman, S. Cathepsin cysteine proteases in cardiovascular disease. FASEB J. 2007, 21, 3029–3041. [Google Scholar] [CrossRef]
- Turk, V.; Stoka, V.; Vasiljeva, O.; Renko, M.; Sun, T.; Turk, B.; Turk, D. Cysteine cathepsins: From structure, function and regulation to new frontiers. Biochim. Biophys. Acta BBA Proteins Proteom. 2012, 1824, 68–88. [Google Scholar] [CrossRef] [Green Version]
- Yasuhara, O.; Hanai, K.; Ohkubo, I.; Sasaki, M.; McGeer, P.L.; Kimura, H. Expression of cystatin C in rat, monkey and human brains. Brain Res. 1993, 628, 85–92. [Google Scholar] [CrossRef]
- Ren, Y.; Zhu, W.; Cui, F.; Yang, F.; Chen, Z.; Ling, L.; Huang, X. Measurement of cystatin C levels in the cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Int. J. Clin. Exp. Pathol. 2015, 8, 5419–5426. [Google Scholar]
- Vidak, E.; Javoršek, U.; Vizovišek, M.; Turk, B. Cysteine cathepsins and their extracellular roles: Shaping the microenvironment. Cells 2019, 8, 264. [Google Scholar] [CrossRef] [Green Version]
- Winklhofer, K.F.; Tatzelt, J.; Haass, C. The two faces of protein misfolding: Gain- and loss-of-function in neurodegenerative diseases. EMBO J. 2008, 27, 336–349. [Google Scholar] [CrossRef]
- Lloret, A.; Fuchsberger, T.; Giraldo, E.; Viña, J. Molecular mechanisms linking amyloid β toxicity and Tau hyperphosphorylation in Alzheimer’s disease. Free Radic. Biol. Med. 2015, 83, 186–191. [Google Scholar] [CrossRef]
- Mehra, S.; Sahay, S.; Maji, S.K. α-Synuclein misfolding and aggregation: Implications in Parkinson’s disease pathogenesis. Biochim. Biophys. Acta BBA Proteins Proteom. 2019, 1867, 890–908. [Google Scholar] [CrossRef]
- Cohen, D.H.; Feiner, H.; Jensson, O.; Frangione, B. Amyloid fibril in hereditary cerebral hemorrhage with amyloidosis (HCHWA) is related to the gastroentero-pancreatic neuroendocrine protein, gamma trace. J. Exp. Med. 1983, 158, 623–628. [Google Scholar] [CrossRef] [PubMed]
- Palsdottir, A.; Snorradottir, A.O.; Thorsteinsson, L. Hereditary cystatin C amyloid angiopathy: Genetic, clinical, and pathological aspects. Brain Pathol. 2006, 16, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Abrahamson, M.; Grubb, A. Increased body temperature accelerates aggregation of the Leu-68--> Gln mutant cystatin C, the amyloid-forming protein in hereditary cystatin C amyloid angiopathy. Proc. Natl. Acad. Sci. USA 1994, 91, 1416–1420. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tizon, B.; Ribe, E.M.; Mi, W.; Troy, C.M.; Levy, E. Cystatin C protects neuronal cells from amyloid-β-induced toxicity. J. Alzheimer’s Dis. 2010, 19, 885–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nagai, A.; Kobayashi, S.; Shimode, K.; Imaoka, K.; Umegae, N.; Fujihara, S.; Nakamura, M. No mutations in cystatin C gene in cerebral amyloid angiopathy with cystatin C deposition. Mol. Chem. Neuropathol. 1998, 33, 63–78. [Google Scholar] [CrossRef]
- Wada, Y.; Nagai, A.; Sheikh, A.M.; Onoda, K.; Terashima, M.; Shiota, Y.; Araki, A.; Yamaguchi, S. Co-localization of cystatin C and prosaposin in cultured neurons and in anterior horn neurons with amyotrophic lateral sclerosis. J. Neurol. Sci. 2018, 384, 67–74. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, S.; Komine, O.; Endo, F.; Wakasugi, K.; Yamanaka, K. Intracerebroventricular administration of Cystatin C ame-liorates disease in SOD1-linked amyotrophic lateral sclerosis mice. J. Neurochem. 2018, 145, 80–89. [Google Scholar] [CrossRef]
- Ekiel, I.; Abrahamson, M. Folding-related dimerization of human cystatin, C. J. Biol. Chem. 1996, 271, 1314–1321. [Google Scholar] [CrossRef] [Green Version]
- Mitaki, S.; Nagai, A.; Sheikh, A.M.; Terashima, M.; Isomura, M.; Nabika, T.; Yamaguchi, S. Contribution of cystatin C gene polymorphisms to cerebral white matter lesions. Cerebrovasc. Dis. 2011, 32, 489–496. [Google Scholar] [CrossRef]
