
The RhoGEF Trio: A Protein with a Wide Range of Functions in the Vascular Endothelium

 , and
, and 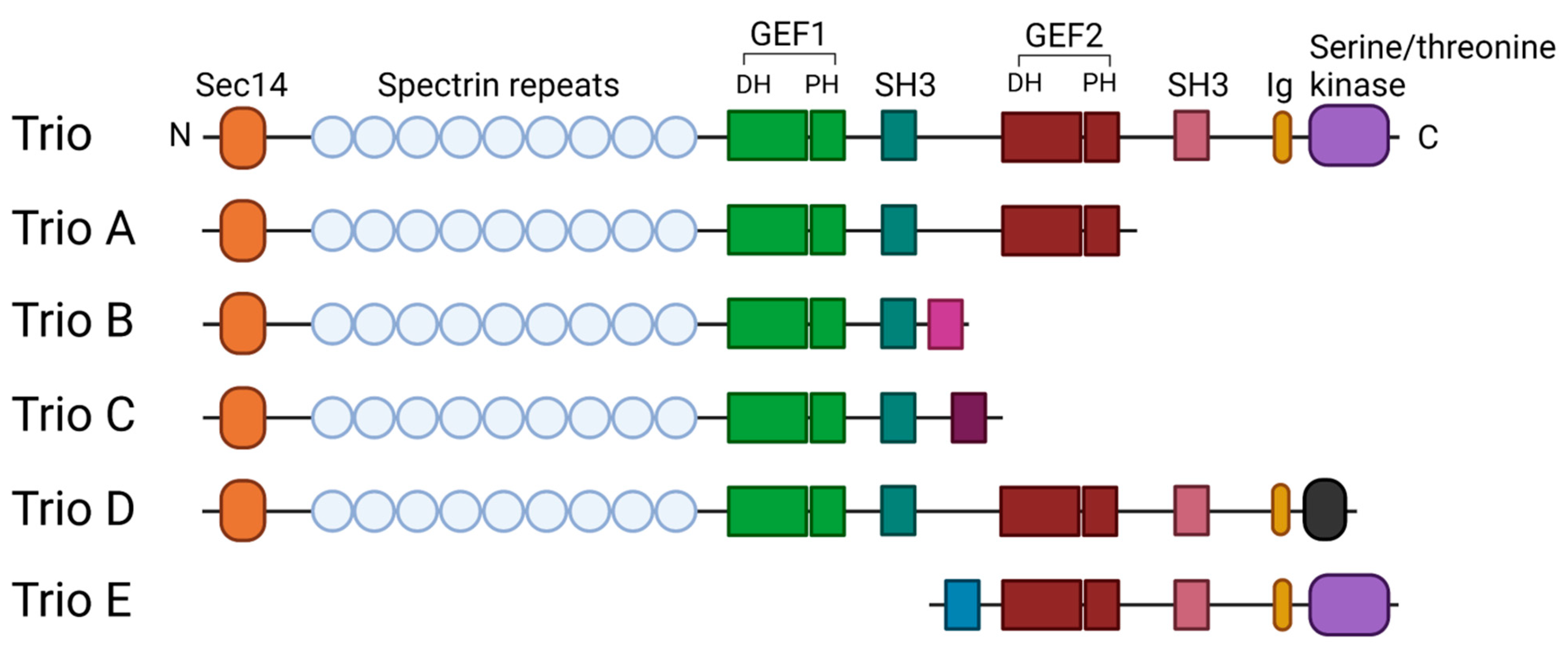{kind=link}
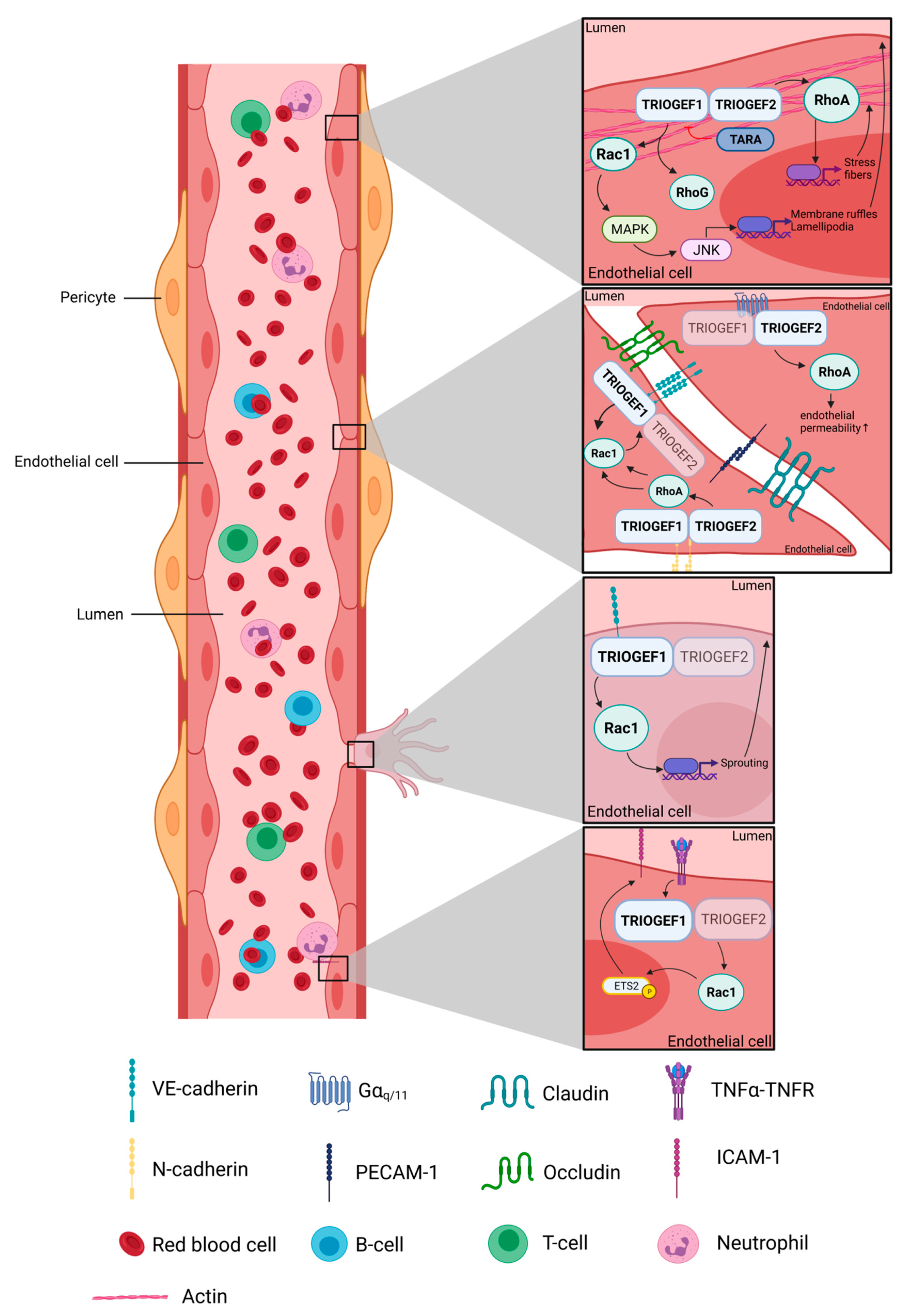{kind=link}
Abstract
:1. Introduction
2. Trio Localization
3. Trio Remodels the Actin Cytoskeleton
4. The GEF Domains of Trio Differentially Regulate Endothelial Barrier Function
4.1. Stabilization of the Barrier
4.2. Destabilization of the Endothelial Barrier by Trio
5. Sprouting Angiogenesis and Vascular Remoddeling Require the GEF1 Domain of Trio
6. Trio Mediates Leukocyte Adhesion for Transendothelial Migration during Inflammation
7. The Potential Functions of Trio during Inflammation
8. Clinical Implications of Trio Mutants
9. Trio Inhibitors for Disease Prevention
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Lum, H.; Malik, A.B. Regulation of vascular endothelial barrier function. Am. J. Physiol.-Lung Cell. Mol. Physiol. 1994, 267, L223–L241. [Google Scholar] [CrossRef]
- Rahimi, N. Defenders and challengers of endothelial barrier function. Front. Immunol. 2017, 8, 1847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vandenbroucke, E.; Mehta, D.; Minshall, R.; Malik, A.B. Regulation of endothelial junctional permeability. Ann. N. Y. Acad. Sci. 2008, 1123, 134–145. [Google Scholar] [CrossRef] [PubMed]
- Wojciak-Stothard, B.; Ridley, A.J. Rho GTPases and the regulation of endothelial permeability. Vascul. Pharmacol. 2002, 39, 187–199. [Google Scholar] [CrossRef]
- Van Rijssel, J.; Van Buul, J.D. The many faces of the guanine-nucleotide exchange factor trio. Cell Adhes. Migr. 2012, 6, 482–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debant, A.; Serra-PagèS, C.; Seipel, K.; O’Brien, S.; Tang, M.; Park, S.H.; Streuli, M. The multidomain protein Trio binds the LAR transmembrane tyrosine phosphatase, contains a protein kinase domain, and has separate rac-specific and rho-specific guanine nucleotide exchange factor domains. Proc. Natl. Acad. Sci. USA 1996, 93, 5466–5471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellanger, J.M.; Lazaro, J.B.; Diriong, S.; Fernandez, A.; Lamb, N.; Debant, A. The two guanine nucleotide exchange factor domains of Trio link the Rac1 and the RhoA pathways in vivo. Oncogene 1998, 16, 147–152. [Google Scholar] [CrossRef] [Green Version]
- Blangy, A.; Vignal, E.; Schmidt, S.; Debant, A.; Gauthier-Rouvière, C.; Fort, P. TrioGEF1 controls Rac- and Cdc42-dependent cell structures through the direct activation of RhoG. J. Cell Sci. 2000, 113, 729–739. [Google Scholar] [CrossRef]
- Saito, K.; Tautz, L.; Mustelin, T. The lipid-binding SEC14 domain. Biochim. Biophys. Acta-Mol. Cell Biol. Lipids 2007, 1771, 719–726. [Google Scholar] [CrossRef]
- Seipel, K.; Medley, Q.G.; Kedersha, N.L.; Zhang, X.A.; O’Brien, S.P.; Serra-Pages, C.; Hemler, M.E.; Streuli, M. Trio amino-terminal guanine nucleotide exchange factor domain expression promotes actin cytoskeleton reorganization, cell migration and anchorage-independent cell growth. J. Cell Sci. 1999, 112, 1825–1834. [Google Scholar] [CrossRef]
- Medley, Q.G.; Buchbinder, E.G.; Tachibana, K.; Ngo, H.; Serra-Pagès, C.; Streuli, M. Signaling between focal adhesion kinase and trio. J. Biol. Chem. 2003, 278, 13265–13270. [Google Scholar] [CrossRef] [Green Version]
