
Tau Protein Interaction Partners and Their Roles in Alzheimer’s Disease and Other Tauopathies


Abstract
:1. Introduction
2. Roles of Tau Protein in Physiology and Pathology
2.1. Tau and Axonal Transport
2.2. Tau Protein in Synapses
2.3. Tau Protein in the Nucleus of Neurons
2.4. Big Tau
2.5. Extracellular Tau Protein
3. Tau Interaction Partners (TIPs), Their Biological Functions and Related Molecular Pathways
3.1. Tau as a Substrate for Kinases and Phosphatases
3.2. Tau-Interacting Partners Involved in Acetylation and Deacetylation of Proteins
3.3. Tau-Interacting Partners Involved in Glycosylation
3.4. Interactions of Tau with Ubiquitin-Proteasome System and Chaperone System
3.4.1. Tau-Interacting Proteins Involved in the Ubiquitin-Proteasome Pathway
3.4.2. Tau-Interacting Proteins Involved in Chaperone System
3.5. Interactions of Tau with Proteins Regulating Programmed Cell Death
3.6. Proteolytic Cleavage and Truncation of Tau Protein
3.7. Proteins Involved in the Regulation of the Cytoskeleton and Intracellular Transport
3.8. Tau-Interacting Proteins Involved in DNA Replication and Transcription
3.9. Tau-Interacting Proteins Involved in RNA Processing and Translation
3.10. Signal Transduction Mediators Interacting with Tau
4. Current Therapeutic Approaches Targeting Tau and Its Interacting Partners
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Uversky, V.N. Intrinsic disorder, protein–protein interactions, and disease. Adv. Protein Chem. Struct. Biol. 2018, 110, 85–121. [Google Scholar]
- Weingarten, M.D.; Lockwood, A.H.; Hwo, S.-Y.; Kirschner, M.W. A protein factor essential for microtubule assembly. Proc. Natl. Acad. Sci. USA 1975, 72, 1858–1862. [Google Scholar] [CrossRef] [Green Version]
- Cleveland, D.W.; Hwo, S.-Y.; Kirschner, M.W. Physical and chemical properties of purified tau factor and the role of tau in microtubule assembly. J. Mol. Biol. 1977, 116, 227–247. [Google Scholar] [CrossRef]
- Ferreira, A.; Busciglio, J.; Cáceres, A. Microtubule formation and neurite growth in cerebellar macroneurons which develop in vitro: Evidence for the involvement of the microtubule-associated proteins, MAP-1a, HMW-MAP2 and Tau. Dev. Brain Res. 1989, 49, 215–228. [Google Scholar] [CrossRef]
- Takei, Y.; Teng, J.; Harada, A.; Hirokawa, N. Defects in axonal elongation and neuronal migration in mice with disrupted tau and map1b genes. J. Cell Biol. 2000, 150, 989–1000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sotiropoulos, I.; Galas, M.C.; Silva, J.M.; Skoulakis, E.; Wegmann, S.; Maina, M.B.; Blum, D.; Sayas, C.L.; Mandelkow, E.M.; Mandelkow, E.; et al. Atypical, non-standard functions of the microtubule associated Tau protein. Acta Neuropathol. Commun. 2017, 5, 91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Mandelkow, E. Tau in physiology and pathology. Nat. Rev. Neurosci. 2016, 17, 5–21. [Google Scholar] [CrossRef]
- Binder, L.I.; Frankfurter, A.; Rebhun, L.I. The distribution of tau in the mammalian central nervous system. J. Cell Biol. 1985, 101, 1371–1378. [Google Scholar] [CrossRef] [Green Version]
- LoPresti, P.; Szuchet, S.; Papasozomenos, S.C.; Zinkowski, R.P.; Binder, L.I. Functional implications for the microtubule-associated protein tau: Localization in oligodendrocytes. Proc. Natl. Acad. Sci. USA 1995, 92, 10369–10373. [Google Scholar] [CrossRef] [Green Version]
- Müller, R.; Heinrich, M.; Heck, S.; Blohm, D.; Richter-Landsberg, C. Expression of microtubule-associated proteins MAP2 and tau in cultured rat brain oligodendrocytes. Cell Tissue Res. 1997, 288, 239–249. [Google Scholar] [CrossRef]
- Komori, T. Tau-positive dial Inclusions in Progressive Supranuclear Palsy, Corticobasal Degeneration and Pick’s Disease. Brain Pathol. 1999, 9, 663–679. [Google Scholar] [CrossRef] [PubMed]
- Maragakis, N.J.; Rothstein, J.D. Mechanisms of disease: Astrocytes in neurodegenerative disease. Nat. Clin. Pract. Neurol. 2006, 2, 679–689. [Google Scholar] [CrossRef] [PubMed]
- Trojanowski, J.Q.; Schuck, T.; Schmidt, M.L.; Lee, V. Distribution of tau proteins in the normal human central and peripheral nervous system. J. Histochem. Cytochem. 1989, 37, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Dugger, B.N.; Hoffman, B.R.; Scroggins, A.; Serrano, G.E.; Adler, C.H.; Shill, H.A.; Belden, C.M.; Sabbagh, M.N.; Caviness, J.N.; Dunckley, E.D. Tau immunoreactivity in peripheral tissues of human aging and select tauopathies. Neurosci. Lett. 2019, 696, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Goedert, M.; Jakes, R. Expression of separate isoforms of human tau protein: Correlation with the tau pattern in brain and effects on tubulin polymerization. EMBO J. 1990, 9, 4225–4230. [Google Scholar] [CrossRef]
- Kosik, K.S.; Orecchio, L.D.; Bakalis, S.; Neve, R.L. Developmentally regulated expression of specific tau sequences. Neuron 1989, 2, 1389–1397. [Google Scholar] [CrossRef]
- Buee, L.; Bussiere, T.; Buee-Scherrer, V.; Delacourte, A.; Hof, P.R. Tau protein isoforms, phosphorylation and role in neurodegenerative disorders. Brain Res. Brain Res. Rev. 2000, 33, 95–130. [Google Scholar] [CrossRef]
- Guo, T.; Noble, W.; Hanger, D.P. Roles of tau protein in health and disease. Acta Neuropathol. 2017, 133, 665–704. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, G.; Cowan, N.; Kirschner, M. The primary structure and heterogeneity of tau protein from mouse brain. Science 1988, 239, 285–288. [Google Scholar] [CrossRef] [PubMed]
- Kadavath, H.; Hofele, R.V.; Biernat, J.; Kumar, S.; Tepper, K.; Urlaub, H.; Mandelkow, E.; Zweckstetter, M. Tau stabilizes microtubules by binding at the interface between tubulin heterodimers. Proc. Natl. Acad. Sci. USA 2015, 112, 7501–7506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawson, H.N.; Ferreira, A.; Eyster, M.V.; Ghoshal, N.; Binder, L.I.; Vitek, M.P. Inhibition of neuronal maturation in primary hippocampal neurons from tau deficient mice. J. Cell Sci. 2001, 114 Pt 6, 1179–1187. [Google Scholar] [CrossRef]
- Harada, A.; Oguchi, K.; Okabe, S.; Kuno, J.; Terada, S.; Ohshima, T.; Sato-Yoshitake, R.; Takei, Y.; Noda, T.; Hirokawa, N. Altered microtubule organization in small-calibre axons of mice lacking tau protein. Nature 1994, 369, 488–491. [Google Scholar] [CrossRef] [PubMed]
- Tan, D.C.S.; Yao, S.; Ittner, A.; Bertz, J.; Ke, Y.D.; Ittner, L.M.; Delerue, F. Generation of a New Tau Knockout (tauDeltaex1) Line Using CRISPR/Cas9 Genome Editing in Mice. J. Alzheimer’s Dis. 2018, 62, 571–578. [Google Scholar] [CrossRef]
- Terwel, D.; Lasrado, R.; Snauwaert, J.; Vandeweert, E.; Van Haesendonck, C.; Borghgraef, P.; Van Leuven, F. Changed conformation of mutant Tau-P301L underlies the moribund tauopathy, absent in progressive, nonlethal axonopathy of Tau-4R/2N transgenic mice. J. Biol. Chem. 2005, 280, 3963–3973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tucker, K.L.; Meyer, M.; Barde, Y.A. Neurotrophins are required for nerve growth during development. Nat. Neurosci. 2001, 4, 29–37. [Google Scholar] [CrossRef]
- DiTella, M.; Feiguin, F.; Carri, N.; Kosik, K.; Caceres, A. MAP-1B/TAU functional redundancy during laminin-enhanced axonal growth. J. Cell Sci. 1996, 109, 467–477. [Google Scholar] [CrossRef]
- Lei, P.; Ayton, S.; Moon, S.; Zhang, Q.; Volitakis, I.; Finkelstein, D.I.; Bush, A.I. Motor and cognitive deficits in aged tau knockout mice in two background strains. Mol. Neurodegener. 2014, 9, 29. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, T.; Van der Jeugd, A.; Blum, D.; Galas, M.C.; D’Hooge, R.; Buee, L.; Balschun, D. Cognition and hippocampal synaptic plasticity in mice with a homozygous tau deletion. Neurobiol. Aging 2014, 35, 2474–2478. [Google Scholar] [CrossRef]
- Hong, X.P.; Peng, C.X.; Wei, W.; Tian, Q.; Liu, Y.H.; Yao, X.Q.; Zhang, Y.; Cao, F.Y.; Wang, Q.; Wang, J.Z. Essential role of tau phosphorylation in adult hippocampal neurogenesis. Hippocampus 2010, 20, 1339–1349. [Google Scholar] [CrossRef]
- Lei, P.; Ayton, S.; Finkelstein, D.I.; Spoerri, L.; Ciccotosto, G.D.; Wright, D.K.; Wong, B.X.; Adlard, P.A.; Cherny, R.A.; Lam, L.Q.; et al. Tau deficiency induces parkinsonism with dementia by impairing APP-mediated iron export. Nat. Med. 2012, 18, 291–295. [Google Scholar] [CrossRef]
- Kimura, T.; Whitcomb, D.J.; Jo, J.; Regan, P.; Piers, T.; Heo, S.; Brown, C.; Hashikawa, T.; Murayama, M.; Seok, H.; et al. Microtubule-associated protein tau is essential for long-term depression in the hippocampus. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2014, 369, 20130144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Regan, P.; Piers, T.; Yi, J.H.; Kim, D.H.; Huh, S.; Park, S.J.; Ryu, J.H.; Whitcomb, D.J.; Cho, K. Tau phosphorylation at serine 396 residue is required for hippocampal LTD. J. Neurosci. 2015, 35, 4804–4812. [Google Scholar] [CrossRef] [Green Version]
- Ittner, L.M.; Ke, Y.D.; Delerue, F.; Bi, M.; Gladbach, A.; van Eersel, J.; Wolfing, H.; Chieng, B.C.; Christie, M.J.; Napier, I.A.; et al. Dendritic function of tau mediates amyloid-beta toxicity in Alzheimer’s disease mouse models. Cell 2010, 142, 387–397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leroy, K.; Ando, K.; Laporte, V.; Dedecker, R.; Suain, V.; Authelet, M.; Heraud, C.; Pierrot, N.; Yilmaz, Z.; Octave, J.N.; et al. Lack of tau proteins rescues neuronal cell death and decreases amyloidogenic processing of APP in APP/PS1 mice. Am. J. Pathol. 2012, 181, 1928–1940. [Google Scholar] [CrossRef] [PubMed]
- Roberson, E.D.; Halabisky, B.; Yoo, J.W.; Yao, J.; Chin, J.; Yan, F.; Wu, T.; Hamto, P.; Devidze, N.; Yu, G.Q.; et al. Amyloid-beta/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J. Neurosci. 2011, 31, 700–711. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roberson, E.D.; Scearce-Levie, K.; Palop, J.J.; Yan, F.; Cheng, I.H.; Wu, T.; Gerstein, H.; Yu, G.Q.; Mucke, L. Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model. Science 2007, 316, 750–754. [Google Scholar] [CrossRef] [Green Version]
- Bi, M.; Gladbach, A.; van Eersel, J.; Ittner, A.; Przybyla, M.; van Hummel, A.; Chua, S.W.; van der Hoven, J.; Lee, W.S.; Muller, J.; et al. Tau exacerbates excitotoxic brain damage in an animal model of stroke. Nat. Commun. 2017, 8, 473. [Google Scholar] [CrossRef] [Green Version]
