
Dimer Interface in Natural Variant NK1 Is Dispensable for HGF-Dependent Met Receptor Activation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Preparation of Wild-Type and Mutant HGF and NK1 Proteins
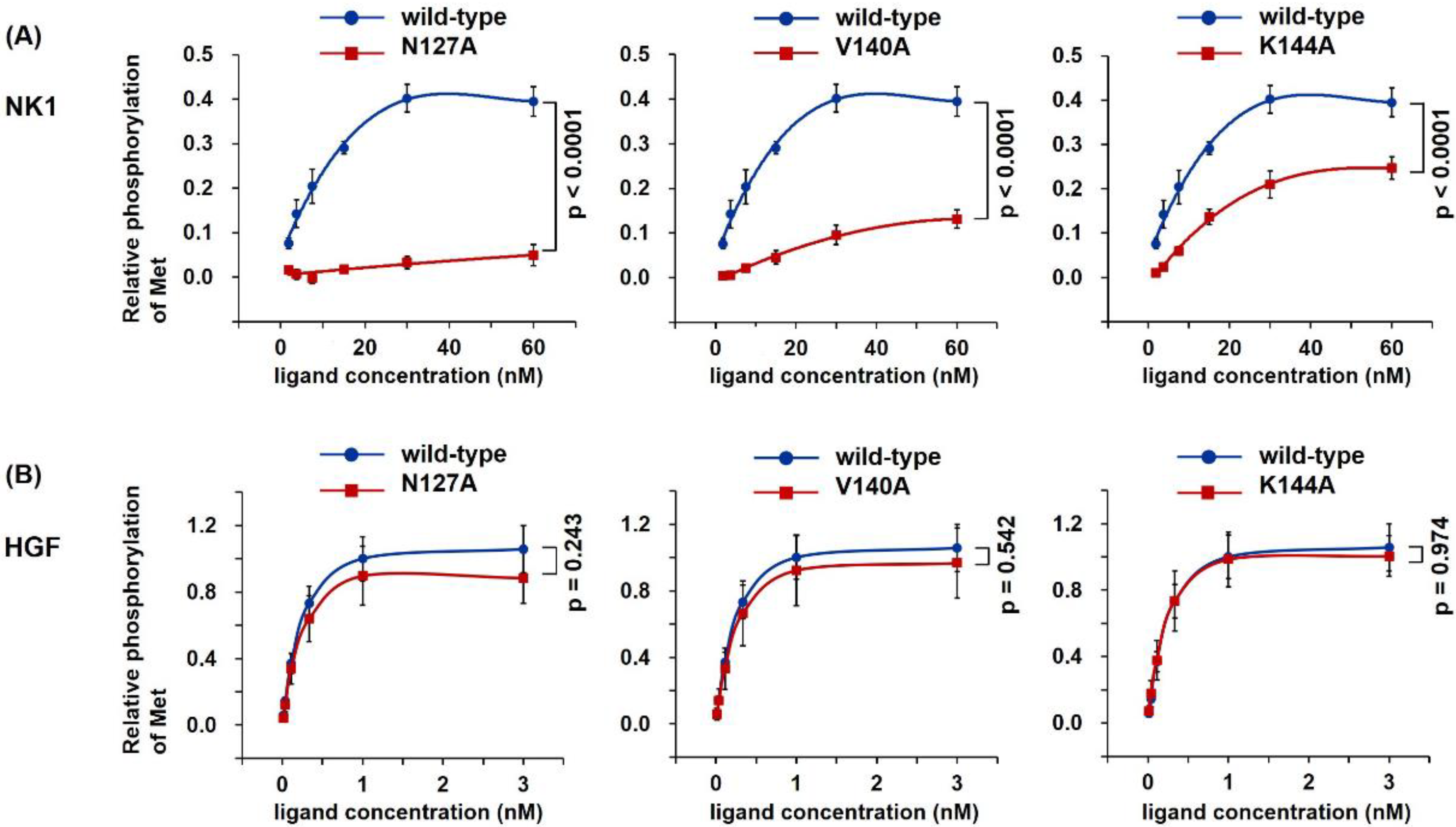
2.2. Loss of Met Activating Activity in NK1 Mutants but Not HGF Mutants
2.3. Distinct Activation of the Met-Related Signaling Pathway by NK1 and HGF Mutants
2.4. Loss of Biological Activity to Promote Cell Motility in NK1 Mutants but Not HGF Mutants
2.5. Competitive Action of NK1 Mutant on Cell Scattering
3. Discussion
4. Materials and Methods
4.1. DNA Construction
4.2. Recombinant Protein Preparation and Analysis
4.3. Cell-Based Met Phosphorylation Assay
4.4. Western Blot Analysis
4.5. Met Receptor Dimerization in Live Cells
4.6. Migration Assay
4.7. Cell-scattering Assay
4.8. Cell Proliferation Assay
4.9. Statistics
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| HGF | hepatocyte growth factor |
| SAXS | small-angle X-ray scattering |
| cryo-EM | cryo-electron microscopy |
| scHGF | single-chain HGF |
| tcHGF | two-chain HGF |
| Gab1 | GRB2-associated-binding protein 1 |
| Erk | Extracellular signal-regulated kinase |
| Akt | Protein kinase B |
| GAPDH | Glyceraldehyde-3-phosphate dehydrogenase |
References
- Sakai, K.; Aoki, S.; Matsumoto, K. Hepatocyte growth factor and Met in drug discovery. J. Biochem. 2015, 157, 271–284. [Google Scholar] [CrossRef] [Green Version]
- Trusolino, L.; Bertotti, A.; Comoglio, P.M. MET signalling: Principles and functions in development, organ regeneration and cancer. Nat. Rev. Mol. Cell Biol. 2010, 11, 834–848. [Google Scholar] [CrossRef] [PubMed]
- De Silva, D.M.; Roy, A.; Kato, T.; Cecchi, F.; Lee, Y.H.; Matsumoto, K.; Bottaro, D.P. Targeting the hepatocyte growth factor/Met pathway in cancer. Biochem. Soc. Trans. 2017, 45, 855–870. [Google Scholar] [CrossRef] [Green Version]
- Kitamura, K.; Nagoshi, N.; Tsuji, O.; Matsumoto, M.; Okano, H.; Nakamura, M. Application of hepatocyte growth factor for acute spinal cord injury: The road from basic studies to human treatment. Int. J. Mol. Sci. 2019, 20, 1054. [Google Scholar] [CrossRef] [Green Version]
- Powell, R.J.; Goodney, P.; Mendelsohn, F.O.; Moen, E.K.; Annex, B.H. Safety and efficacy of patient specific intramuscular injection of HGF plasmid gene therapy on limb perfusion and wound healing in patients with ischemic lower extremity ulceration: Results of the HGF-0205 trial. J. Vasc. Surg. 2010, 52, 1525–1530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolf, J.; Seto, T.; Han, J.; Reguart, N.; Garon, E.B.; Groen, H.J.M.; Tan, D.S.W.; Hida, T.; De Jonge, M.; Orlov, S.V.; et al. Capmatinib in MET exon 14-mutated or MET-amplified non-small-cell lung cancer. N. Engl. J. Med. 2020, 383, 944–957. [Google Scholar] [CrossRef] [PubMed]