- Medley, Q.G.; Serra-Pagès, C.; Iannotti, E.; Seipel, K.; Tang, M.; O’Brien, S.P.; Streuli, M. The trio guanine nucleotide exchange factor is a RhoA target. Binding of RhoA to the trio immunoglobulin-like domain. J. Biol. Chem. 2000, 275, 36116–36123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McPherson, C.E.; Eipper, B.A.; Mains, R.E. Multiple novel isoforms of Trio are expressed in the developing rat brain. Gene 2005, 347, 125–135. [Google Scholar] [CrossRef] [PubMed]
- Portales-Casamar, E.; Briançon-Marjollet, A.; Fromont, S.; Triboulet, R.; Debant, A. Identification of novel neuronal isoforms of the Rho-GEF Trio. Biol. Cell 2006, 98, 183–193. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, S.; Debant, A. Function and regulation of the Rho guanine nucleotide exchange factor Trio. Small GTPases 2014, 5, e983880. [Google Scholar] [CrossRef] [Green Version]
- Kalucka, J.; de Rooij, L.P.M.H.; Goveia, J.; Rohlenova, K.; Dumas, S.J.; Meta, E.; Conchinha, N.V.; Taverna, F.; Teuwen, L.A.; Veys, K.; et al. Single-Cell Transcriptome Atlas of Murine Endothelial Cells. Cell 2020, 180, 764–779. [Google Scholar] [CrossRef] [PubMed]
- Aspenström, P. Fast-cycling Rho GTPases. Small GTPases 2020, 11, 248–255. [Google Scholar] [CrossRef] [PubMed]
- Müller, P.M.; Rademacher, J.; Bagshaw, R.D.; Wortmann, C.; Barth, C.; van Unen, J.; Alp, K.M.; Giudice, G.; Eccles, R.L.; Heinrich, L.E.; et al. Systems analysis of RhoGEF and RhoGAP regulatory proteins reveals spatially organized RAC1 signalling from integrin adhesions. Nat. Cell Biol. 2020, 22, 498–511. [Google Scholar] [CrossRef]
- Pertz, O. Spatio-temporal Rho GTPase signaling-Where are we now? J. Cell Sci. 2010, 123, 1841–1850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benink, H.A.; Bement, W.M. Concentric zones of active RhoA and Cdc42 around single cell wounds. J. Cell Biol. 2005, 168, 429–439. [Google Scholar] [CrossRef] [Green Version]
- MacHacek, M.; Hodgson, L.; Welch, C.; Elliott, H.; Pertz, O.; Nalbant, P.; Abell, A.; Johnson, G.L.; Hahn, K.M.; Danuser, G. Coordination of Rho GTPase activities during cell protrusion. Nature 2009, 461, 99–103. [Google Scholar] [CrossRef] [Green Version]
- Bravo-Cordero, J.J.; Oser, M.; Chen, X.; Eddy, R.; Hodgson, L.; Condeelis, J. A novel spatiotemporal RhoC activation pathway locally regulates cofilin activity at invadopodia. Curr. Biol. 2011, 21, 635–644. [Google Scholar] [CrossRef] [Green Version]
- Fritz, R.D.; Letzelter, M.; Reimann, A.; Martin, K.; Fusco, L.; Ritsma, L.; Ponsioen, B.; Fluri, E.; Schulte-Merker, S.; Van Rheenen, J.; et al. A versatile toolkit to produce sensitive FRET biosensors to visualize signaling in time and space. Sci. Signal. 2013, 6, rs12. [Google Scholar] [CrossRef]
- Graessl, M.; Koch, J.; Calderon, A.; Kamps, D.; Banerjee, S.; Mazel, T.; Schulze, N.; Jungkurth, J.K.; Patwardhan, R.; Solouk, D.; et al. An excitable Rho GTPase signaling network generates dynamic subcellular contraction patterns. J. Cell Biol. 2017, 216, 4271–4285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timmerman, I.; Heemskerk, N.; Kroon, J.; Schaefer, A.; van Rijssel, J.; Hoogenboezem, M.; van Unen, J.; Goedhart, J.; Gadella, T.W.J.; Yin, T.; et al. A local VE-cadherin and Trio-based signaling complex stabilizes endothelial junctions through Rac1. J. Cell Sci. 2015, 128, 3041–3054. [Google Scholar] [CrossRef] [Green Version]