- De Vos, S.L.; Goncharoff, D.K.; Chen, G.; Kebodeaux, C.S.; Yamada, K.; Stewart, F.R.; Schuler, D.R.; Maloney, S.E.; Wozniak, D.F.; Rigo, F.; et al. Antisense reduction of tau in adult mice protects against seizures. J. Neurosci. 2013, 33, 12887–12897. [Google Scholar] [CrossRef] [Green Version]
- Gheyara, A.L.; Ponnusamy, R.; Djukic, B.; Craft, R.J.; Ho, K.; Guo, W.; Finucane, M.M.; Sanchez, P.E.; Mucke, L. Tau reduction prevents disease in a mouse model of Dravet syndrome. Ann. Neurol. 2014, 76, 443–456. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Braak, E. Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol. Aging 1995, 16, 271–278. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Lin, W.-L.; Lewis, J.; Yen, S.-H.; Hutton, M.; Dickson, D.W. Filamentous tau in oligodendrocytes and astrocytes of transgenic mice expressing the human tau isoform with the P301L mutation. Am. J. Pathol. 2003, 162, 213–218. [Google Scholar] [CrossRef] [Green Version]
- Perea, J.R.; López, E.; Díez-Ballesteros, J.C.; Ávila, J.; Hernández, F.; Bolós, M. Extracellular monomeric tau is internalized by astrocytes. Front. Neurosci. 2019, 13, 442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacs, G.G. Tauopathies. Handb. Clin. Neurol. 2018, 145, 355–368. [Google Scholar]
- De Calignon, A.; Fox, L.M.; Pitstick, R.; Carlson, G.A.; Bacskai, B.J.; Spires-Jones, T.L.; Hyman, B.T. Caspase activation precedes and leads to tangles. Nature 2010, 464, 1201–1204. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.A.; Poirier, M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004, 10, S10–S17. [Google Scholar] [CrossRef]
- Cowan, C.M.; Mudher, A. Are tau aggregates toxic or protective in tauopathies? Front. Neurol. 2013, 4, 114. [Google Scholar] [CrossRef] [Green Version]
- Kitazawa, M.; Oddo, S.; Yamasaki, T.R.; Green, K.N.; La Ferla, F.M. Lipopolysaccharide-induced inflammation exacerbates tau pathology by a cyclin-dependent kinase 5-mediated pathway in a transgenic model of Alzheimer’s disease. J. Neurosci. 2005, 25, 8843–8853. [Google Scholar] [CrossRef] [Green Version]
- Hansen, D.V.; Hanson, J.E.; Sheng, M. Microglia in Alzheimer’s disease. J. Cell Biol. 2018, 217, 459–472. [Google Scholar] [CrossRef]
- Friedman, B.A.; Srinivasan, K.; Ayalon, G.; Meilandt, W.J.; Lin, H.; Huntley, M.A.; Cao, Y.; Lee, S.-H.; Haddick, P.C.; Ngu, H. Diverse brain myeloid expression profiles reveal distinct microglial activation states and aspects of Alzheimer’s disease not evident in mouse models. Cell Rep. 2018, 22, 832–847. [Google Scholar] [CrossRef] [Green Version]
- Rademakers, R.; Cruts, M.; van Broeckhoven, C. The role of tau (MAPT) in frontotemporal dementia and related tauopathies. Hum. Mutat. 2004, 24, 277–295. [Google Scholar] [CrossRef] [PubMed]
- Tolnay, M.; Probst, A. Argyrophilic grain disease. Handb. Clin. Neurol. 2008, 89, 553–563. [Google Scholar] [PubMed]
- Mahapatra, R.K.; Edwards, M.J.; Schott, J.M.; Bhatia, K.P. Corticobasal degeneration. Lancet Neurol. 2004, 3, 736–743. [Google Scholar] [CrossRef]
- Arai, T.; Ikeda, K.; Akiyama, H.; Nonaka, T.; Hasegawa, M.; Ishiguro, K.; Iritani, S.; Tsuchiya, K.; Iseki, E.; Yagishita, S.; et al. Identification of amino-terminally cleaved tau fragments that distinguish progressive supranuclear palsy from corticobasal degeneration. Ann. Neurol. 2004, 55, 72–79. [Google Scholar] [CrossRef]
- Hauw, J.J.; Verny, M.; Delaere, P.; Cervera, P.; He, Y.; Duyckaerts, C. Constant neurofibrillary changes in the neocortex in progressive supranuclear palsy. Basic differences with Alzheimer’s disease and aging. Neurosci. Lett. 1990, 119, 182–186. [Google Scholar] [CrossRef]
- Brandt, R.; Léger, J.; Lee, G. Interaction of tau with the neural plasma membrane mediated by tau’s amino-terminal projection domain. J. Cell Biol. 1995, 131, 1327–1340. [Google Scholar] [CrossRef]
- Brandt, R.; Trushina, N.I.; Bakota, L. Much More Than a Cytoskeletal Protein: Physiological and Pathological Functions of the Non-microtubule Binding Region of Tau. Front. Neurol. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Dixit, R.; Ross, J.L.; Goldman, Y.E.; Holzbaur, E.L. Differential regulation of dynein and kinesin motor proteins by tau. Science 2008, 319, 1086–1089. [Google Scholar] [CrossRef] [Green Version]
- Konzack, S.; Thies, E.; Marx, A.; Mandelkow, E.M.; Mandelkow, E. Swimming against the tide: Mobility of the microtubule-associated protein tau in neurons. J. Neurosci. 2007, 27, 9916–9927. [Google Scholar] [CrossRef]
- Utton, M.A.; Noble, W.J.; Hill, J.E.; Anderton, B.H.; Hanger, D.P. Molecular motors implicated in the axonal transport of tau and α-synuclein. J. Cell Sci. 2005, 118, 4645–4654. [Google Scholar] [CrossRef] [Green Version]
- Magnani, E.; Fan, J.; Gasparini, L.; Golding, M.; Williams, M.; Schiavo, G.; Goedert, M.; Amos, L.A.; Spillantini, M.G. Interaction of tau protein with the dynactin complex. EMBO J. 2007, 26, 4546–4554. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, S.; Tanaka, T.; Soeda, Y.; Takashima, A. Enhanced tau protein translation by hyper-excitation. Front. Aging Neurosci. 2019, 11, 322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, K.; Holth, J.K.; Liao, F.; Stewart, F.R.; Mahan, T.E.; Jiang, H.; Cirrito, J.R.; Patel, T.K.; Hochgräfe, K.; Mandelkow, E.-M. Neuronal activity regulates extracellular tau in vivo. J. Exp. Med. 2014, 211, 387–393. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Balaji, V.; Kaniyappan, S.; Krüger, L.; Irsen, S.; Tepper, K.; Chandupatla, R.; Maetzler, W.; Schneider, A.; Mandelkow, E. The release and trans-synaptic transmission of Tau via exosomes. Mol. Neurodegener. 2017, 12, 1–25. [Google Scholar] [CrossRef] [Green Version]
- Sato, C.; Barthelemy, N.R.; Mawuenyega, K.G.; Patterson, B.W.; Gordon, B.A.; Jockel-Balsarotti, J.; Sullivan, M.; Crisp, M.J.; Kasten, T.; Kirmess, K.M.; et al. Tau Kinetics in Neurons and the Human Central Nervous System. Neuron 2018, 97, 1284–1298.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pooler, A.M.; Phillips, E.C.; Lau, D.H.; Noble, W.; Hanger, D.P. Physiological release of endogenous tau is stimulated by neuronal activity. EMBO Rep. 2013, 14, 389–394. [Google Scholar] [CrossRef]
- Araque, A.; Parpura, V.; Sanzgiri, R.P.; Haydon, P.G. Tripartite synapses: Glia, the unacknowledged partner. Trends Neurosci. 1999, 22, 208–215. [Google Scholar] [CrossRef]
- Perea, G.; Navarrete, M.; Araque, A. Tripartite synapses: Astrocytes process and control synaptic information. Trends Neurosci. 2009, 32, 421–431. [Google Scholar] [CrossRef]
- Halassa, M.M.; Fellin, T.; Haydon, P.G. The tripartite synapse: Roles for gliotransmission in health and disease. Trends Mol. Med. 2007, 13, 54–63. [Google Scholar] [CrossRef]
- Piacentini, R.; Li Puma, D.D.; Mainardi, M.; Lazzarino, G.; Tavazzi, B.; Arancio, O.; Grassi, C. Reduced gliotransmitter release from astrocytes mediates tau-induced synaptic dysfunction in cultured hippocampal neurons. Glia 2017, 65, 1302–1316. [Google Scholar] [CrossRef] [PubMed]
- Tremblay, M.-È.; Lowery, R.L.; Majewska, A.K. Microglial interactions with synapses are modulated by visual experience. PLoS Biol. 2010, 8, e1000527. [Google Scholar] [CrossRef] [Green Version]
- Wake, H.; Moorhouse, A.J.; Jinno, S.; Kohsaka, S.; Nabekura, J. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J. Neurosci. 2009, 29, 3974–3980. [Google Scholar] [CrossRef] [Green Version]
- Maphis, N.; Xu, G.; Kokiko-Cochran, O.N.; Jiang, S.; Cardona, A.; Ransohoff, R.M.; Lamb, B.T.; Bhaskar, K. Reactive microglia drive tau pathology and contribute to the spreading of pathological tau in the brain. Brain 2015, 138, 1738–1755. [Google Scholar] [CrossRef]
- Mondragon-Rodriguez, S.; Trillaud-Doppia, E.; Dudilot, A.; Bourgeois, C.; Lauzon, M.; Leclerc, N.; Boehm, J. Interaction of endogenous tau protein with synaptic proteins is regulated by N-methyl-D-aspartate receptor-dependent tau phosphorylation. J. Biol. Chem. 2012, 287, 32040–32053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tai, H.C.; Serrano-Pozo, A.; Hashimoto, T.; Frosch, M.P.; Spires-Jones, T.L.; Hyman, B.T. The synaptic accumulation of hyperphosphorylated tau oligomers in Alzheimer disease is associated with dysfunction of the ubiquitin-proteasome system. Am. J. Pathol. 2012, 181, 1426–1435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suetsugu, S.; Toyooka, K.; Senju, Y. Subcellular Membrane Curvature Mediated by the BAR Domain Superfamily Proteins. Semin. Cell Dev. Biol. 2010, 21, 340–349. [Google Scholar] [CrossRef]
- Sinsky, J.; Majerova, P.; Kovac, A.; Kotlyar, M.; Jurisica, I.; Hanes, J. Physiological tau interactome in brain and its link to tauopathies. J. Proteome Res. 2020, 19, 2429–2442. [Google Scholar] [CrossRef] [PubMed]
- Safari, F.; Suetsugu, S. The BAR domain superfamily proteins from subcellular structures to human diseases. Membranes 2012, 2, 91–117. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Lv, K.; Li, Z.; Yu, A.C.; Chen, J.; Teng, J. PACSIN1, a Tau-interacting protein, regulates axonal elongation and branching by facilitating microtubule instability. J. Biol. Chem. 2012, 287, 39911–39924. [Google Scholar] [CrossRef] [Green Version]
- He, H.J.; Wang, X.S.; Pan, R.; Wang, D.L.; Liu, M.N.; He, R.Q. The proline-rich domain of tau plays a role in interactions with actin. BMC Cell Biol. 2009, 10, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Elie, A.; Prezel, E.; Guerin, C.; Denarier, E.; Ramirez-Rios, S.; Serre, L.; Andrieux, A.; Fourest-Lieuvin, A.; Blanchoin, L.; Arnal, I. Tau co-organizes dynamic microtubule and actin networks. Sci. Rep. 2015, 5, 9964. [Google Scholar] [CrossRef] [Green Version]
- Pallas-Bazarra, N.; Draffin, J.; Cuadros, R.; Antonio Esteban, J.; Avila, J. Tau is required for the function of extrasynaptic NMDA receptors. Sci. Rep. 2019, 9, 9116. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Li, T.; He, R.; Bartlett, P.F.; Gotz, J. Visualizing the microtubule-associated protein tau in the nucleus. Sci. China Life Sci. 2014, 57, 422–431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Gotz, J. Profiling murine tau with 0N, 1N and 2N isoform-specific antibodies in brain and peripheral organs reveals distinct subcellular localization, with the 1N isoform being enriched in the nucleus. PLoS ONE 2013, 8, e84849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maina, M.B.; Bailey, L.J.; Wagih, S.; Biasetti, L.; Pollack, S.J.; Quinn, J.P.; Thorpe, J.R.; Doherty, A.J.; Serpell, L.C. The involvement of tau in nucleolar transcription and the stress response. Acta Neuropathol. Commun. 2018, 6, 70. [Google Scholar] [CrossRef] [Green Version]