- Lindholt, J.S.; Erlandsen, E.J.; Henneberg, E.W. Cystatin C deficiency is associated with the progression of small abdominal aortic aneurysms. Br. J. Surg. 2001, 88, 1472–1475. [Google Scholar] [CrossRef]
- Shi, G.-P.; Sukhova, G.K.; Grubb, A.; Ducharme, A.; Rhode, L.H.; Lee, R.T.; Ridker, P.M.; Libby, P.; Chapman, H.A. Cystatin C deficiency in human atherosclerosis and aortic aneurysms. J. Clin. Investig. 1999, 104, 1191–1197. [Google Scholar] [CrossRef] [Green Version]
- Nishimura, Y.; Honda, K.; Yuzaki, M.; Tajima, K.; Nakamura, R.; Nakanishi, Y.; Kaneko, M.; Agematsu, K.; Nagashima, M. Serum cystatin C level as a biomarker of aortic plaque in patients with an aortic arch aneurysm. J. Atheroscler. Thromb. 2021, 28, 506–513. [Google Scholar] [CrossRef]
- Lv, B.-J.; Lindholt, J.; Cheng, X.; Wang, J.; Shi, G.-P. Plasma cathepsin S and cystatin C levels and risk of abdominal aortic aneurysm: A randomized population-based study. PLoS ONE 2012, 7, e41813. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Sukhova, G.K.; Sun, J.-S.; Xu, W.-H.; Libby, P.; Shi, G.-P. Lysosomal cysteine proteases in atherosclerosis. Arter. Thromb. Vasc. Biol. 2004, 24, 1359–1366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, S.; Cai, J.; Lu, R.; Wu, J.; Zhang, M.; Zhou, X. Association between serum cystatin C level and total magnetic resonance imaging burden of cerebral small vessel disease in patients with acute lacunar stroke. J. Stroke Cerebrovasc. Dis. 2017, 26, 186–191. [Google Scholar] [CrossRef] [PubMed]
- Maniwa, K.; Yano, S.; Sheikh, A.M.; Onoda, K.; Mitaki, S.; Isomura, M.; Mishima, S.; Yamaguchi, S.; Nabika, T.; Nagai, A. Association between cystatin C gene polymorphism and the prevalence of white matter lesion in elderly healthy subjects. Sci. Rep. 2020, 10, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lashuel, H.A.; Hartley, D.; Petre, B.M.; Walz, T.; Lansbury, P.T., Jr. Neurodegenerative disease: Amyloid pores from pathogenic mutations. Nature 2002, 418, 291. [Google Scholar] [CrossRef] [PubMed]
- Maruyama, K.; Kametani, F.; Ikeda, S.; Ishihara, T.; Yanagisawa, N. Characterization of amyloid fibril protein from a case of cerebral amyloid angiopathy showing immunohistochemical reactivity for both beta protein and cystatin C. Neurosci. Lett. 1992, 144, 38–42. [Google Scholar] [CrossRef]
- Levy, E.; Jaskolski, M.; Grubb, A. The role of cystatin C in cerebral amyloid angiopathy and stroke: Cell biology and animal models. Brain Pathol. 2006, 16, 60–70. [Google Scholar] [CrossRef] [PubMed]
- Aoki, T.; Kataoka, H.; Ishibashi, R.; Nozaki, K.; Hashimoto, N. Cathepsin B, K, and S are expressed in cerebral aneurysms and promote the progression of cerebral aneurysms. Stroke 2008, 39, 2603–2610. [Google Scholar] [CrossRef] [PubMed]
- Perlenfein, T.J.; Mehlhoff, J.D.; Murphy, R.M. Insights into the mechanism of cystatin C oligomer and amyloid formation and its interaction with beta-amyloid. J. Biol. Chem. 2017, 292, 11485–11498. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janowski, R.; Kozak, M.; Jankowska, E.; Grzonka, Z.; Grubb, A.; Abrahamson, M.; Jaskolski, M. Human cystatin C, an amyloidogenic protein, dimerizes through three-dimensional domain swapping. Nat. Struct. Biol. 2001, 8, 316–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chrabaszczewska, M.; Sieradzan, A.K.; Rodziewicz-Motowidło, S.; Grubb, A.; Dobson, C.M.; Kumita, J.R.; Kozak, M. Structural characterization of covalently stabilized human cystatin C oligomers. Int. J. Mol. Sci. 2020, 21, 5860. [Google Scholar] [CrossRef]
- Wilkening, A.; Rüb, C.; Sylvester, M.; Voos, W. Analysis of heat-induced protein aggregation in human mitochondria. J. Biol. Chem. 2018, 293, 11537–11552. [Google Scholar] [CrossRef] [Green Version]
- Jahn, T.; Radford, S.E. Folding versus aggregation: Polypeptide conformations on competing pathways. Arch. Biochem. Biophys. 2008, 469, 100–117. [Google Scholar] [CrossRef] [Green Version]