- Vaqué, J.P.; Dorsam, R.T.; Feng, X.; Iglesias-Bartolome, R.; Forsthoefel, D.J.; Chen, Q.; Debant, A.; Seeger, M.A.; Ksander, B.R.; Teramoto, H.; et al. A Genome-wide RNAi Screen Reveals a Trio-Regulated Rho GTPase Circuitry Transducing Mitogenic Signals Initiated by G Protein-Coupled Receptors. Mol. Cell 2013, 49, 94–108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yano, T.; Yamazaki, Y.; Adachi, M.; Okawa, K.; Fort, P.; Uji, M.; Tsukita, S.; Tsukita, S. Tara up-regulates E-cadherin transcription by binding to the Trio RhoGEF and inhibiting Rac signaling. J. Cell Biol. 2011, 193, 319–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klems, A.; van Rijssel, J.; Ramms, A.S.; Wild, R.; Hammer, J.; Merkel, M.; Derenbach, L.; Préau, L.; Hinkel, R.; Suarez-Martinez, I.; et al. The GEF Trio controls endothelial cell size and arterial remodeling downstream of Vegf signaling in both zebrafish and cell models. Nat. Commun. 2020, 11, 5319. [Google Scholar] [CrossRef] [PubMed]
- Kroon, J.; Heemskerk, N.; Kalsbeek, M.J.T.; De Waard, V.; Van Rijssel, J.; Van Buul, J.D. Flow-induced endothelial cell alignment requires the RhoGEF Trio as a scaffold protein to polarize active Rac1 distribution. Mol. Biol. Cell 2017, 28, 1745–1753. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.-J.; Nishikawa, K.; Yuda, H.; Wang, Y.-L.; Osaka, H.; Fukazawa, N.; Naito, A.; Kudo, Y.; Wada, K.; Aoki, S. Solo/Trio8, a Membrane-Associated Short Isoform of Trio, Modulates Endosome Dynamics andNeurite Elongation. Mol. Cell. Biol. 2006, 26, 6923–6935. [Google Scholar] [CrossRef] [Green Version]
- Atlas, T. Human Protein Trio. Available online: https://www.proteinatlas.org/ENSG00000038382-TRIO (accessed on 16 August 2021).
- Sirokmány, G.; Szidonya, L.; Káldi, K.; Gáborik, Z.; Ligeti, E.; Geiszt, M. Sec14 homology domain targets p50RhoGAP to endosomes and provides a link between Rab and Rho GTPases. J. Biol. Chem. 2006, 281, 6096–6105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valdivia, A.; Goicoechea, S.M.; Awadia, S.; Zinn, A.; Garcia-Mata, R. Regulation of circular dorsal ruffles, macropinocytosis, and cell migration by RhoG and its exchange factor, Trio. Mol. Biol. Cell 2017, 28, 1768–1781. [Google Scholar] [CrossRef] [PubMed]
- Skowronek, K.R.; Guo, F.; Zheng, Y.; Nassar, N. The C-terminal basic tail of RhoG assists the guanine nucleotide exchange factor Trio in binding to phospholipids. J. Biol. Chem. 2004, 279, 37895–37907. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, H.; Negishi, M. RhoG activates Rac1 by direct interaction with the Dock180-binding protein Elmo. Nature 2003, 424, 461–464. [Google Scholar] [CrossRef]
- Van Rijssel, J.; Hoogenboezem, M.; Wester, L.; Hordijk, P.L.; van Buul, J.D. The N-terminal DH-PH domain of trio induces cell spreading and migration by regulating lamellipodia dynamics in a Rac1-dependent fashion. PLoS ONE 2012, 7, e29912. [Google Scholar] [CrossRef] [Green Version]
- Van Rijssel, J.; Kroon, J.; Hoogenboezem, M.; Van Alphen, F.P.J.; De Jong, R.J.; Kostadinova, E.; Geerts, D.; Hordijk, P.L.; Van Buul, J.D. The Rho-guanine nucleotide exchange factor Trio controls leukocyte transendothelial migration by promoting docking structure formation. Mol. Biol. Cell 2012, 23, 2831–2844. [Google Scholar] [CrossRef] [PubMed]