- Sjoberg, M.K.; Shestakova, E.; Mansuroglu, Z.; Maccioni, R.B.; Bonnefoy, E. Tau protein binds to pericentromeric DNA: A putative role for nuclear tau in nucleolar organization. J. Cell Sci. 2006, 119 Pt 10, 2025–2034. [Google Scholar] [CrossRef] [Green Version]
- Thurston, V.C.; Zinkowski, R.P.; Binder, L.I. Tau as a nucleolar protein in human nonneural cells in vitro and in vivo. Chromosoma 1996, 105, 20–30. [Google Scholar] [CrossRef]
- Wei, Y.; Qu, M.H.; Wang, X.S.; Chen, L.; Wang, D.L.; Liu, Y.; Hua, Q.; He, R.Q. Binding to the minor groove of the double-strand, tau protein prevents DNA from damage by peroxidation. PLoS ONE 2008, 3, e2600. [Google Scholar] [CrossRef] [Green Version]
- Kampers, T.; Friedhoff, P.; Biernat, J.; Mandelkow, E.M.; Mandelkow, E. RNA stimulates aggregation of microtubule-associated protein tau into Alzheimer-like paired helical filaments. FEBS Lett. 1996, 399, 344–349. [Google Scholar] [CrossRef]
- Violet, M.; Delattre, L.; Tardivel, M.; Sultan, A.; Chauderlier, A.; Caillierez, R.; Talahari, S.; Nesslany, F.; Lefebvre, B.; Bonnefoy, E.; et al. A major role for Tau in neuronal DNA and RNA protection in vivo under physiological and hyperthermic conditions. Front. Cell Neurosci. 2014, 8, 84. [Google Scholar] [CrossRef] [Green Version]
- Sultan, A.; Nesslany, F.; Violet, M.; Begard, S.; Loyens, A.; Talahari, S.; Mansuroglu, Z.; Marzin, D.; Sergeant, N.; Humez, S.; et al. Nuclear tau, a key player in neuronal DNA protection. J. Biol. Chem. 2011, 286, 4566–4575. [Google Scholar] [CrossRef] [Green Version]
- Mansuroglu, Z.; Benhelli-Mokrani, H.; Marcato, V.; Sultan, A.; Violet, M.; Chauderlier, A.; Delattre, L.; Loyens, A.; Talahari, S.; Begard, S.; et al. Loss of Tau protein affects the structure, transcription and repair of neuronal pericentromeric heterochromatin. Sci. Rep. 2016, 6, 33047. [Google Scholar] [CrossRef] [Green Version]
- Qi, H.; Cantrelle, F.X.; Benhelli-Mokrani, H.; Smet-Nocca, C.; Buee, L.; Lippens, G.; Bonnefoy, E.; Galas, M.C.; Landrieu, I. Nuclear magnetic resonance spectroscopy characterization of interaction of Tau with DNA and its regulation by phosphorylation. Biochemistry 2015, 54, 1525–1533. [Google Scholar] [CrossRef]
- Eftekharzadeh, B.; Daigle, J.G.; Kapinos, L.E.; Coyne, A.; Schiantarelli, J.; Carlomagno, Y.; Cook, C.; Miller, S.J.; Dujardin, S.; Amaral, A.S. Tau protein disrupts nucleocytoplasmic transport in Alzheimer’s disease. Neuron 2018, 99, 925–940.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goedert, M.; Spillantini, M.; Crowther, R. Cloning of a big tau microtubule-associated protein characteristic of the peripheral nervous system. Proc. Natl. Acad. Sci. USA 1992, 89, 1983–1987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taleghany, N.; Oblinger, M. Regional distribution and biochemical characteristics of high molecular weight tau in the nervous system. J. Neurosci. Res. 1992, 33, 257–265. [Google Scholar] [CrossRef]
- Fischer, I.; Baas, P.W. Resurrecting the mysteries of big tau. Trends Neurosci. 2020, 43, 493–504. [Google Scholar] [CrossRef] [PubMed]
- Mercken, M.; Fischer, I.; Kosik, K.; Nixon, R. Three distinct axonal transport rates for tau, tubulin, and other microtubule-associated proteins: Evidence for dynamic interactions of tau with microtubules in vivo. J. Neurosci. 1995, 15, 8259–8267. [Google Scholar] [CrossRef]
- Boyne, L.; Tessler, A.; Murray, M.; Fischer, I. Distribution of Big tau in the central nervous system of the adult and developing rat. J. Comp. Neurol. 1995, 358, 279–293. [Google Scholar] [CrossRef]
- Frappier, T.F.; Georgieff, I.S.; Brown, K.; Shelanski, M.L. τ Regulation of Microtubule-Microtubule Spacing and Bundling. J. Neurochem. 1994, 63, 2288–2294. [Google Scholar] [CrossRef]
- Kanaan, N.M.; Morfini, G.; Pigino, G.; LaPointe, N.E.; Andreadis, A.; Song, Y.; Leitman, E.; Binder, L.I.; Brady, S.T. Phosphorylation in the amino terminus of tau prevents inhibition of anterograde axonal transport. Neurobiol. Aging 2012, 33, 826.e15–826.e30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frost, B.; Jacks, R.L.; Diamond, M.I. Propagation of tau misfolding from the outside to the inside of a cell. J. Biol. Chem. 2009, 284, 12845–12852. [Google Scholar] [CrossRef] [Green Version]
- Clavaguera, F.; Duyckaerts, C.; Haïk, S. Prion-like properties of Tau assemblies. Curr. Opin. Neurobiol. 2020, 61, 49–57. [Google Scholar] [CrossRef]
- Banati, R.B.; Gehrmann, J.; Schubert, P.; Kreutzberg, G.W. Cytotoxicity of microglia. Glia 1993, 7, 111–118. [Google Scholar] [CrossRef]
- Paolicelli, R.C.; Bergamini, G.; Rajendran, L. Cell-to-cell communication by extracellular vesicles: Focus on microglia. Neuroscience 2019, 405, 148–157. [Google Scholar] [CrossRef] [PubMed]
- Asai, H.; Ikezu, S.; Tsunoda, S.; Medalla, M.; Luebke, J.; Haydar, T.; Wolozin, B.; Butovsky, O.; Kügler, S.; Ikezu, T. Depletion of microglia and inhibition of exosome synthesis halt tau propagation. Nat. Neurosci. 2015, 18, 1584. [Google Scholar] [CrossRef]
- Ebneth, A.; Godemann, R.; Stamer, K.; Illenberger, S.; Trinczek, B.; Mandelkow, E.-M.; Mandelkow, E. Overexpression of tau protein inhibits kinesin-dependent trafficking of vesicles, mitochondria, and endoplasmic reticulum: Implications for Alzheimer’s disease. J. Cell Biol. 1998, 143, 777–794. [Google Scholar] [CrossRef] [Green Version]
- Denk, F.; Wade-Martins, R. Knock-out and transgenic mouse models of tauopathies. Neurobiol. Aging 2009, 30, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Blair, L.J.; Frauen, H.D.; Zhang, B.; Nordhues, B.A.; Bijan, S.; Lin, Y.-C.; Zamudio, F.; Hernandez, L.D.; Sabbagh, J.J.; Selenica, M.-L.B. Tau depletion prevents progressive blood-brain barrier damage in a mouse model of tauopathy. Acta Neuropathol. Commun. 2015, 3, 1–22. [Google Scholar] [CrossRef] [Green Version]
- Ledesma, M.D.; Bonay, P.; Avila, J. τ Protein from Alzheimer’s disease patients is glycated at its tubulin-binding domain. J. Neurochem. 1995, 65, 1658–1664. [Google Scholar] [CrossRef]
- Du Yan, S.; Yan, S.F.; Chen, X.; Fu, J.; Chen, M.; Kuppusamy, P.; Smith, M.A.; Perry, G.; Godman, G.C.; Nawroth, P. Non-enzymatically glycated tau in Alzheimer’s disease induces neuronal oxidant stress resulting in cytokine gene expression and release of amyloid β-peptide. Nat. Med. 1995, 1, 693–699. [Google Scholar] [CrossRef]
- Gamblin, T.C.; Chen, F.; Zambrano, A.; Abraha, A.; Lagalwar, S.; Guillozet, A.L.; Lu, M.; Fu, Y.; Garcia-Sierra, F.; LaPointe, N. Caspase cleavage of tau: Linking amyloid and neurofibrillary tangles in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2003, 100, 10032–10037. [Google Scholar] [CrossRef] [Green Version]
- Rissman, R.A.; Poon, W.W.; Blurton-Jones, M.; Oddo, S.; Torp, R.; Vitek, M.P.; LaFerla, F.M.; Rohn, T.T.; Cotman, C.W. Caspase-cleavage of tau is an early event in Alzheimer disease tangle pathology. J. Clin. Investig. 2004, 114, 121–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jadhav, S.; Zilka, N.; Novak, M. Protein truncation as a common denominator of human neurodegenerative foldopathies. Mol. Neurobiol. 2013, 48, 516–532. [Google Scholar] [CrossRef] [PubMed]
- Guo, Q.; Xie, J. AATF inhibits aberrant production of amyloid β peptide 1-42 by interacting directly with Par-4. J. Biol. Chem. 2004, 279, 4596–4603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Köpke, E.; Tung, Y.-C.; Shaikh, S.; Alonso, A.d.C.; Iqbal, K.; Grundke-Iqbal, I. Microtubule-associated protein tau. Abnormal phosphorylation of a non-paired helical filament pool in Alzheimer disease. J. Biol. Chem. 1993, 268, 24374–24384. [Google Scholar] [CrossRef]
- Novak, M. Truncated tau protein as a new marker for Alzheimer’s disease. Acta Virol. 1994, 38, 173–189. [Google Scholar] [PubMed]
- Yen, S.-h.; Liu, W.-K.; Hall, F.L.; Yan, S.-D.; Stern, D.; Dickson, D.W. Alzheimer neurofibrillary lesions: Molecular nature and potential roles of different components. Neurobiol. Aging 1995, 16, 381–387. [Google Scholar] [CrossRef]
- Armstrong, R.A.; Lantos, P.L.; Cairns, N.J. What determines the molecular composition of abnormal protein aggregates in neurodegenerative disease? Neuropathology 2008, 28, 351–365. [Google Scholar] [CrossRef]
- Goldman, R.D.; Steinert, P.M. Cellular and Molecular Biology of Intermediate Filaments; Springer Science & Business Media: New York, NY, USA, 2013. [Google Scholar]
- Samson, A.L.; Ho, B.; Au, A.E.; Schoenwaelder, S.M.; Smyth, M.J.; Bottomley, S.P.; Kleifeld, O.; Medcalf, R.L. Physicochemical properties that control protein aggregation also determine whether a protein is retained or released from necrotic cells. Open Biol. 2016, 6, 160098. [Google Scholar] [CrossRef]
- Kosik, K.S.; Duffy, L.K.; Dowling, M.M.; Abraham, C.; McCluskey, A.; Selkoe, D.J. Microtubule-associated protein 2: Monoclonal antibodies demonstrate the selective incorporation of certain epitopes into Alzheimer neurofibrillary tangles. Proc. Natl. Acad. Sci. USA 1984, 81, 7941–7945. [Google Scholar] [CrossRef] [Green Version]
- Hasegawa, M.; Arai, T.; Ihara, Y. Immunochemical evidence that fragments of phosphorylated MAP5 (MAP1B) are bound to neurofibrillary tangles in Alzheimer’s disease. Neuron 1990, 4, 909–918. [Google Scholar] [CrossRef]
- Takahashi, H.; Hirokawa, K.; Ando, S.; Obata, K. Immunohistological study on brains of Alzheimer’s disease using antibodies to fetal antigens, C-series gangliosides and microtubule-associated protein 5. Acta Neuropathol. 1991, 81, 626–631. [Google Scholar] [CrossRef]
- Anderton, B.H.; Breinburg, D.; Downes, M.J.; Green, P.J.; Tomlinson, B.; Ulrich, J.; Wood, J.N.; Kahn, J. Monoclonal antibodies show that neurofibrillary tangles and neurofilaments share antigenic determinants. Nature 1982, 298, 84–86. [Google Scholar] [CrossRef]
- Snow, A.; Mar, H.; Nochlin, D.; Sekiguchi, R.; Kimata, K.; Koike, Y.; Wight, T. Early accumulation of heparan sulfate in neurons and in the beta-amyloid protein-containing lesions of Alzheimer’s disease and Down’s syndrome. Am. J. Pathol. 1990, 137, 1253. [Google Scholar]