- Jahn, T.; Parker, M.J.; Homans, S.W.; Radford, S. Amyloid formation under physiological conditions proceeds via a native-like folding intermediate. Nat. Struct. Mol. Biol. 2006, 13, 195–201. [Google Scholar] [CrossRef]
- Tsiolaki, P.L.; Louros, N.N.; Hamodrakas, S.J.; Iconomidou, V.A. Exploring the ‘aggregation-prone’ core of human cystatin C: A structural study. J. Struct. Biol. 2015, 191, 272–280. [Google Scholar] [CrossRef]
- Haghighi-Poodeh, S.; Navidpour, L.; Yaghmaei, P.; Ebrahim-Habibi, A. Monocyclic phenolic compounds stabilize human insulin and suppress its amorphous aggregation: In vitro and in vivo study. Biochem. Biophys. Res. Commun. 2019, 518, 362–367. [Google Scholar] [CrossRef] [PubMed]
- Wallin, H.; Bjarnadottir, M.; Vogel, L.K.; Wassélius, J.; Ekström, U.; Abrahamson, M. Cystatins—Extra- and intracellular cysteine protease inhibitors: High-level secretion and uptake of cystatin C in human neuroblastoma cells. Biochimie 2010, 92, 1625–1634. [Google Scholar] [CrossRef] [PubMed]
- Ekström, U.; Wallin, H.; Lorenzo, J.; Holmqvist, B.; Abrahamson, M.; Avilés, F.X. Internalization of cystatin C in human cell lines. FEBS J. 2008, 275, 4571–4582. [Google Scholar] [CrossRef]
- Holmes, B.B.; Diamond, M.I. Cellular mechanisms of protein aggregate propagation. Curr. Opin. Neurol. 2012, 25, 721–726. [Google Scholar] [CrossRef] [Green Version]
- Wesén, E.; Jeffries, G.D.M.; Dzebo, M.M.; Esbjörner, E.K. Endocytic uptake of monomeric amyloid-β peptides is clathrin- and dynamin-independent and results in selective accumulation of Aβ(1–42) compared to Aβ(1–40). Sci. Rep. 2017, 7, 2021. [Google Scholar] [CrossRef] [PubMed]
- Beck, A.P.; Meyerholz, D.K. Evolving challenges to model human diseases for translational research. Cell Tissue Res. 2020, 380, 305–311. [Google Scholar] [CrossRef]
- Almy, F.S.; Christopher, M.M.; King, D.P.; Brown, S.A. Evaluation of cystatin c as an endogenous marker of glomerular filtration rate in dogs. J. Vet. Intern. Med. 2002, 16, 45–51. [Google Scholar] [CrossRef]
- Kayed, R.; Head, E.; Thompson, J.L.; McIntire, T.M.; Milton, S.C.; Cotman, C.W.; Glabe, C.G. Common structure of soluble amyloid oligomers implies common mechanism of pathogenesis. Science 2003, 300, 486–489. [Google Scholar] [CrossRef] [Green Version]
- Tabassum, S.; Sheikh, A.M.; Yano, S.; Ikeue, T.; Handa, M.; Nagai, A. A carboxylated Zn-phthalocyanine inhibits fibril formation of Alzheimer’s amyloid beta peptide. FEBS J. 2015, 282, 463–476. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, A.M.; Yano, S.; Mitaki, S.; Haque, M.A.; Yamaguchi, S.; Nagai, A. A mesenchymal stem cell line (B10) increases angiogenesis in a rat MCAO model. Exp. Neurol. 2019, 311, 182–193. [Google Scholar] [CrossRef] [PubMed]
- Sheikh, A.; Yano, S.; Tabassum, S.; Omura, K.; Araki, A.; Mitaki, S.; Ito, Y.; Huang, S.; Nagai, A. Alteration of neural stem cell functions in ataxia and male sterility mice: A possible role of β-tubulin glutamylation in neurodegeneration. Cells 2021, 10, 155. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sheikh, A.M.; Wada, Y.; Tabassum, S.; Inagaki, S.; Mitaki, S.; Yano, S.; Nagai, A. Aggregation of Cystatin C Changes Its Inhibitory Functions on Protease Activities and Amyloid β Fibril Formation. Int. J. Mol. Sci. 2021, 22, 9682. https://doi.org/10.3390/ijms22189682
Sheikh AM, Wada Y, Tabassum S, Inagaki S, Mitaki S, Yano S, Nagai A. Aggregation of Cystatin C Changes Its Inhibitory Functions on Protease Activities and Amyloid β Fibril Formation. International Journal of Molecular Sciences. 2021; 22(18):9682. https://doi.org/10.3390/ijms22189682
Chicago/Turabian StyleSheikh, Abdullah Md., Yasuko Wada, Shatera Tabassum, Satoshi Inagaki, Shingo Mitaki, Shozo Yano, and Atsushi Nagai. 2021. "Aggregation of Cystatin C Changes Its Inhibitory Functions on Protease Activities and Amyloid β Fibril Formation" International Journal of Molecular Sciences 22, no. 18: 9682. https://doi.org/10.3390/ijms22189682