- Martin, K.; Reimann, A.; Fritz, R.D.; Ryu, H.; Jeon, N.L.; Pertz, O. Spatio-temporal co-ordination of RhoA, Rac1 and Cdc42 activation during prototypical edge protrusion and retraction dynamics. Sci. Rep. 2016, 6, 21901. [Google Scholar] [CrossRef] [Green Version]
- Seipel, K.; O’Brien, S.P.; Lannotti, E.; Medley, Q.G.; Streuli, M. Tara, a novel F-actin binding protein, associates with the Trio guanine nucleotide exchange factor and regulates actin cytoskeletal organization. J. Cell Sci. 2001, 114, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Bellanger, J.M.; Estrach, S.; Schmidt, S.; Briaņcon-Marjollet, A.; Zugasti, O.; Fromont, S.; Debant, A. Different regulation of the Trio Dbl-Homology domains by their associated PH domains. Biol. Cell 2003, 95, 625–634. [Google Scholar] [CrossRef]
- Dejana, E. Endothelial cell-cell junctions: Happy together. Nat. Rev. Mol. Cell Biol. 2004, 5, 261–270. [Google Scholar] [CrossRef]
- Campbell, H.K.; Maiers, J.L.; DeMali, K.A. Interplay between tight junctions & adherens junctions. Exp. Cell Res. 2017, 358, 39–44. [Google Scholar] [CrossRef] [PubMed]
- Horwitz, A.R. The origins of the molecular era of adhesion research. Nat. Rev. Mol. Cell Biol. 2012, 13, 805–811. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charrasse, S.; Comunale, F.; Fortier, M.; Portales-Casamar, E.; Debant, A.; Gauthier-Rouvière, C. M-cadherin activates Rac1 GTPase through the Rho-GEF Trio during myoblast fusion. Mol. Biol. Cell 2007, 18, 1734–1743. [Google Scholar] [CrossRef] [PubMed]
- Kruse, K.; Lee, Q.S.; Sun, Y.; Klomp, J.; Yang, X.; Huang, F.; Sun, M.Y.; Zhao, S.; Hong, Z.; Vogel, S.M.; et al. N-cadherin signaling via Trio assembles adherens junctions to restrict endothelial permeability. J. Cell Biol. 2019, 218, 299–316. [Google Scholar] [CrossRef]
- Payne, L.B.; Zhao, H.; James, C.C.; Darden, J.; McGuire, D.; Taylor, S.; Smyth, J.W.; Chappell, J.C. The pericyte microenvironment during vascular development. Microcirculation 2019, 26, e12554. [Google Scholar] [CrossRef]
- Wang, Z.-G.; Cheng, Y.; Yu, X.-C.; Ye, L.-B.; Xia, Q.-H.; Johnson, N.R.; Wei, X.; Chen, D.-Q.; Cao, G.; Fu, X.-B.; et al. bFGF Protects Against Blood-Brain Barrier Damage Through Junction Protein Regulation via PI3K-Akt-Rac1 Pathway Following Traumatic Brain Injury. Mol. Neurobiol. 2016, 53, 7298–7311. [Google Scholar] [CrossRef] [Green Version]
- Hahn, C.; Schwartz, M.A. Mechanotransduction in vascular physiology and atherogenesis. Nat. Rev. Mol. Cell Biol. 2009, 10, 53–62. [Google Scholar] [CrossRef] [Green Version]
- Polacheck, W.J.; Kutys, M.L.; Yang, J.; Eyckmans, J.; Wu, Y.; Vasavada, H.; Hirschi, K.K.; Chen, C.S. A non-canonical Notch complex regulates adherens junctions and vascular barrier function. Nature 2017, 552, 258–262. [Google Scholar] [CrossRef] [Green Version]
- Mikelis, C.M.; Simaan, M.; Ando, K.; Fukuhara, S.; Sakurai, A.; Amornphimoltham, P.; Masedunskas, A.; Weigert, R.; Chavakis, T.; Adams, R.H.; et al. RhoA and ROCK mediate histamine-induced vascular leakage and anaphylactic shock. Nat. Commun. 2015, 6, 6725. [Google Scholar] [CrossRef] [Green Version]
- Rojas, R.J.; Yohe, M.E.; Gershburg, S.; Kawano, T.; Kozasa, T.; Sondek, J. Gαq directly activates p63RhoGEF and trio via a conserved extension of the Dbl homology-associated pleckstrin homology domain. J. Biol. Chem. 2007, 282, 29201–29210. [Google Scholar] [CrossRef] [Green Version]