- Perry, G.; Siedlak, S.L.; Richey, P.; Kawai, M.; Cras, P.; Kalaria, R.N.; Galloway, P.G.; Scardina, J.M.; Cordell, B.; Greenberg, B.D. Association of heparan sulfate proteoglycan with the neurofibrillary tangles of Alzheimer’s disease. J. Neurosci. 1991, 11, 3679–3683. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.; Herman, M.M.; Liu, J.; Katsetos, C.D.; Wills, M.R.; Savory, J. Neurofibrillary lesions in experimental aluminum-induced encephalopathy and Alzheimer’s disease share immunoreactivity for amyloid precursor protein, Aβ, α1-antichymotrypsin and ubiquitin-protein conjugates. Brain Res. 1997, 771, 213–220. [Google Scholar] [CrossRef]
- Perry, G.; Richey, P.L.; Siedlak, S.L.; Smith, M.A.; Mulvihill, P.; DeWitt, D.A.; Barnett, J.; Greenberg, B.D.; Kalaria, R.N. Immunocytochemical evidence that the beta-protein precursor is an integral component of neurofibrillary tangles of Alzheimer’s disease. Am. J. Pathol. 1993, 143, 1586. [Google Scholar] [PubMed]
- Baum, L.; Masliah, E.; Iimoto, D.S.; Hansen, L.A.; Halliday, W.C.; Saitoh, T. Casein kinase II is associated with neurofibrillary tangles but is not an intrinsic component of paired helical filaments. Brain Res. 1992, 573, 126–132. [Google Scholar] [CrossRef]
- Trojanowski, J.Q.; Mawal-Dewan, M.; Schmidt, M.L.; Martin, J.; Lee, V.M.-Y. Localization of the mitogen activated protein kinase ERK2 in Alzheimer’s disease neurofibrillary tangles and senile plaque neurites. Brain Res. 1993, 618, 333–337. [Google Scholar] [CrossRef]
- Hanger, D.P.; Hughes, K.; Woodgett, J.R.; Brion, J.-P.; Anderton, B.H. Glycogen synthase kinase-3 induces Alzheimer’s disease-like phosphorylation of tau: Generation of paired helical filament epitopes and neuronal localisation of the kinase. Neurosci. Lett. 1992, 147, 58–62. [Google Scholar] [CrossRef]
- Shimohama, S.; Homma, Y.; Suenaga, T.; Fujimoto, S.; Taniguchi, T.; Araki, W.; Yamaoka, Y.; Takenawa, T.; Kimura, J. Aberrant accumulation of phospholipase C-delta in Alzheimer brains. Am. J. Pathol. 1991, 139, 737. [Google Scholar]
- Smith, M.A.; Taneda, S.; Richey, P.L.; Miyata, S.; Yan, S.-D.; Stern, D.; Sayre, L.M.; Monnier, V.M.; Perry, G. Advanced Maillard reaction end products are associated with Alzheimer disease pathology. Proc. Natl. Acad. Sci. USA 1994, 91, 5710–5714. [Google Scholar] [CrossRef] [Green Version]
- Yan, S.; Chen, X.; Schmidt, A.; Brett, J.; Godman, G.; Zou, Y.; Scott, C.; Caputo, C.; Frappier, T.; Smith, M. Glycated tau protein in Alzheimer disease: A mechanism for induction of oxidant stress. Proc. Natl. Acad. Sci. USA 1994, 91, 7787–7791. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, N.; Fukatsu, R.; Tsuzuki, K.; Hayashi, Y.; Yoshida, T.; Fujii, N.; Koike, T.; Wakayama, I.; Yanagihara, R.; Garruto, R. Advanced glycation end products in Alzheimer’s disease and other neurodegenerative diseases. Am. J. Pathol. 1998, 153, 1149–1155. [Google Scholar] [CrossRef]
- Gella, A.; Durany, N. Oxidative stress in Alzheimer disease. Cell Adhes. Migr. 2009, 3, 88–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGeer, P.; Akiyama, H.; Itagaki, S.; McGeer, E. Activation of the classical complement pathway in brain tissue of Alzheimer patients. Neurosci. Lett. 1989, 107, 341–346. [Google Scholar] [CrossRef]
- Itagaki, S.; Akiyama, H.; Saito, H.; McGeer, P.L. Ultrastructural localization of complement membrane attack complex (MAC)-like immunoreactivity in brains of patients with Alzheimer’s disease. Brain Res. 1994, 645, 78–84. [Google Scholar] [CrossRef]
- Akiyama, H.; Kawamata, T.; Dedhar, S.; McGeer, P. Immunohistochemical localization of vitronectin, its receptor and beta-3 integrin in Alzheimer brain tissue. J. Neuroimmunol. 1991, 32, 19–28. [Google Scholar] [CrossRef]
- Namba, Y.; Tomonaga, M.; Kawasaki, H.; Otomo, E.; Ikeda, K. Apolipoprotein E immunoreactivity in cerebral amyloid deposits and neurofibrillary tangles in Alzheimer’s disease and kuru plaque amyloid in Creutzfeldt-Jakob disease. Brain Res. 1991, 541, 163–166. [Google Scholar] [CrossRef]
- Yamaguchi, H.; Nakazato, Y.; Shoji, M.; Okamoto, K.; Ihara, Y.; Morimatsu, M.; Hirai, S. Secondary deposition of beta amyloid within extracellular neurofibrillary tangles in Alzheimer-type dementia. Am. J. Pathol. 1991, 138, 699. [Google Scholar]
- Mori, H.; Kondo, J.; Ihara, Y. Ubiquitin is a component of paired helical filaments in Alzheimer’s disease. Science 1987, 235, 1641–1644. [Google Scholar] [CrossRef]
- Iwatsubo, T.; Hasegawa, M.; Esaki, Y.; Ihara, Y. Lack of ubiquitin immunoreactivities at both ends of neuropil threads. Possible bidirectional growth of neuropil threads. Am. J. Pathol. 1992, 140, 277. [Google Scholar]
- Nakamura, M.; Kaneko, S.; Dickson, D.W.; Kusaka, H. Aberrant accumulation of BRCA1 in Alzheimer disease and other tauopathies. J. Neuropathol. Exp. Neurol. 2020, 79, 22–33. [Google Scholar] [CrossRef] [PubMed]
- Arima, K.; Nakamura, M.; Sunohara, N.; Nishio, T.; Ogawa, M.; Hirai, S.; Kawai, M.; Ikeda, K. Immunohistochemical and ultrastructural characterization of neuritic clusters around ghost tangles in the hippocampal formation in progressive supranuclear palsy brains. Acta Neuropathol. 1999, 97, 565–576. [Google Scholar] [CrossRef] [PubMed]
- Kalaria, R.; Golde, T.; Kroon, S.; Perry, G. Serine protease inhibitor antithrombin III and its messenger RNA in the pathogenesis of Alzheimer’s disease. Am. J. Pathol. 1993, 143, 886. [Google Scholar] [PubMed]
- Kawamata, T.; Tooyama, I.; Yamada, T.; Walker, D.G.; McGeer, P.L. Lactotransferrin immunocytochemistry in Alzheimer and normal human brain. Am. J. Pathol. 1993, 142, 1574. [Google Scholar] [PubMed]
- Oughtred, R.; Rust, J.; Chang, C.; Breitkreutz, B.J.; Stark, C.; Willems, A.; Boucher, L.; Leung, G.; Kolas, N.; Zhang, F. The BioGRID database: A comprehensive biomedical resource of curated protein, genetic, and chemical interactions. Protein Sci. 2021, 30, 187–200. [Google Scholar] [CrossRef]
- Fahey, M.E.; Bennett, M.J.; Mahon, C.; Jäger, S.; Pache, L.; Kumar, D.; Shapiro, A.; Rao, K.; Chanda, S.K.; Craik, C.S. GPS-Prot: A web-based visualization platform for integrating host-pathogen interaction data. BMC Bioinform. 2011, 12, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Salwinski, L.; Miller, C.S.; Smith, A.J.; Pettit, F.K.; Bowie, J.U.; Eisenberg, D. The database of interacting proteins: 2004 update. Nucleic Acids Res. 2004, 32 (Suppl. 1), D449–D451. [Google Scholar] [CrossRef] [Green Version]
- Orchard, S.; Ammari, M.; Aranda, B.; Breuza, L.; Briganti, L.; Broackes-Carter, F.; Campbell, N.H.; Chavali, G.; Chen, C.; Del-Toro, N. The MIntAct project—IntAct as a common curation platform for 11 molecular interaction databases. Nucleic Acids Res. 2014, 42, D358–D363. [Google Scholar] [CrossRef] [Green Version]
- Licata, L.; Briganti, L.; Peluso, D.; Perfetto, L.; Iannuccelli, M.; Galeota, E.; Sacco, F.; Palma, A.; Nardozza, A.P.; Santonico, E. MINT, the molecular interaction database: 2012 update. Nucleic Acids Res. 2012, 40, D857–D861. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P. STRING v11: Protein–protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kotlyar, M.; Pastrello, C.; Malik, Z.; Jurisica, I. IID 2018 update: Context-specific physical protein–protein interactions in human, model organisms and domesticated species. Nucleic Acids Res. 2019, 47, D581–D589. [Google Scholar] [CrossRef] [PubMed]
- Ksiezak-Reding, H.; Liu, W.-K.; Yen, S.-H. Phosphate analysis and dephosphorylation of modified tau associated with paired helical filaments. Brain Res. 1992, 597, 209–219. [Google Scholar] [CrossRef]
- Biernat, J.; Gustke, N.; Drewes, G.; Mandelkow, E.M.; Mandelkow, E. Phosphorylation of Ser262 strongly reduces binding of tau to microtubules: Distinction between PHF-like immunoreactivity and microtubule binding. Neuron 1993, 11, 153–163. [Google Scholar] [CrossRef]
- Goedert, M.; Jakes, R.; Crowther, R.A.; Cohen, P.; Vanmechelen, E.; Vandermeeren, M.; Cras, P. Epitope mapping of monoclonal antibodies to the paired helical filaments of Alzheimer’s disease: Identification of phosphorylation sites in tau protein. Biochem. J. 1994, 301 Pt 3, 871–877. [Google Scholar] [CrossRef] [Green Version]
- Zheng-Fischhofer, Q.; Biernat, J.; Mandelkow, E.M.; Illenberger, S.; Godemann, R.; Mandelkow, E. Sequential phosphorylation of Tau by glycogen synthase kinase-3beta and protein kinase A at Thr212 and Ser214 generates the Alzheimer-specific epitope of antibody AT100 and requires a paired-helical-filament-like conformation. Eur. J. Biochem. 1998, 252, 542–552. [Google Scholar] [CrossRef] [PubMed]
- Wegmann, S.; Biernat, J.; Mandelkow, E. A current view on Tau protein phosphorylation in Alzheimer’s disease. Curr. Opin. Neurobiol. 2021, 69, 131–138. [Google Scholar] [CrossRef]
- Hasegawa, M.; Morishima-Kawashima, M.; Takio, K.; Suzuki, M.; Titani, K.A.; Ihara, Y. Protein sequence and mass spectrometric analyses of tau in the Alzheimer’s disease brain. J. Biol. Chem. 1992, 267, 17047–17054. [Google Scholar] [CrossRef]
- Brici, D.; Götz, J.; Nisbet, R.M. A novel antibody targeting tau phosphorylated at serine 235 detects neurofibrillary tangles. J. Alzheimer’s Dis. 2018, 61, 899–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neddens, J.; Temmel, M.; Flunkert, S.; Kerschbaumer, B.; Hoeller, C.; Loeffler, T.; Niederkofler, V.; Daum, G.; Attems, J.; Hutter-Paier, B. Phosphorylation of different tau sites during progression of Alzheimer’s disease. Acta Neuropathol. Commun. 2018, 6, 1–15. [Google Scholar] [CrossRef]
- Hanger, D.P.; Betts, J.C.; Loviny, T.L.; Blackstock, W.P.; Anderton, B.H. New phosphorylation sites identified in hyperphosphorylated tau (paired helical filament-tau) from Alzheimer’s disease brain using nanoelectrospray mass spectrometry. J. Neurochem. 1998, 71, 2465–2476. [Google Scholar] [CrossRef] [PubMed]
- Matsuo, E.S.; Shin, R.W.; Billingsley, M.L.; Van deVoorde, A.; O’Connor, M.; Trojanowski, J.Q.; Lee, V.M. Biopsy-derived adult human brain tau is phosphorylated at many of the same sites as Alzheimer’s disease paired helical filament tau. Neuron 1994, 13, 989–1002. [Google Scholar] [CrossRef]