- Paik, P.K.; Felip, E.; Veillon, R.; Sakai, H.; Cortot, A.B.; Garassino, M.C.; Mazieres, J.; Viteri, S.; Senellart, H.; Van Meerbeeck, J.; et al. Tepotinib in non-small-cell lung cancer with MET exon 14 skipping mutations. N. Engl. J. Med. 2020, 383, 931–943. [Google Scholar] [CrossRef]
- Ito, K.; Sakai, K.; Suzuki, Y.; Ozawa, N.; Hatta, T.; Natsume, T.; Matsumoto, K.; Suga, H. Artificial human Met agonists based on macrocycle scaffolds. Nat. Commun. 2015, 6, 6373. [Google Scholar] [CrossRef] [Green Version]
- Ueki, R.; Ueki, A.; Kanda, N.; Sando, S. Oligonucleotide-based mimetics of hepatocyte growth factor. Angew. Chem. 2016, 55, 579–582. [Google Scholar] [CrossRef]
- Sakai, K.; Passioura, T.; Sato, H.; Ito, K.; Furuhashi, H.; Umitsu, M.; Takagi, J.; Kato, Y.; Mukai, H.; Warashina, S.; et al. Macrocyclic peptide-based inhibition and imaging of hepatocyte growth factor. Nat. Chem. Biol. 2019, 15, 598–606. [Google Scholar] [CrossRef]
- Ueki, R.; Uchida, S.; Kanda, N.; Yamada, N.; Ueki, A.; Akiyama, M.; Toh, K.; Cabral, H.; Sando, S. A chemically unmodified agonistic DNA with growth factor functionality for in vivo therapeutic application. Sci. Adv. 2020, 6, eaay2801. [Google Scholar] [CrossRef] [Green Version]
- Miyazawa, K.; Tsubouchi, H.; Naka, D.; Takahashi, K.; Okigaki, M.; Arakaki, N.; Nakayama, H.; Hirono, S.; Sakiyama, O.; Takahashi, K.; et al. Molecular cloning and sequence analysis of cDNA for human hepatocyte growth factor. Biochem. Biophys. Res. Commun. 1989, 163, 967–973. [Google Scholar] [CrossRef]
- Nakamura, T.; Nishizawa, T.; Hagiya, M.; Seki, T.; Shimonishi, M.; Sugimura, A.; Tashiro, K.; Shimizu, S. Molecular cloning and expression of human hepatocyte growth factor. Nature 1989, 342, 440–443. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Kataoka, H.; Date, K.; Nakamura, T. Cooperative interaction between α- and β- chains of hepatocyte growth factor on c-Met receptor confers ligand-induced receptor tyrosine phosphorylation and multiple biological responses. J. Biol. Chem. 1998, 273, 22913–22920. [Google Scholar] [CrossRef] [Green Version]
- Youles, M.; Holmes, O.; Petoukhov, M.V.; Nessen, M.A.; Stivala, S.; Svergun, D.I.; Gherardi, E. Engineering the NK1 fragment of hepatocyte growth factor/scatter factor as a MET receptor antagonist. J. Mol. Biol. 2008, 377, 616–622. [Google Scholar] [CrossRef]
- Tolbert, W.D.; Daugherty-Holtrop, J.; Gherardi, E.; Woude, G.V.; Xu, H.E. Structural basis for agonism and antagonism of hepatocyte growth factor. Proc. Natl. Acad. Sci. USA 2010, 107, 13264–13269. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stamos, J.; Lazarus, R.A.; Yao, X.; Kirchhofer, D.; Wiesmann, C. Crystal structure of the HGF β-chain in complex with the Sema domain of the Met receptor. EMBO J. 2004, 23, 2325–2335. [Google Scholar] [CrossRef] [PubMed]
- Holmes, O.; Pillozzi, S.; Deakin, J.A.; Carafoli, F.; Kemp, L.; Butler, P.J.G.; Lyon, M.; Gherardi, E. Insights into the structure/function of hepatocyte growth factor/scatter factor from studies with individual domains. J. Mol. Biol. 2007, 367, 395–408. [Google Scholar] [CrossRef]