- Bandekar, S.J.; Arang, N.; Tully, E.S.; Tang, B.A.; Barton, B.L.; Li, S.; Gutkind, J.S.; Tesmer, J.J.G. Structure of the C-terminal guanine nucleotide exchange factor module of Trio in an autoinhibited conformation reveals its oncogenic potential. Sci. Signal. 2019, 12, eaav2449. [Google Scholar] [CrossRef]
- Dela Paz, N.G.; Melchior, B.; Frangos, J.A. Shear stress induces Gαq/11 activation independently of G protein-coupled receptor activation in endothelial cells. Am. J. Physiol.-Cell Physiol. 2017, 312, C428–C437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, Y.; Hla, T. S1P control of endothelial integrity. Curr. Top. Microbiol. Immunol. 2014, 378, 85–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Rijssel, J.; Timmerman, I.; Van Alphen, F.P.J.; Hoogenboezem, M.; Korchynskyi, O.; Geerts, D.; Geissler, J.; Reedquist, K.A.; Niessen, H.W.M.; Van Buul, J.D. The Rho-GEF Trio regulates a novel pro-inflammatory pathway through the transcription factor Ets2. Biol. Open 2013, 2, 569–579. [Google Scholar] [CrossRef] [Green Version]
- Lucke, S.; Levkau, B. Endothelial functions of sphingosine-1-phosphate. Cell. Physiol. Biochem. 2010, 26, 87–96. [Google Scholar] [CrossRef]
- Hernández-García, R.; Iruela-Arispe, M.L.; Reyes-Cruz, G.; Vázquez-Prado, J. Endothelial RhoGEFs: A systematic analysis of their expression profiles in VEGF-stimulated and tumor endothelial cells. Vascul. Pharmacol. 2015, 74, 60–72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hakeda-Suzuki, S.; Ng, J.; Tzu, J.; Dietzl, G.; Sun, Y.; Harms, M.; Nardine, T.; Luo, L.; Dickson, B.J. Rac function and regulation during Drosophila development. Nature 2002, 416, 438–442. [Google Scholar] [CrossRef]
- Eelen, G.; Treps, L.; Li, X.; Carmeliet, P. Basic and Therapeutic Aspects of Angiogenesis Updated. Circ. Res. 2020, 127, 310–329. [Google Scholar] [CrossRef]
- O’Brien, S.P.; Seipel, K.; Medley, Q.G.; Bronson, R.; Segal, R.; Streuli, M. Skeletal muscle deformity and neuroanl disorder in Trio exchange factor-deficient mouse embryos. Proc. Natl. Acad. Sci. USA 2000, 97, 12074–12078. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, W.; Palmby, T.R.; Gavard, J.; Amornphimoltham, P.; Zheng, Y.; Gui, J.S. An essential role for Rac1 in endothelial cell function and vascular development. FASEB J. 2008, 22, 1829–1838. [Google Scholar] [CrossRef]
- Paatero, I.; Sauteur, L.; Lee, M.; Lagendijk, A.K.; Heutschi, D.; Wiesner, C.; Guzmán, C.; Bieli, D.; Hogan, B.M.; Affolter, M.; et al. Junction-based lamellipodia drive endothelial cell rearrangements in vivo via a VE-cadherin-F-actin based oscillatory cell-cell interaction. Nat. Commun. 2018, 9, 3545. [Google Scholar] [CrossRef] [PubMed]
- Szymborska, A.; Gerhardt, H. Hold me, but not too tight—Endothelial cell–cell junctions in angiogenesis. Cold Spring Harb. Perspect. Biol. 2018, 10, a029223. [Google Scholar] [CrossRef] [PubMed]
- Schnoor, M. Endothelial Actin-Binding Proteins and Actin Dynamics in Leukocyte Transendothelial Migration. J. Immunol. 2015, 194, 3535–3541. [Google Scholar] [CrossRef] [Green Version]
- Van Steen, A.C.I.; van der Meer, W.J.; Hoefer, I.E.; van Buul, J.D. Actin remodelling of the endothelium during transendothelial migration of leukocytes. Atherosclerosis 2020, 315, 102–110. [Google Scholar] [CrossRef]