- Drepper, F.; Biernat, J.; Kaniyappan, S.; Meyer, H.E.; Mandelkow, E.M.; Warscheid, B.; Mandelkow, E. A combinatorial native MS and LC-MS/MS approach reveals high intrinsic phosphorylation of human Tau but minimal levels of other key modifications. J. Biol. Chem. 2020, 295, 18213–18225. [Google Scholar] [CrossRef]
- Mair, W.; Muntel, J.; Tepper, K.; Tang, S.; Biernat, J.; Seeley, W.W.; Kosik, K.S.; Mandelkow, E.; Steen, H.; Steen, J.A. FLEXITau: Quantifying post-translational modifications of tau protein in vitro and in human disease. Anal. Chem. 2016, 88, 3704–3714. [Google Scholar] [CrossRef] [Green Version]
- Tepper, K.; Biernat, J.; Kumar, S.; Wegmann, S.; Timm, T.; Hübschmann, S.; Redecke, L.; Mandelkow, E.-M.; Müller, D.J.; Mandelkow, E. Oligomer formation of tau protein hyperphosphorylated in cells. J. Biol. Chem. 2014, 289, 34389–34407. [Google Scholar] [CrossRef] [Green Version]
- Martin, L.; Latypova, X.; Wilson, C.M.; Magnaudeix, A.; Perrin, M.-L.; Yardin, C.; Terro, F. Tau protein kinases: Involvement in Alzheimer’s disease. Ageing Res. Rev. 2013, 12, 289–309. [Google Scholar] [CrossRef]
- Liu, F.; Grundke-Iqbal, I.; Iqbal, K.; Gong, C.X. Contributions of protein phosphatases PP1, PP2A, PP2B and PP5 to the regulation of tau phosphorylation. Eur. J. Neurosci. 2005, 22, 1942–1950. [Google Scholar] [CrossRef]
- Martin, L.; Latypova, X.; Wilson, C.M.; Magnaudeix, A.; Perrin, M.-L.; Terro, F. Tau protein phosphatases in Alzheimer’s disease: The leading role of PP2A. Ageing Res. Rev. 2013, 12, 39–49. [Google Scholar] [CrossRef]
- Alvarez, A.R.; Sandoval, P.C.; Leal, N.R.; Castro, P.U.; Kosik, K.S. Activation of the neuronal c-Abl tyrosine kinase by amyloid-β-peptide and reactive oxygen species. Neurobiol. Dis. 2004, 17, 326–336. [Google Scholar] [CrossRef]
- Franke, T.F.; Kaplan, D.R.; Cantley, L.C. PI3K: Downstream AKTion blocks apoptosis. Cell 1997, 88, 435–437. [Google Scholar] [CrossRef] [Green Version]
- Litersky, J.M.; Johnson, G.V.; Jakes, R.; Goedert, M.; Lee, M.; Seubert, P. Tau protein is phosphorylated by cyclic AMP-dependent protein kinase and calcium/calmodulin-dependent protein kinase II within its microtubule-binding domains at Ser-262 and Ser-356. Biochem. J. 1996, 316, 655–660. [Google Scholar] [CrossRef] [Green Version]
- Singh, T.J.; Wang, J.-Z.; Novak, M.; Kontzekova, E.; Grundke-Iqbal, I.; Iqbal, K. Calcium/calmodulin-dependent protein kinase II phosphorylates tau at Ser-262 but only partially inhibits its binding to microtubules. FEBS Lett. 1996, 387, 145–148. [Google Scholar] [CrossRef] [Green Version]
- Patrick, G.N.; Zukerberg, L.; Nikolic, M.; de La Monte, S.; Dikkes, P.; Tsai, L.-H. Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 1999, 402, 615–622. [Google Scholar] [CrossRef]
- Piedrahita, D.; Hernández, I.; López-Tobón, A.; Fedorov, D.; Obara, B.; Manjunath, B.; Boudreau, R.L.; Davidson, B.; LaFerla, F.; Gallego-Gómez, J.C. Silencing of CDK5 reduces neurofibrillary tangles in transgenic Alzheimer’s mice. J. Neurosci. 2010, 30, 13966–13976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amniai, L.; Barbier, P.; Sillen, A.; Wieruszeski, J.-M.; Peyrot, V.; Lippens, G.; Landrieu, I. Alzheimer disease specific phosphoepitopes of Tau interfere with assembly of tubulin but not binding to microtubules. FASEB J. 2009, 23, 1146–1152. [Google Scholar] [CrossRef] [PubMed]
- Flajolet, M.; He, G.; Heiman, M.; Lin, A.; Nairn, A.C.; Greengard, P. Regulation of Alzheimer’s disease amyloid-β formation by casein kinase I. Proc. Natl. Acad. Sci. USA 2007, 104, 4159–4164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwab, C.; DeMaggio, A.J.; Ghoshal, N.; Binder, L.I.; Kuret, J.; McGeer, P.L. Casein kinase 1 delta is associated with pathological accumulation of tau in several neurodegenerative diseases. Neurobiol. Aging 2000, 21, 503–510. [Google Scholar] [CrossRef]
- Kimura, R.; Kamino, K.; Yamamoto, M.; Nuripa, A.; Kida, T.; Kazui, H.; Hashimoto, R.; Tanaka, T.; Kudo, T.; Yamagata, H. The DYRK1A gene, encoded in chromosome 21 Down syndrome critical region, bridges between β-amyloid production and tau phosphorylation in Alzheimer disease. Hum. Mol. Genet. 2007, 16, 15–23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryoo, S.-R.; Jeong, H.K.; Radnaabazar, C.; Yoo, J.-J.; Cho, H.-J.; Lee, H.-W.; Kim, I.-S.; Cheon, Y.-H.; Ahn, Y.S.; Chung, S.-H. DYRK1A-mediated Hyperphosphorylation of Tau. J. Biol. Chem. 2007, 282, 34850–34857. [Google Scholar] [CrossRef] [Green Version]
- Taira, N.; Nihira, K.; Yamaguchi, T.; Miki, Y.; Yoshida, K. DYRK2 is targeted to the nucleus and controls p53 via Ser46 phosphorylation in the apoptotic response to DNA damage. Mol. Cell 2007, 25, 725–738. [Google Scholar] [CrossRef]
- Briner, A.; Götz, J.; Polanco, J.C. Fyn kinase controls tau aggregation in vivo. Cell Rep. 2020, 32, 108045. [Google Scholar] [CrossRef]
- Li, C.; Götz, J. Pyk2 is a novel tau tyrosine kinase that is regulated by the tyrosine kinase fyn. J. Alzheimer’s Dis. 2018, 64, 205–221. [Google Scholar] [CrossRef] [Green Version]
- Chin, J.; Palop, J.J.; Yu, G.-Q.; Kojima, N.; Masliah, E.; Mucke, L. Fyn kinase modulates synaptotoxicity, but not aberrant sprouting, in human amyloid precursor protein transgenic mice. J. Neurosci. 2004, 24, 4692–4697. [Google Scholar] [CrossRef] [PubMed]
- Takashima, A. GSK-3 is essential in the pathogenesis of Alzheimer’s disease. J. Alzheimer’s Dis. 2006, 9, 309–317. [Google Scholar] [CrossRef] [PubMed]
- Hernández, F.; de Barreda, E.G.; Fuster-Matanzo, A.; Lucas, J.J.; Avila, J. GSK3: A possible link between beta amyloid peptide and tau protein. Exp. Neurol. 2010, 223, 322–325. [Google Scholar] [CrossRef] [PubMed]
- Mines, M.A.; Beurel, E.; Jope, R.S. Regulation of cell survival mechanisms in Alzheimer’s disease by glycogen synthase kinase-3. Int. J. Alzheimer’s Dis. 2011, 2011. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hata, R.; Masumura, M.; Akatsu, H.; Li, F.; Fujita, H.; Nagai, Y.; Yamamoto, T.; Okada, H.; Kosaka, K.; Sakanaka, M. Up-regulation of calcineurin Aβ mRNA in the Alzheimer’s disease brain: Assessment by cDNA microarray. Biochem. Biophys. Res. Commun. 2001, 284, 310–316. [Google Scholar] [CrossRef] [PubMed]
- Scales, T.M.; Derkinderen, P.; Leung, K.-Y.; Byers, H.L.; Ward, M.A.; Price, C.; Bird, I.N.; Perera, T.; Kellie, S.; Williamson, R. Tyrosine phosphorylation of tau by the SRC family kinases lck and fyn. Mol. Neurodegener. 2011, 6, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Ujiie, S.; Hatano, T.; Kubo, S.-I.; Imai, S.; Sato, S.; Uchihara, T.; Yagishita, S.; Hasegawa, K.; Kowa, H.; Sakai, F. LRRK2 I2020T mutation is associated with tau pathology. Parkinsonism Relat. Disord. 2012, 18, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Ohta, E.; Nihira, T.; Uchino, A.; Imaizumi, Y.; Okada, Y.; Akamatsu, W.; Takahashi, K.; Hayakawa, H.; Nagai, M.; Ohyama, M. I2020T mutant LRRK2 iPSC-derived neurons in the Sagamihara family exhibit increased Tau phosphorylation through the AKT/GSK-3β signaling pathway. Hum. Mol. Genet. 2015, 24, 4879–4900. [Google Scholar] [CrossRef] [PubMed]
- Henderson, M.X.; Changolkar, L.; Trojanowski, J.Q.; Lee, V.M. LRRK2 Kinase Activity Does Not Alter Cell-Autonomous Tau Pathology Development in Primary Neurons. J. Parkinson’s Dis. 2021, 11, 1187–1196. [Google Scholar] [CrossRef] [PubMed]
- Bailey, R.M.; Covy, J.P.; Melrose, H.L.; Rousseau, L.; Watkinson, R.; Knight, J.; Miles, S.; Farrer, M.J.; Dickson, D.W.; Giasson, B.I. LRRK2 phosphorylates novel tau epitopes and promotes tauopathy. Acta Neuropathol. 2013, 126, 809–827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, X.; Raina, A.K.; Rottkamp, C.A.; Aliev, G.; Perry, G.; Boux, H.; Smith, M.A. Activation and redistribution of c-jun N-terminal kinase/stress activated protein kinase in degenerating neurons in Alzheimer’s disease. J. Neurochem. 2001, 76, 435–441. [Google Scholar] [CrossRef]
- Shen, C.; Chen, Y.; Liu, H.; Zhang, K.; Zhang, T.; Lin, A.; Jing, N. Hydrogen peroxide promotes Aβ production through JNK-dependent activation of γ-secretase. J. Biol. Chem. 2008, 283, 17721–17730. [Google Scholar] [CrossRef] [PubMed]
- Atzori, C.; Ghetti, B.; Piva, R.; Srinivasan, A.N.; Zolo, P.; Delisle, M.B.; Mirra, S.S.; Migheli, A. Activation of the JNK/p38 pathway occurs in diseases characterized by tau protein pathology and is related to tau phosphorylation but not to apoptosis. J. Neuropathol. Exp. Neurol. 2001, 60, 1190–1197. [Google Scholar] [CrossRef] [Green Version]
- Swatton, J.E.; Sellers, L.A.; Faull, R.L.; Holland, A.; Iritani, S.; Bahn, S. Increased MAP kinase activity in Alzheimer’s and Down syndrome but not in schizophrenia human brain. Eur. J. Neurosci. 2004, 19, 2711–2719. [Google Scholar] [CrossRef]
- Chin, J.Y.; Knowles, R.B.; Schneider, A.; Drewes, G.; Mandelkow, E.-M.; Hyman, B.T. Microtubule-affinity regulating kinase (MARK) is tightly associated with neurofibrillary tangles in Alzheimer brain: A fluorescence resonance energy transfer study. J. Neuropathol. Exp. Neurol. 2000, 59, 966–971. [Google Scholar] [CrossRef]
- Oba, T.; Saito, T.; Asada, A.; Shimizu, S.; Iijima, K.M.; Ando, K. MARK4 with an Alzheimer’s disease-related mutation promotes tau hyperphosphorylation directly and indirectly and exacerbates neurodegeneration. bioRxiv 2020. [Google Scholar] [CrossRef]
- Kawamata, T.; Taniguchi, T.; Mukai, H.; Kitagawa, M.; Hashimoto, T.; Maeda, K.; Ono, Y.; Tanaka, C. A protein kinase, PKN, accumulates in Alzheimer neurofibrillary tangles and associated endoplasmic reticulum-derived vesicles and phosphorylates tau protein. J. Neurosci. 1998, 18, 7402–7410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taniguchi, T.; Kawamata, T.; Mukai, H.; Hasegawa, H.; Isagawa, T.; Yasuda, M.; Hashimoto, T.; Terashima, A.; Nakai, M.; Ono, Y. Phosphorylation of tau is regulated by PKN. J. Biol. Chem. 2001, 276, 10025–10031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, P.H.; Hager, H.; Nielsen, M.S.; Højrup, P.; Gliemann, J.; Jakes, R. α-Synuclein binds to tau and stimulates the protein kinase A-catalyzed tau phosphorylation of serine residues 262 and 356. J. Biol. Chem. 1999, 274, 25481–25489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, S.J.; Zhang, J.Y.; Li, H.L.; Fang, Z.Y.; Wang, Q.; Deng, H.M.; Gong, C.X.; Grundke-Iqbal, I.; Iqbal, K.; Wang, J.Z. Tau becomes a more favorable substrate for GSK-3 when it is prephosphorylated by PKA in rat brain. J. Biol. Chem. 2004, 279, 50078–50088. [Google Scholar] [CrossRef] [Green Version]