- Cioce, V.; Csaky, K.G.; Chan, A.M.; Bottaro, D.P.; Taylor, W.G.; Jensen, R.; Aaronson, S.A.; Rubin, J.S. Hepatocyte growth factor (HGF)/NK1 is a naturally occurring HGF/scatter factor variant with partial agonist/antagonist activity. J. Biol. Chem. 1996, 271, 13110–13115. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwall, R.H.; Chang, L.Y.; Godowski, P.J.; Kahn, D.W.; Hillan, K.J.; Bauer, K.D.; Zioncheck, T.F. Heparin induces dimerization and confers proliferative activity onto the hepatocyte growth factor antagonists NK1 and NK2. J. Cell Biol. 1996, 133, 709–718. [Google Scholar] [CrossRef]
- Uchikawa, E.; Chen, Z.; Xiao, G.; Zhang, X.; Bai, X. Structural basis of the activation of c-MET receptor. Nat. Commun. 2021, 12, 4074. [Google Scholar] [CrossRef] [PubMed]
- Gherardi, E.; Sandin, S.; Petoukhov, M.V.; Finch, J.; Youles, M.E.; Ofverstedt, L.; Miguel, R.N.; Blundell, T.L.; Vande Woude, G.F.; Skoglund, U.; et al. Structural basis of hepatocyte growth factor/scatter factor and MET signalling. Proc. Natl. Acad. Sci. USA 2006, 103, 4046–4051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Umitsu, M.; Sakai, K.; Tamura-Kawakami, K.; Matsumoto, K.; Takagi, J. The constitutive high-affinity Met-binding site in the kringle domain is dispensable for the signalling activity of hepatocyte growth factor. J. Biochem. 2020, 167, 577–586. [Google Scholar] [CrossRef]
- Chirgadze, D.Y.; Hepple, J.P.; Zhou, H.; Byrd, R.A.; Blundell, T.L.; Gherardi, E. Crystal structure of the NK1 fragment of HGF/SF suggests a novel mode for growth factor dimerization and receptor binding. Nat. Struct. Biol. 1999, 6, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Tolbert, W.D.; Daugherty, J.; Gao, C.F.; Xie, Q.; Miranti, C.; Gherardi, E.; Vande Woude, G.; Xu, H.E. A mechanistic basis for converting a receptor tyrosine kinase agonist to an antagonist. Proc. Natl. Acad. Sci. USA 2007, 104, 14592–14597. [Google Scholar] [CrossRef] [Green Version]
- Fukuta, K.; Matsumoto, K.; Nakamura, T. Multiple biological responses are induced by glycosylation-deficient hepatocyte growth factor. Biochem. J. 2005, 388, 555–562. [Google Scholar] [CrossRef]
- Miao, W.; Sakai, K.; Ozawa, N.; Nishiuchi, T.; Suzuki, Y.; Ito, K.; Morioka, T.; Umitsu, M.; Takagi, J.; Suga, H.; et al. Cellular signaling and gene expression profiles evoked by a bivalent macrocyclic peptide that serves as an artificial MET receptor agonist. Sci. Rep. 2018, 8, 16492. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montesano, R.; Soriano, J.V.; Malinda, K.M.; Ponce, M.L.; Bafico, A.; Kleinman, H.K.; Bottaro, D.P.; Aaronson, S.A. Differential effects of hepatocyte growth factor isoforms on epithelial and endothelial tubulogenesis. Cell Growth Differ. 1998, 9, 355–365. [Google Scholar]