- Heemskerk, N.; Van Rijssel, J.; Van Buul, J.D. Rho-GTPase signaling in leukocyte extravasation: An endothelial point of view. Cell Adhes. Migr. 2014, 8, 67–75. [Google Scholar] [CrossRef] [Green Version]
- Qiao, J.; Huang, F.; Lum, H. PKA inhibits RhoA activation: A protection mechanism against endothelial barrier dysfunction. Am. J. Physiol.-Lung Cell. Mol. Physiol. 2003, 284, L972–L980. [Google Scholar] [CrossRef] [Green Version]
- Van Buul, J.D.; Hordijk, P.L. Signaling in Leukocyte Transendothelial Migration. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 824–833. [Google Scholar] [CrossRef]
- Yao, L.; Romero, M.J.; Toque, H.A.; Yang, G.; Caldwell, R.B.; Caldwell, R.W. The role of RhoA/Rho kinase pathway in endothelial dysfunction. J. Cardiovasc. Dis. Res. 2010, 1, 165–170. [Google Scholar] [CrossRef] [Green Version]
- Feng, X.; Degese, M.S.; Iglesias-Bartolome, R.; Vaque, J.P.; Molinolo, A.A.; Rodrigues, M.; Zaidi, M.R.; Ksander, B.R.; Merlino, G.; Sodhi, A.; et al. Hippo-independent activation of YAP by the GNAQ uveal melanoma oncogene through a Trio-regulated Rho GTPase Signaling Circuitry. Cancer Cell 2014, 25, 831–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonoshita, M.; Itatani, Y.; Kakizaki, F.; Sakimura, K.; Terashima, T.; Katsuyama, Y.; Sakai, Y.; Taketo, M.M. Promotion of colorectal cancer invasion and metastasis through activation of NOTCH–DAB1–ABL–RHOGEF protein TRIO. Cancer Discov. 2015, 5, 198–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lane, J.; Martin, T.A.; Mansel, R.E.; Jiang, W.G. The expression and prognostic value of the guanine nucleotide exchange factors (GEFs) Trio, Vav1 and TIAM-1 in human breast cancer. Int. Semin. Surg. Oncol. 2008, 5, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foley, J.H.; Conway, E.M. Cross Talk Pathways between Coagulation and Inflammation. Circ. Res. 2016, 118, 1392–1408. [Google Scholar] [CrossRef] [PubMed]
- Saibeni, S.; Saladino, V.; Chantarangkul, V.; Villa, F.; Bruno, S.; Vecchi, M.; de Franchis, R.; Sei, C.; Tripodi, A. Increased thrombin generation in inflammatory bowel diseases. Thromb. Res. 2010, 125, 278–282. [Google Scholar] [CrossRef] [PubMed]
- Popović, M.; Smiljanić, K.; Dobutović, B.; Syrovets, T.; Simmet, T.; Isenović, E.R. Thrombin and vascular inflammation. Mol. Cell. Biochem. 2012, 359, 301–313. [Google Scholar] [CrossRef] [PubMed]
- Gavard, J.; Gutkind, J.S. Protein kinase C-related kinase and ROCK are required for thrombin-induced endothelial cell permeability downstream from Gα12/13 and Gα11/q. J. Biol. Chem. 2008, 283, 29888–29896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunningham, K.S.; Gotlieb, A.I. The role of shear stress in the pathogenesis of atherosclerosis. Lab. Investig. 2005, 85, 9–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sega, F.V.D.; Fortini, F.; Aquila, G.; Campo, G.; Vaccarezza, M.; Rizzo, P. Notch signaling regulates immune responses in atherosclerosis. Front. Immunol. 2019, 10, 1130. [Google Scholar] [CrossRef] [Green Version]
- Kanekura, K.; Hashimoto, Y.; Niikura, T.; Aiso, S.; Matsuoka, M.; Nishimoto, I. Alsin, the Product of ALS2 Gene, Suppresses SOD1 Mutant Neurotoxicity through RhoGEF Domain by Interacting with SOD1 Mutants. J. Biol. Chem. 2004, 279, 19247–19256. [Google Scholar] [CrossRef] [Green Version]