- Jicha, G.A.; Weaver, C.; Lane, E.; Vianna, C.; Kress, Y.; Rockwood, J.; Davies, P. cAMP-dependent protein kinase phosphorylations on tau in Alzheimer’s disease. J. Neurosci. 1999, 19, 7486–7494. [Google Scholar] [CrossRef]
- Shi, J.; Qian, W.; Yin, X.; Iqbal, K.; Grundke-Iqbal, I.; Gu, X.; Ding, F.; Gong, C.-X.; Liu, F. Cyclic AMP-dependent protein kinase regulates the alternative splicing of tau exon 10: A mechanism involved in tau pathology of Alzheimer disease. J. Biol. Chem. 2011, 286, 14639–14648. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.F.; Li, H.L.; Li, X.C.; Zhang, Q.; Tian, Q.; Wang, Q.; Xu, H.; Wang, J.Z. Effects of endogenous β-amyloid overproduction on tau phosphorylation in cell culture. J. Neurochem. 2006, 98, 1167–1175. [Google Scholar] [CrossRef]
- Isagawa, T.; Mukai, H.; Oishi, K.; Taniguchi, T.; Hasegawa, H.; Kawamata, T.; Tanaka, C.; Ono, Y. Dual effects of PKNα and protein kinase C on phosphorylation of tau protein by glycogen synthase kinase-3β. Biochem. Biophys. Res. Commun. 2000, 273, 209–212. [Google Scholar] [CrossRef]
- Dourlen, P.; Fernandez-Gomez, F.; Dupont, C.; Grenier-Boley, B.; Bellenguez, C.; Obriot, H.; Caillierez, R.; Sottejeau, Y.; Chapuis, J.; Bretteville, A. Functional screening of Alzheimer risk loci identifies PTK2B as an in vivo modulator and early marker of Tau pathology. Mol. Psychiatry 2017, 22, 874–883. [Google Scholar] [CrossRef]
- Caccamo, A.; Branca, C.; Talboom, J.S.; Shaw, D.M.; Turner, D.; Ma, L.; Messina, A.; Huang, Z.; Wu, J.; Oddo, S. Reducing ribosomal protein S6 kinase 1 expression improves spatial memory and synaptic plasticity in a mouse model of Alzheimer’s disease. J. Neurosci. 2015, 35, 14042–14056. [Google Scholar] [CrossRef] [Green Version]
- An, W.-L.; Cowburn, R.F.; Li, L.; Braak, H.; Alafuzoff, I.; Iqbal, K.; Iqbal, I.-G.; Winblad, B.; Pei, J.-J. Up-regulation of phosphorylated/activated p70 S6 kinase and its relationship to neurofibrillary pathology in Alzheimer’s disease. Am. J. Pathol. 2003, 163, 591–607. [Google Scholar] [CrossRef]
- Pei, J.-J.; Björkdahl, C.; Zhang, H.; Zhou, X.; Winblad, B. p70 S6 kinase and tau in Alzheimer’s disease. J. Alzheimer’s Dis. 2008, 14, 385–392. [Google Scholar] [CrossRef]
- Elahi, M.; Motoi, Y.; Shimonaka, S.; Ishiguro, K.; Imai, Y.; Hattori, N. High-fat diet-induced activation of SGK1 contributes to Alzheimer’s disease pathogenesis by promoting tau pathology. bioRxiv 2020. [Google Scholar] [CrossRef]
- Liu, F.; Gong, C.-X. Tau exon 10 alternative splicing and tauopathies. Mol. Neurodegener. 2008, 3, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Wu, F.; Iqbal, K.; Gong, C.-X.; Hu, W.; Liu, F. Subacute to chronic Alzheimer-like alterations after controlled cortical impact in human tau transgenic mice. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Schweig, J.E.; Yao, H.; Beaulieu-Abdelahad, D.; Ait-Ghezala, G.; Mouzon, B.; Crawford, F.; Mullan, M.; Paris, D. Alzheimer’s disease pathological lesions activate the spleen tyrosine kinase. Acta Neuropathol. Commun. 2017, 5, 1–25. [Google Scholar] [CrossRef]
- Schweig, J.E.; Yao, H.; Jin, C.; Crawford, F.; Mullan, M.; Paris, D. Neuronal Spleen tyrosine kinase (SYK) mediates cytokine release in Transgenic Tau P301S mice organotypic brain slice cultures. Neurosci. Lett. 2020, 729, 134992. [Google Scholar] [CrossRef] [PubMed]
- Schweig, J.E.; Yao, H.; Coppola, K.; Jin, C.; Crawford, F.; Mullan, M.; Paris, D. Spleen tyrosine kinase (SYK) blocks autophagic Tau degradation in vitro and in vivo. J. Biol. Chem. 2019, 294, 13378–13395. [Google Scholar] [CrossRef]
- Xu, J.; Sato, S.; Okuyama, S.; Swan, R.J.; Jacobsen, M.T.; Strunk, E.; Ikezu, T. Tau-tubulin kinase 1 enhances prefibrillar tau aggregation and motor neuron degeneration in P301L FTDP-17 tau-mutant mice. FASEB J. 2010, 24, 2904–2915. [Google Scholar] [CrossRef] [Green Version]
- Dillon, G.M.; Henderson, J.L.; Bao, C.; Joyce, J.A.; Calhoun, M.; Amaral, B.; King, K.W.; Bajrami, B.; Rabah, D. Acute inhibition of the CNS-specific kinase TTBK1 significantly lowers tau phosphorylation at several disease relevant sites. PLoS ONE 2020, 15, e0228771. [Google Scholar] [CrossRef]
- Gong, C.X.; Singh, T.J.; Grundke-Iqbal, I.; Iqbal, K. Phosphoprotein phosphatase activities in Alzheimer disease brain. J. Neurochem. 1993, 61, 921–927. [Google Scholar] [CrossRef] [PubMed]
- Kins, S.; Crameri, A.; Evans, D.R.; Hemmings, B.A.; Nitsch, R.M.; Götz, J. Reduced protein phosphatase 2A activity induces hyperphosphorylation and altered compartmentalization of tau in transgenic mice. J. Biol. Chem. 2001, 276, 38193–38200. [Google Scholar] [CrossRef] [PubMed]
- McKenzie-Nickson, S.; Chan, J.; Perez, K.; Hung, L.W.; Cheng, L.; Sedjahtera, A.; Gunawan, L.; Adlard, P.A.; Hayne, D.J.; McInnes, L.E. Modulating protein phosphatase 2A rescues disease phenotype in neurodegenerative tauopathies. ACS Chem. Neurosci. 2018, 9, 2731–2740. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Ortiz, E.; Hahm, B.K.; Armstrong, D.L.; Rossie, S. Protein phosphatase 5 protects neurons against amyloid-β toxicity. J. Neurochem. 2009, 111, 391–402. [Google Scholar] [CrossRef] [Green Version]
- Gong, C.X.; Liu, F.; Wu, G.; Rossie, S.; Wegiel, J.; Li, L.; Grundke-Iqbal, I.; Iqbal, K. Dephosphorylation of microtubule-associated protein tau by protein phosphatase 5. J. Neurochem. 2004, 88, 298–310. [Google Scholar] [CrossRef]
- Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Rossie, S.; Gong, C.-X. Dephosphorylation of tau by protein phosphatase 5: Impairment in Alzheimer’s disease. J. Biol. Chem. 2005, 280, 1790–1796. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Liu, G.; Leugers, C.J.; Mueller, J.D.; Francis, M.B.; Hefti, M.M.; Schneider, J.A.; Lee, G. Tau interacts with SHP2 in neuronal systems and in Alzheimer’s disease brains. J. Cell Sci. 2019, 132, jcs229054. [Google Scholar] [CrossRef]
- Chao, Y.; Xing, Y.; Chen, Y.; Xu, Y.; Lin, Z.; Li, Z.; Jeffrey, P.D.; Stock, J.B.; Shi, Y. Structure and mechanism of the phosphotyrosyl phosphatase activator. Mol. Cell 2006, 23, 535–546. [Google Scholar] [CrossRef]
- Luo, Y.; Nie, Y.-J.; Shi, H.-R.; Ni, Z.-F.; Wang, Q.; Wang, J.-Z.; Liu, G.-P. PTPA activates protein phosphatase-2A through reducing its phosphorylation at tyrosine-307 with upregulation of protein tyrosine phosphatase 1B. Biochim. Biophys. Acta Mol. Cell Res. 2013, 1833, 1235–1243. [Google Scholar] [CrossRef] [Green Version]
- Jinwal, U.K.; Trotter, J.H.; Abisambra, J.F.; Koren, J.; Lawson, L.Y.; Vestal, G.D.; O’Leary, J.C.; Johnson, A.G.; Jin, Y.; Jones, J.R. The Hsp90 kinase co-chaperone Cdc37 regulates tau stability and phosphorylation dynamics. J. Biol. Chem. 2011, 286, 16976–16983. [Google Scholar] [CrossRef] [Green Version]
- Cheng, W.; Chen, W.; Wang, P.; Chu, J. Asiatic acid protects differentiated PC12 cells from Aβ25–35-induced apoptosis and tau hyperphosphorylation via regulating PI3K/Akt/GSK-3β signaling. Life Sci. 2018, 208, 96–101. [Google Scholar] [CrossRef]
- Malek-Ahmadi, M.; Beach, T.; Obradov, A.; Sue, L.; Belden, C.; Davis, K.; Walker, D.G.; Lue, L.; Adem, A.; Sabbagh, M.N. Increased Alzheimer’s disease neuropathology is associated with type 2 diabetes and ApoE ε4 carrier status. Curr. Alzheimer Res. 2013, 10, 654–659. [Google Scholar] [CrossRef]
- Curtis, D.; Bakaya, K.; Sharma, L.; Bandyopadhyay, S. Weighted burden analysis of exome-sequenced late-onset Alzheimer’s cases and controls provides further evidence for a role for PSEN1 and suggests involvement of the PI3K/Akt/GSK-3β and WNT signalling pathways. Ann. Hum. Genet. 2020, 84, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, C.H.; Garwood, C.J.; Wray, S.; Price, C.; Kellie, S.; Perera, T.; Zvelebil, M.; Yang, A.; Sheppard, P.W.; Varndell, I.M.; et al. Phosphorylation regulates tau interactions with Src homology 3 domains of phosphatidylinositol 3-kinase, phospholipase Cgamma1, Grb2, and Src family kinases. J. Biol. Chem. 2008, 283, 18177–18186. [Google Scholar] [CrossRef] [Green Version]
- Baudier, J.; Cole, R.D. Interactions between the microtubule-associated tau proteins and S100b regulate tau phosphorylation by the Ca2+/calmodulin-dependent protein kinase II. J. Biol. Chem. 1988, 263, 5876–5883. [Google Scholar] [CrossRef]
- Yamaguchi, F.; Umeda, Y.; Shimamoto, S.; Tsuchiya, M.; Tokumitsu, H.; Tokuda, M.; Kobayashi, R. S100 proteins modulate protein phosphatase 5 function: A link between CA2+ signal transduction and protein dephosphorylation. J. Biol. Chem. 2012, 287, 13787–13798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huttunen, H.J.; Kuja-Panula, J.; Sorci, G.; Agneletti, A.L.; Donato, R.; Rauvala, H. Coregulation of neurite outgrowth and cell survival by amphoterin and S100 proteins through receptor for advanced glycation end products (RAGE) activation. J. Biol. Chem. 2000, 275, 40096–40105. [Google Scholar] [CrossRef] [Green Version]
- Esposito, G.; Scuderi, C.; Lu, J.; Savani, C.; De Filippis, D.; Iuvone, T.; Steardo, L., Jr.; Sheen, V.; Steardo, L. S100B induces tau protein hyperphosphorylation via Dickopff-1 up-regulation and disrupts the Wnt pathway in human neural stem cells. J. Cell. Mol. Med. 2008, 12, 914–927. [Google Scholar] [CrossRef]
- Van Eldik, L.J.; Wainwright, M.S. The Janus face of glial-derived S100B: Beneficial and detrimental functions in the brain. Restor. Neurol. Neurosci. 2003, 21, 97–108. [Google Scholar]
- Brown, J.L.; Roberts, W.K. Evidence that approximately eighty per cent of the soluble proteins from Ehrlich ascites cells are Nalpha-acetylated. J. Biol. Chem. 1976, 251, 1009–1014. [Google Scholar] [CrossRef]
- Allfrey, V.G.; Mirsky, A.E. Structural Modifications of Histones and their Possible Role in the Regulation of RNA Synthesis. Science 1964, 144, 559. [Google Scholar] [CrossRef] [PubMed]
- Levenson, J.M.; O’Riordan, K.J.; Brown, K.D.; Trinh, M.A.; Molfese, D.L.; Sweatt, J.D. Regulation of histone acetylation during memory formation in the hippocampus. J. Biol. Chem. 2004, 279, 40545–40559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Behnia, R.; Panic, B.; Whyte, J.R.; Munro, S. Targeting of the Arf-like GTPase Arl3p to the Golgi requires N-terminal acetylation and the membrane protein Sys1p. Nat. Cell Biol. 2004, 6, 405–413. [Google Scholar] [CrossRef] [PubMed]