- Chan, A.M.; Rubin, J.S.; Bottaro, D.P.; Hirschfield, D.W.; Chedid, M.; Aaronson, S.A. Identification of a competitive HGF antagonist encoded by an alternative transcript. Science 1991, 254, 1382–1385. [Google Scholar] [CrossRef]
- Stahl, S.J.; Wingfield, P.T.; Kaufman, J.D.; Pannell, L.K.; Cioce, V.; Sakata, H.; Taylor, W.G.; Rubin, J.S.; Bottaro, D.P. Functional and biophysical characterization of recombinant human hepatocyte growth factor isoforms produced in Escherichia coli. Biochem. J. 1997, 326, 763–772. [Google Scholar] [CrossRef] [Green Version]
- Cecchi, F.; Lih, C.; Lee, Y.H. Walsh, W.; Rabe, D.C.; Williams, P.M.; Bottaro, D.P. Expression array analysis of the hepatocyte growth factor invasive program. Clin. Exp. Metastasis. 2015, 32, 659–676. [Google Scholar] [CrossRef]
- Miao, W.; Sakai, K.; Imamura, R.; Ito, K.; Suga, H.; Sakuma, T.; Yamamoto, T.; Matsumoto, K. MET activation by a macrocyclic peptide agonist that couples to biological responses differently from HGF in a context-dependent manner. Int. J. Mol. Sci. 2018, 19, 3141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gherardi, E.; Birchmeier, W.; Birchmeier, C.; Vande Woude, G. Targeting MET in cancer: Rationale and progress. Nat. Rev. Cancer 2012, 12, 89–103. [Google Scholar] [CrossRef] [PubMed]
- Cecchi, F.; Rabe, D.C.; Bottaro, D.P. Targeting the HGF/Met signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 553–572. [Google Scholar] [CrossRef] [Green Version]
- Gaddy, D.F.; Riedel, M.J.; Pejawar-Gaddy, S.; Kieffer, T.J.; Robbins, P.D. In vivo expression of HGF/NK1 and GLP-1 from dsAAV vectors enhances pancreatic β-cell proliferation and improves pathology in the db/db mouse model of diabetes. Diabetes 2010, 59, 3108–3116. [Google Scholar] [CrossRef] [Green Version]
- Lokker, N.A.; Mark, M.R.; Luis, E.A.; Bennett, G.L.; Robbins, K.A.; Baker, J.B.; Godowski, P.J. Structure-function analysis of hepatocyte growth factor: Identification of variants that lack mitogenic activity yet retain high affinity receptor binding. EMBO J. 1992, 11, 2503–2510. [Google Scholar] [CrossRef] [PubMed]
- Naka, D.; Ishii, T.; Yoshiyama, Y.; Miyazawa, K.; Hara, H.; Hishida, T.; Kidamura, N. Activation of hepatocyte growth factor by proteolytic conversion of a single chain form to a heterodimer. J. Biol. Chem. 1992, 267, 20114–20119. [Google Scholar] [CrossRef]
- Fukushima, T.; Uchiyama, S.; Tanaka, H.; Kataoka, H. Hepatocyte growth factor activator: A proteinase linking tissue injury with repair. Int. J. Mol. Sci. 2018, 19, 3435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirchhofer, D.; Yao, X.; Peek, M.; Eigenbrot, C.; Lipari, M.T.; Billeci, K.L.; Maun, H.R.; Moran, P.; Santell, L.; Wiesmann, C.; et al. Structural and functional basis of the serine protease-like hepatocyte growth factor β-chain in Met binding and signaling. J. Biol. Chem. 2004, 279, 39915–39924. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landgraf, K.E.; Santell, L.; Billeci, K.L.; Quan, C.; Young, J.C.; Maun, H.R.; Kirchhofer, D.; Lazarus, R.A. Allosteric peptide activators of pro-hepatocyte growth factor stimulate Met signaling. J. Biol. Chem. 2010, 285, 40362–40372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landgraf, K.E.; Steffek, M.; Quan, C.; Tom, J.; Yu, C.; Santell, L.; Maun, H.R.; Eigenbrot, C.; Lazarus, R.A. An allosteric switch for pro-HGF/Met signaling using zymogen activator peptides. Nat. Chem. Biol. 2014, 10, 567–573. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, K.; Takehara, T.; Inoue, H.; Hagiya, M.; Shimizu, S.; Nakamura, T. Deletion of kringle domains or the N-terminal hairpin structure in hepatocyte growth factor results in marked decreases in related biological activities. Biochem. Biophys. Res. Commun. 1991, 181, 691–699. [Google Scholar] [CrossRef]
- Koschut, D.; Richert, L.; Pace, G.; Niemann, H.H.; Mély, Y.; Orian-Rousseau, V. Live cell imaging shows hepatocyte growth factor-induced Met dimerization. Biochim. Biophys. Acta 2016, 1863, 1552–1558. [Google Scholar] [CrossRef]
- Prat, M.; Crepaldi, T.; Pennacchietti, S.; Bussolino, F.; Comoglio, P.M. Agonistic monoclonal antibodies against the Met receptor dissect the biological responses to HGF. J. Cell Sci. 1998, 111, 237–247. [Google Scholar] [CrossRef]
- Ohashi, K.; Marion, P.L.; Nakai, H.; Meuse, L.; Cullen, J.M.; Bordier, B.B.; Schwall, R.; Greenberg, H.B.; Glenn, J.S.; Kay, M.A. Sustained survival of human hepatocytes in mice: A model for in vivo infection with human hepatitis B and hepatitis delta viruses. Nat. Med. 2000, 6, 327–331. [Google Scholar] [CrossRef] [PubMed]
- Rafferty, B.; Maile, O.; Rigsby, P.; Gaines Das, R.E.; Robinson, C.J. International Standards for hepatocyte growth factor/scatter factor: Initial assessment of candidate materials and their evaluation by multicentre collaborative study. J. Immunol. Methods 2001, 258, 1–11. [Google Scholar] [CrossRef]
- Hayata, D.; Fukuta, K.; Matsumoto, K.; Adachi, E.; Hanada, K.; Adachi, K.; Nakamura, T. Generation of engineered recombinant hepatocyte growth factor cleaved and activated by Genenase I. J. Biotechnol. 2008, 133, 478–485. [Google Scholar] [CrossRef] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tahira, Y.; Sakai, K.; Sato, H.; Imamura, R.; Matsumoto, K. Dimer Interface in Natural Variant NK1 Is Dispensable for HGF-Dependent Met Receptor Activation. Int. J. Mol. Sci. 2021, 22, 9240. https://doi.org/10.3390/ijms22179240
Tahira Y, Sakai K, Sato H, Imamura R, Matsumoto K. Dimer Interface in Natural Variant NK1 Is Dispensable for HGF-Dependent Met Receptor Activation. International Journal of Molecular Sciences. 2021; 22(17):9240. https://doi.org/10.3390/ijms22179240
Chicago/Turabian StyleTahira, Yumiko, Katsuya Sakai, Hiroki Sato, Ryu Imamura, and Kunio Matsumoto. 2021. "Dimer Interface in Natural Variant NK1 Is Dispensable for HGF-Dependent Met Receptor Activation" International Journal of Molecular Sciences 22, no. 17: 9240. https://doi.org/10.3390/ijms22179240