- Orrico, A.; Galli, L.; Cavaliere, M.L.; Garavelli, L.; Fryns, J.P.; Crushell, E.; Rinaldi, M.M.; Medeira, A.; Sorrentino, V. Phenotypic and molecular characterisation of the Aarskog-Scott syndrome: A survey of the clinical variability in light of FGD1 mutation analysis in 46 patients. Eur. J. Hum. Genet. 2004, 12, 16–23. [Google Scholar] [CrossRef] [Green Version]
- Ellenbroek, S.I.J.; Collard, J.G. Rho GTPases: Functions and association with cancer. Clin. Exp. Metastasis 2007, 24, 657–672. [Google Scholar] [CrossRef]
- Vega, F.M.; Ridley, A.J. Rho GTPases in cancer cell biology. FEBS Lett. 2008, 582, 2093–2101. [Google Scholar] [CrossRef] [Green Version]
- Briançon-Marjollet, A.; Ghogha, A.; Nawabi, H.; Triki, I.; Auziol, C.; Fromont, S.; Piché, C.; Enslen, H.; Chebli, K.; Cloutier, J.-F.; et al. Trio Mediates Netrin-1-Induced Rac1 Activation in Axon Outgrowth and Guidance. Mol. Cell. Biol. 2008, 28, 2314–2323. [Google Scholar] [CrossRef] [Green Version]
- Iyer, S.C.; Wang, D.; Iyer, E.P.R.; Trunnell, S.A.; Meduri, R.; Shinwari, R.; Sulkowski, M.J.; Cox, D.N. The rhogef trio functions in sculpting class specific dendrite morphogenesis in drosophila sensory neurons. PLoS ONE 2012, 7, e33634. [Google Scholar] [CrossRef] [Green Version]
- Barbosa, S.; Greville-Heygate, S.; Bonnet, M.; Godwin, A.; Fagotto-Kaufmann, C.; Kajava, A.V.; Laouteouet, D.; Mawby, R.; Wai, H.A.; Dingemans, A.J.M.; et al. Opposite Modulation of RAC1 by Mutations in TRIO Is Associated with Distinct, Domain-Specific Neurodevelopmental Disorders. Am. J. Hum. Genet. 2020, 106, 338–355. [Google Scholar] [CrossRef] [Green Version]
- Pengelly, R.J.; Greville-Heygate, S.; Schmidt, S.; Seaby, E.G.; Jabalameli, M.R.; Mehta, S.G.; Parker, M.J.; Goudie, D.; Fagotto-Kaufmann, C.; Mercer, C.; et al. Mutations specific to the Rac-GEF domain of TRIO cause intellectual disability and microcephaly. J. Med. Genet. 2016, 53, 735–742. [Google Scholar] [CrossRef] [Green Version]
- Sadybekov, A.; Tian, C.; Arnesano, C.; Katritch, V.; Herring, B.E. An autism spectrum disorder-related de novo mutation hotspot discovered in the GEF1 domain of Trio. Nat. Commun. 2017, 8, 601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katrancha, S.M.; Wu, Y.; Zhu, M.; Eipper, B.A.; Koleske, A.J.; Mains, R.E. Neurodevelopmental disease-associated de novo mutations and rare sequence variants affect TRIO GDP/GTP exchange factor activity. Hum. Mol. Genet. 2017, 26, 4728–4740. [Google Scholar] [CrossRef] [PubMed]
- Salhia, B.; Tran, N.L.; Chan, A.; Wolf, A.; Nakada, M.; Rutka, F.; Ennis, M.; McDonough, W.S.; Berens, M.E.; Symons, M.; et al. The guanine nucleotide exchange factors Trio, Ect2, and Vav3 mediate the invasive behavior of glioblastoma. Am. J. Pathol. 2008, 173, 1828–1838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molenaar, J.J.; Koster, J.; Zwijnenburg, D.A.; Van Sluis, P.; Valentijn, L.J.; Van Der Ploeg, I.; Hamdi, M.; Van Nes, J.; Westerman, B.A.; Van Arkel, J.; et al. Sequencing of neuroblastoma identifies chromothripsis and defects in neuritogenesis genes. Nature 2012, 483, 589–593. [Google Scholar] [CrossRef] [PubMed]