- Scott, D.C.; Monda, J.K.; Bennett, E.J.; Harper, J.W.; Schulman, B.A. N-terminal acetylation acts as an avidity enhancer within an interconnected multiprotein complex. Science 2011, 334, 674–678. [Google Scholar] [CrossRef] [Green Version]
- Ferreon, J.C.; Jain, A.; Choi, K.-J.; Tsoi, P.S.; MacKenzie, K.R.; Jung, S.Y.; Ferreon, A.C. Acetylation disfavors tau phase separation. Int. J. Mol. Sci. 2018, 19, 1360. [Google Scholar] [CrossRef] [Green Version]
- Cohen, T.J.; Guo, J.L.; Hurtado, D.E.; Kwong, L.K.; Mills, I.P.; Trojanowski, J.Q.; Lee, V.M. The acetylation of tau inhibits its function and promotes pathological tau aggregation. Nat. Commun. 2011, 2, 252. [Google Scholar] [CrossRef] [Green Version]
- Cohen, T.J.; Friedmann, D.; Hwang, A.W.; Marmorstein, R.; Lee, V.M. The microtubule-associated tau protein has intrinsic acetyltransferase activity. Nat. Struct. Mol. Biol. 2013, 20, 756–762. [Google Scholar] [CrossRef] [Green Version]
- Irwin, D.J.; Cohen, T.J.; Grossman, M.; Arnold, S.E.; Xie, S.X.; Lee, V.M.; Trojanowski, J.Q. Acetylated tau, a novel pathological signature in Alzheimer’s disease and other tauopathies. Brain 2012, 135 Pt 3, 807–818. [Google Scholar] [CrossRef] [Green Version]
- Min, S.W.; Cho, S.H.; Zhou, Y.; Schroeder, S.; Haroutunian, V.; Seeley, W.W.; Huang, E.J.; Shen, Y.; Masliah, E.; Mukherjee, C.; et al. Acetylation of tau inhibits its degradation and contributes to tauopathy. Neuron 2010, 67, 953–966. [Google Scholar] [CrossRef] [Green Version]
- Cohen, T.J.; Constance, B.H.; Hwang, A.W.; James, M.; Yuan, C.-X. Intrinsic tau acetylation is coupled to auto-proteolytic tau fragmentation. PLoS ONE 2016, 11, e0158470. [Google Scholar] [CrossRef] [PubMed]
- Cook, C.; Carlomagno, Y.; Gendron, T.F.; Dunmore, J.; Scheffel, K.; Stetler, C.; Davis, M.; Dickson, D.; Jarpe, M.; DeTure, M.; et al. Acetylation of the KXGS motifs in tau is a critical determinant in modulation of tau aggregation and clearance. Hum. Mol. Genet. 2014, 23, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Ding, H.; Dolan, P.J.; Johnson, G.V. Histone deacetylase 6 interacts with the microtubule-associated protein tau. J. Neurochem. 2008, 106, 2119–2130. [Google Scholar] [CrossRef] [Green Version]
- Ogryzko, V.V.; Schiltz, R.L.; Russanova, V.; Howard, B.H.; Nakatani, Y. The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell 1996, 87, 953–959. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Lin, R.J.; Xie, W.; Wilpitz, D.; Evans, R.M. Regulation of hormone-induced histone hyperacetylation and gene activation via acetylation of an acetylase. Cell 1999, 98, 675–686. [Google Scholar] [CrossRef] [Green Version]
- Rouaux, C.; Jokic, N.; Mbebi, C.; Boutillier, S.; Loeffler, J.P.; Boutillier, A.L. Critical loss of CBP/p300 histone acetylase activity by caspase-6 during neurodegeneration. EMBO J. 2003, 22, 6537–6549. [Google Scholar] [CrossRef] [Green Version]
- Verdel, A.; Curtet, S.; Brocard, M.-P.; Rousseaux, S.; Lemercier, C.; Yoshida, M.; Khochbin, S. Active maintenance of mHDA2/mHDAC6 histone-deacetylase in the cytoplasm. Curr. Biol. 2000, 10, 747–749. [Google Scholar] [CrossRef] [Green Version]
- Sen, A.; Nelson, T.J.; Alkon, D.L. ApoE4 and Aβ oligomers reduce BDNF expression via HDAC nuclear translocation. J. Neurosci. 2015, 35, 7538–7551. [Google Scholar] [CrossRef] [PubMed]
- Parmigiani, R.; Xu, W.; Venta-Perez, G.; Erdjument-Bromage, H.; Yaneva, M.; Tempst, P.; Marks, P. HDAC6 is a specific deacetylase of peroxiredoxins and is involved in redox regulation. Proc. Natl. Acad. Sci. USA 2008, 105, 9633–9638. [Google Scholar] [CrossRef] [Green Version]
- Cook, C.; Gendron, T.F.; Scheffel, K.; Carlomagno, Y.; Dunmore, J.; DeTure, M.; Petrucelli, L. Loss of HDAC6, a novel CHIP substrate, alleviates abnormal tau accumulation. Hum. Mol. Genet. 2012, 21, 2936–2945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, B.; West, E.J.; Van, K.C.; Gurkoff, G.G.; Zhou, J.; Zhang, X.-M.; Kozikowski, A.P.; Lyeth, B.G. HDAC inhibitor increases histone H3 acetylation and reduces microglia inflammatory response following traumatic brain injury in rats. Brain Res. 2008, 1226, 181–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.J.; Rowe, M.; Ren, M.; Hong, J.S.; Chen, P.S.; Chuang, D.M. Histone deacetylase inhibitors exhibit anti-inflammatory and neuroprotective effects in a rat permanent ischemic model of stroke: Multiple mechanisms of action. J Pharm. Exp. 2007, 321, 892–901. [Google Scholar] [CrossRef] [PubMed]
- Vaziri, H.; Dessain, S.K.; Ng Eaton, E.; Imai, S.I.; Frye, R.A.; Pandita, T.K.; Guarente, L.; Weinberg, R.A. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 2001, 107, 149–159. [Google Scholar] [CrossRef] [Green Version]
- Langley, E.; Pearson, M.; Faretta, M.; Bauer, U.M.; Frye, R.A.; Minucci, S.; Pelicci, P.G.; Kouzarides, T. Human SIR2 deacetylates p53 and antagonizes PML/p53-induced cellular senescence. EMBO J. 2002, 21, 2383–2396. [Google Scholar] [CrossRef] [Green Version]
- Julien, C.; Tremblay, C.; Emond, V.; Lebbadi, M.; Salem, N., Jr.; Bennett, D.A.; Calon, F. Sirtuin 1 reduction parallels the accumulation of tau in Alzheimer disease. J. Neuropathol. Exp. Neurol. 2009, 68, 48–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakhary, S.M.; Ayubcha, D.; Dileo, J.N.; Jose, R.; Leheste, J.R.; Horowitz, J.M.; Torres, G. Distribution analysis of deacetylase SIRT1 in rodent and human nervous systems. Anat. Rec. 2010, 293, 1024–1032. [Google Scholar] [CrossRef] [Green Version]
- Pallas, M.; Pizarro, J.G.; Gutierrez-Cuesta, J.; Crespo-Biel, N.; Alvira, D.; Tajes, M.; Yeste-Velasco, M.; Folch, J.; Canudas, A.M.; Sureda, F.X.; et al. Modulation of SIRT1 expression in different neurodegenerative models and human pathologies. Neuroscience 2008, 154, 1388–1397. [Google Scholar] [CrossRef]
- Min, S.W.; Sohn, P.D.; Li, Y.; Devidze, N.; Johnson, J.R.; Krogan, N.J.; Masliah, E.; Mok, S.A.; Gestwicki, J.E.; Gan, L. SIRT1 Deacetylates Tau and Reduces Pathogenic Tau Spread in a Mouse Model of Tauopathy. J. Neurosci. 2018, 38, 3680–3688. [Google Scholar] [CrossRef]
- Gao, J.; Wang, W.Y.; Mao, Y.W.; Graff, J.; Guan, J.S.; Pan, L.; Mak, G.; Kim, D.; Su, S.C.; Tsai, L.H. A novel pathway regulates memory and plasticity via SIRT1 and miR-134. Nature 2010, 466, 1105–1109. [Google Scholar] [CrossRef] [Green Version]
- Lazarus, B.D.; Love, D.C.; Hanover, J.A. Recombinant O-GlcNAc transferase isoforms: Identification of O-GlcNAcase, yes tyrosine kinase, and tau as isoform-specific substrates. Glycobiology 2006, 16, 415–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreppel, L.K.; Blomberg, M.A.; Hart, G.W. Dynamic glycosylation of nuclear and cytosolic proteins. Cloning and characterization of a unique O-GlcNAc transferase with multiple tetratricopeptide repeats. J. Biol. Chem. 1997, 272, 9308–9315. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.H.; Si, T.; Wu, W.H.; Hu, J.; Du, J.T.; Zhao, Y.F.; Li, Y.M. O-GlcNAcylation modulates the self-aggregation ability of the fourth microtubule-binding repeat of tau. Biochem. Biophys. Res. Commun. 2008, 375, 59–62. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Iqbal, K.; Grundke-Iqbal, I.; Hart, G.W.; Gong, C.-X. O-GlcNAcylation regulates phosphorylation of tau: A mechanism involved in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2004, 101, 10804–10809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kato, T.; Inui, Y.; Nakamura, A.; Ito, K. Brain fluorodeoxyglucose (FDG) PET in dementia. Ageing Res. Rev. 2016, 30, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Lim, S.; Haque, M.M.; Nam, G.; Ryoo, N.; Rhim, H.; Kim, Y.K. Monitoring of Intracellular Tau Aggregation Regulated by OGA/OGT Inhibitors. Int. J. Mol. Sci. 2015, 16, 20212–20224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koga, H.; Kaushik, S.; Cuervo, A.M. Protein homeostasis and aging: The importance of exquisite quality control. Ageing Res. Rev. 2011, 10, 205–215. [Google Scholar] [CrossRef] [Green Version]
- Morawe, T.; Hiebel, C.; Kern, A.; Behl, C. Protein homeostasis, aging and Alzheimer’s disease. Mol. Neurobiol. 2012, 46, 41–54. [Google Scholar] [CrossRef] [Green Version]
- Glickman, M.H.; Ciechanover, A. The ubiquitin-proteasome proteolytic pathway: Destruction for the sake of construction. Physiol. Rev. 2002, 82, 373–428. [Google Scholar] [CrossRef] [PubMed]
- Dikic, I.; Robertson, M. Ubiquitin ligases and beyond. BMC Biol. 2012, 10, 22. [Google Scholar] [CrossRef] [Green Version]
- Shimura, H.; Schwartz, D.; Gygi, S.P.; Kosik, K.S. CHIP-Hsc70 complex ubiquitinates phosphorylated tau and enhances cell survival. J. Biol. Chem. 2004, 279, 4869–4876. [Google Scholar] [CrossRef] [Green Version]
- Kanack, A.; Vittal, V.; Haver, H.; Keppel, T.; Gundry, R.L.; Klevit, R.E.; Scaglione, K.M. UbcH5 Interacts with Substrates to Participate in Lysine Selection with the E3 Ubiquitin Ligase CHIP. Biochemistry 2020, 59, 2078–2088. [Google Scholar] [CrossRef]
- Ye, Y.; Klenerman, D.; Finley, D. N-terminal ubiquitination of amyloidogenic proteins triggers removal of their oligomers by the proteasome holoenzyme. J. Mol. Biol. 2020, 432, 585–596. [Google Scholar] [CrossRef]
- Ravalin, M.; Theofilas, P.; Basu, K.; Opoku-Nsiah, K.A.; Assimon, V.A.; Medina-Cleghorn, D.; Chen, Y.F.; Bohn, M.F.; Arkin, M.; Grinberg, L.T.; et al. Specificity for latent C termini links the E3 ubiquitin ligase CHIP to caspases. Nat. Chem. Biol. 2019, 15, 786–794. [Google Scholar] [CrossRef]
- Vittal, V.; Shi, L.; Wenzel, D.M.; Scaglione, K.M.; Duncan, E.D.; Basrur, V.; Elenitoba-Johnson, K.S.; Baker, D.; Paulson, H.L.; Brzovic, P.S.; et al. Intrinsic disorder drives N-terminal ubiquitination by Ube2w. Nat. Chem. Biol. 2015, 11, 83–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrucelli, L.; Dickson, D.; Kehoe, K.; Taylor, J.; Snyder, H.; Grover, A.; De Lucia, M.; McGowan, E.; Lewis, J.; Prihar, G.; et al. CHIP and Hsp70 regulate tau ubiquitination, degradation and aggregation. Hum. Mol. Genet. 2004, 13, 703–714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moore, D.J.; West, A.B.; Dikeman, D.A.; Dawson, V.L.; Dawson, T.M. Parkin mediates the degradation-independent ubiquitination of Hsp70. J. Neurochem. 2008, 105, 1806–1819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sassone, J.; Serratto, G.; Valtorta, F.; Silani, V.; Passafaro, M.; Ciammola, A. The synaptic function of parkin. Brain 2017, 140, 2265–2272. [Google Scholar] [CrossRef] [Green Version]