- Yoshizuka, N.; Moriuchi, R.; Mori, T.; Yamada, K.; Hasegawa, S.; Maeda, T.; Shimada, T.; Yamada, Y.; Kamihira, S.; Tomonaga, M.; et al. An alternative transcript derived from the Trio locus encodes a guanosine nucleotide exchange factor with mouse cell-transforming potential. J. Biol. Chem. 2004, 279, 43998–44004. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, K.; Moriuchi, R.; Mori, T.; Okazaki, E.; Kohno, T.; Nagayasu, T.; Matsuyama, T.; Katamine, S. Tgat, a Rho-specific guanine nucleotide exchange factor, activates NF-κB via physical association with IκB kinase complexes. Biochem. Biophys. Res. Commun. 2007, 355, 269–274. [Google Scholar] [CrossRef] [PubMed]
- Moshfegh, Y.; Bravo-Cordero, J.J.; Miskolci, V.; Condeelis, J.; Hodgson, L. A Trio-Rac1-PAK1 signaling axis drives invadopodia disassembly. Nat. Cell Biol. 2014, 16, 574–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Hayre, M.; Vázquez-Prado, J.; Kufareva, I.; Stawiski, E.W.; Handel, T.M.; Seshagiri, S.; Gutkind, J.S. The emerging mutational landscape of G proteins and G-protein-coupled receptors in cancer. Nat. Rev. Cancer 2013, 13, 412–424. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Yang, Y.; Wang, F.; Wei, Q.; Qin, H. Hippo-YAP signaling pathway: A new paradigm for cancer therapy. Int. J. Cancer 2015, 137, 2275–2286. [Google Scholar] [CrossRef] [PubMed]
- Karnoub, A.E.; Symons, M.; Campbell, S.L.; Der, C.J. Molecular basis for Rho GTPase signaling specificity. Breast Cancer Res. Treat. 2004, 84, 61–71. [Google Scholar] [CrossRef] [PubMed]
- Sahai, E.; Marshall, C.J. RHO-GTPases and cancer. Nat. Rev. Cancer 2002, 2, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y. Dbl family guanine nucleotide exchange factors. Trends Biochem. Sci. 2001, 26, 724–732. [Google Scholar] [CrossRef]
- Schmidt, S.; Dirionga, S.; Méry, J.; Fabbriziob, E.; Debant, A. Identification of the first Rho-GEF inhibitor, TRIPalpha, which targets the RhoA-specific GEF domain of Trio. FEBS Lett. 2002, 523, 35–42. [Google Scholar] [CrossRef] [Green Version]
- Blangy, A.; Bouquier, N.; Gauthier-Rouvière, C.; Schmidt, S.; Debant, A.; Leonetti, J.-P.; Fort, P. Identification of TRIO-GEFD1 chemical inhibitors using the yeast exchange assay. Biol. Cell 2006, 98, 511–522. [Google Scholar] [CrossRef] [Green Version]
- Bouquier, N.; Vignal, E.; Charrasse, S.; Weill, M.; Schmidt, S.; Léonetti, J.P.; Blangy, A.; Fort, P. A Cell Active Chemical GEF Inhibitor Selectively Targets the Trio/RhoG/Rac1 Signaling Pathway. Chem. Biol. 2009, 16, 657–666. [Google Scholar] [CrossRef]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kempers, L.; Driessen, A.J.M.; van Rijssel, J.; Nolte, M.A.; van Buul, J.D. The RhoGEF Trio: A Protein with a Wide Range of Functions in the Vascular Endothelium. Int. J. Mol. Sci. 2021, 22, 10168. https://doi.org/10.3390/ijms221810168
Kempers L, Driessen AJM, van Rijssel J, Nolte MA, van Buul JD. The RhoGEF Trio: A Protein with a Wide Range of Functions in the Vascular Endothelium. International Journal of Molecular Sciences. 2021; 22(18):10168. https://doi.org/10.3390/ijms221810168
Chicago/Turabian StyleKempers, Lanette, Amber J. M. Driessen, Jos van Rijssel, Martijn A. Nolte, and Jaap D. van Buul. 2021. "The RhoGEF Trio: A Protein with a Wide Range of Functions in the Vascular Endothelium" International Journal of Molecular Sciences 22, no. 18: 10168. https://doi.org/10.3390/ijms221810168