- Singh, K.; Han, K.; Tilve, S.; Wu, K.; Geller, H.M.; Sack, M.N. Parkin targets NOD2 to regulate astrocyte endoplasmic reticulum stress and inflammation. Glia 2018, 66, 2427–2437. [Google Scholar] [CrossRef]
- Williams, E.T.; Glauser, L.; Tsika, E.; Jiang, H.; Islam, S.; Moore, D.J. Parkin mediates the ubiquitination of VPS35 and modulates retromer-dependent endosomal sorting. Hum. Mol. Genet. 2018, 27, 3189–3205. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Ding, W.; Xu, T.; Ao, X.; Yu, T.; Li, M.; Liu, Y.; Zhang, X.; Hou, L.; Wang, J. Parkin Regulates Programmed Necrosis and Myocardial Ischemia/Reperfusion Injury by Targeting Cyclophilin-D. Antioxid. Redox Signal. 2019, 31, 1177–1193. [Google Scholar] [CrossRef]
- Hatakeyama, S.; Matsumoto, M.; Kamura, T.; Murayama, M.; Chui, D.H.; Planel, E.; Takahashi, R.; Nakayama, K.I.; Takashima, A. U-box protein carboxyl terminus of Hsc70-interacting protein (CHIP) mediates poly-ubiquitylation preferentially on four-repeat Tau and is involved in neurodegeneration of tauopathy. J. Neurochem. 2004, 91, 299–307. [Google Scholar] [CrossRef]
- Flach, K.; Ramminger, E.; Hilbrich, I.; Arsalan-Werner, A.; Albrecht, F.; Herrmann, L.; Goedert, M.; Arendt, T.; Holzer, M. Axotrophin/MARCH7 acts as an E3 ubiquitin ligase and ubiquitinates tau protein in vitro impairing microtubule binding. Biochim. Biophys. Acta 2014, 1842, 1527–1538. [Google Scholar] [CrossRef] [Green Version]
- Babu, J.R.; Geetha, T.; Wooten, M.W. Sequestosome 1/p62 shuttles polyubiquitinated tau for proteasomal degradation. J. Neurochem. 2005, 94, 192–203. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Wang, W.; Wang, Q.; Xie, R.; Landay, A.; Chen, D. The E3 ubiquitin ligase CHIP in normal cell function and in disease conditions. Ann. N. Y. Acad. Sci. 2020, 1460, 3–10. [Google Scholar] [CrossRef]
- Rui, Y.-N.; Xu, Z.; Patel, B.; Chen, Z.; Chen, D.; Tito, A.; David, G.; Sun, Y.; Stimming, E.F.; Bellen, H.J. Huntingtin functions as a scaffold for selective macroautophagy. Nat. Cell Biol. 2015, 17, 262–275. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geetha, T.; Jiang, J.; Wooten, M.W. Lysine 63 polyubiquitination of the nerve growth factor receptor TrkA directs internalization and signaling. Mol. Cell 2005, 20, 301–312. [Google Scholar] [CrossRef]
- Wooten, M.W.; Geetha, T.; Seibenhener, M.L.; Babu, J.R.; Diaz-Meco, M.T.; Moscat, J. The p62 scaffold regulates nerve growth factor-induced NF-kappaB activation by influencing TRAF6 polyubiquitination. J. Biol. Chem. 2005, 280, 35625–35629. [Google Scholar] [CrossRef] [Green Version]
- Seibenhener, M.L.; Geetha, T.; Wooten, M.W. Sequestosome 1/p62-more than just a scaffold. FEBS Lett. 2007, 581, 175–179. [Google Scholar] [CrossRef] [Green Version]
- Bjørkøy, G.; Lamark, T.; Brech, A.; Outzen, H.; Perander, M.; Øvervatn, A.; Stenmark, H.; Johansen, T. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J. Cell Biol. 2005, 171, 603–614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clausen, T.H.; Lamark, T.; Isakson, P.; Finley, K.; Larsen, K.B.; Brech, A.; Overvatn, A.; Stenmark, H.; Bjorkoy, G.; Simonsen, A.; et al. p62/SQSTM1 and ALFY interact to facilitate the formation of p62 bodies/ALIS and their degradation by autophagy. Autophagy 2010, 6, 330–344. [Google Scholar] [CrossRef] [Green Version]
- Hori, T.; Osaka, F.; Chiba, T.; Miyamoto, C.; Okabayashi, K.; Shimbara, N.; Kato, S.; Tanaka, K. Covalent modification of all members of human cullin family proteins by NEDD8. Oncogene 1999, 18, 6829–6834. [Google Scholar] [CrossRef] [Green Version]
- Kamitani, T.; Kito, K.; Fukuda-Kamitani, T.; Yeh, E.T. Targeting of NEDD8 and its conjugates for proteasomal degradation by NUB1. J. Biol. Chem. 2001, 276, 46655–46660. [Google Scholar] [CrossRef] [Green Version]
- Richet, E.; Pooler, A.M.; Rodriguez, T.; Novoselov, S.S.; Schmidtke, G.; Groettrup, M.; Hanger, D.P.; Cheetham, M.E.; van der Spuy, J. NUB1 modulation of GSK3beta reduces tau aggregation. Hum. Mol. Genet. 2012, 21, 5254–5267. [Google Scholar] [CrossRef] [Green Version]
- Guarascio, R.; Salih, D.; Yasvoina, M.; Edwards, F.A.; Cheetham, M.E.; van der Spuy, J. Negative Regulator of Ubiquitin-Like Protein 1 modulates the autophagy-lysosomal pathway via p62 to facilitate the extracellular release of tau following proteasome impairment. Hum. Mol. Genet. 2020, 29, 80–96. [Google Scholar] [CrossRef]
- Bukau, B.; Horwich, A.L. The Hsp70 and Hsp60 chaperone machines. Cell 1998, 92, 351–366. [Google Scholar] [CrossRef] [Green Version]
- Tavaria, M.; Gabriele, T.; Kola, I.; Anderson, R.L. A hitchhiker’s guide to the human Hsp70 family. Cell Stress Chaperones 1996, 1, 23–28. [Google Scholar] [CrossRef] [Green Version]
- Jinwal, U.K.; Akoury, E.; Abisambra, J.F.; O’Leary III, J.C.; Thompson, A.D.; Blair, L.J.; Jin, Y.; Bacon, J.; Nordhues, B.A.; Cockman, M. Imbalance of Hsp70 family variants fosters tau accumulation. FASEB J. 2013, 27, 1450–1459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.-C.; Fu, Z.-Q.; Song, J.; Zhang, J.-Y.; Wei, Y.-P.; Chu, J.; Han, L.; Qu, N.; Wang, J.-Z.; Tian, Q. Bip enhanced the association of GSK-3β with tau during ER stress both in vivo and in vitro. J. Alzheimer’s Dis. 2012, 29, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, M.; Kuret, J.; Lee, G. Two motifs within the tau microtubule-binding domain mediate its association with the hsc70 molecular chaperone. J. Neurosci. Res. 2008, 86, 2763–2773. [Google Scholar] [CrossRef] [Green Version]
- Rauch, J.N.; Zuiderweg, E.R.; Gestwicki, J.E. Non-canonical Interactions between Heat Shock Cognate Protein 70 (Hsc70) and Bcl2-associated Anthanogene (BAG) Co-Chaperones Are Important for Client Release. J. Biol. Chem. 2016, 291, 19848–19857. [Google Scholar] [CrossRef] [Green Version]
- Elliott, E.; Tsvetkov, P.; Ginzburg, I. BAG-1 associates with Hsc70.Tau complex and regulates the proteasomal degradation of Tau protein. J. Biol. Chem. 2007, 282, 37276–37284. [Google Scholar] [CrossRef] [Green Version]
- Patterson, K.R.; Ward, S.M.; Combs, B.; Voss, K.; Kanaan, N.M.; Morfini, G.; Brady, S.T.; Gamblin, T.C.; Binder, L.I. Heat shock protein 70 prevents both tau aggregation and the inhibitory effects of preexisting tau aggregates on fast axonal transport. Biochemistry 2011, 50, 10300–10310. [Google Scholar] [CrossRef] [Green Version]
- Abisambra, J.F.; Jinwal, U.K.; Suntharalingam, A.; Arulselvam, K.; Brady, S.; Cockman, M.; Jin, Y.; Zhang, B.; Dickey, C.A. DnaJA1 antagonizes constitutive Hsp70-mediated stabilization of tau. J. Mol. Biol. 2012, 421, 653–661. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, J.C.; Hartl, F.U. Chaperones and transcriptional regulation by nuclear receptors. Nat. Struct. Biol. 2002, 9, 640–642. [Google Scholar] [CrossRef] [PubMed]
- Oroz, J.; Chang, B.J.; Wysoczanski, P.; Lee, C.-T.; Pérez-Lara, Á.; Chakraborty, P.; Hofele, R.V.; Baker, J.D.; Blair, L.J.; Biernat, J. Structure and pro-toxic mechanism of the human Hsp90/PPIase/Tau complex. Nat. Commun. 2018, 9, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fontaine, S.N.; Rauch, J.N.; Nordhues, B.A.; Assimon, V.A.; Stothert, A.R.; Jinwal, U.K.; Sabbagh, J.J.; Chang, L.; Stevens, S.M.; Zuiderweg, E.R. Isoform-selective genetic inhibition of constitutive cytosolic Hsp70 activity promotes client tau degradation using an altered co-chaperone complement. J. Biol. Chem. 2015, 290, 13115–13127. [Google Scholar] [CrossRef] [Green Version]
- Ali, Y.O.; Allen, H.M.; Yu, L.; Li-Kroeger, D.; Bakhshizadehmahmoudi, D.; Hatcher, A.; McCabe, C.; Xu, J.; Bjorklund, N.; Taglialatela, G. NMNAT2: HSP90 complex mediates proteostasis in proteinopathies. PLoS Biol. 2016, 14, e1002472. [Google Scholar] [CrossRef] [PubMed]
- Jinwal, U.K.; Koren, J.; Borysov, S.I.; Schmid, A.B.; Abisambra, J.F.; Blair, L.J.; Johnson, A.G.; Jones, J.R.; Shults, C.L.; O’Leary, J.C. The Hsp90 cochaperone, FKBP51, increases Tau stability and polymerizes microtubules. J. Neurosci. 2010, 30, 591–599. [Google Scholar] [CrossRef] [Green Version]
- Woodford, M.R.; Sager, R.A.; Marris, E.; Dunn, D.M.; Blanden, A.R.; Murphy, R.L.; Rensing, N.; Shapiro, O.; Panaretou, B.; Prodromou, C.; et al. Tumor suppressor Tsc1 is a new Hsp90 co-chaperone that facilitates folding of kinase and non-kinase clients. EMBO J. 2017, 36, 3650–3665. [Google Scholar] [CrossRef]
- Tortosa, E.; Santa-Maria, I.; Moreno, F.; Lim, F.; Perez, M.; Avila, J. Binding of Hsp90 to tau promotes a conformational change and aggregation of tau protein. J. Alzheimer’s Dis. 2009, 17, 319–325. [Google Scholar] [CrossRef]
- Blair, L.J.; Nordhues, B.A.; Hill, S.E.; Scaglione, K.M.; O’Leary, J.C.; Fontaine, S.N.; Breydo, L.; Zhang, B.; Li, P.; Wang, L. Accelerated neurodegeneration through chaperone-mediated oligomerization of tau. J. Clin. Investig. 2013, 123, 4158–4169. [Google Scholar] [CrossRef] [Green Version]
- Shelton, L.B.; Baker, J.D.; Zheng, D.; Sullivan, L.E.; Solanki, P.K.; Webster, J.M.; Sun, Z.; Sabbagh, J.J.; Nordhues, B.A.; Koren, J. Hsp90 activator Aha1 drives production of pathological tau aggregates. Proc. Natl. Acad. Sci. USA 2017, 114, 9707–9712. [Google Scholar] [CrossRef] [Green Version]
- Freilich, R.; Betegon, M.; Tse, E.; Mok, S.-A.; Julien, O.; Agard, D.A.; Southworth, D.R.; Takeuchi, K.; Gestwicki, J.E. Competing protein-protein interactions regulate binding of Hsp27 to its client protein tau. Nat. Commun. 2018, 9, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimura, H.; Miura-Shimura, Y.; Kosik, K.S. Binding of tau to heat shock protein 27 leads to decreased concentration of hyperphosphorylated tau and enhanced cell survival. J. Biol. Chem. 2004, 279, 17957–17962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poon, S.; Easterbrook-Smith, S.B.; Rybchyn, M.S.; Carver, J.A.; Wilson, M.R. Clusterin is an ATP-independent chaperone with very broad substrate specificity that stabilizes stressed proteins in a folding-competent state. Biochemistry 2000, 39, 15953–15960. [Google Scholar] [CrossRef]
- Shepherd, C.E.; Affleck, A.J.; Bahar, A.Y.; Carew-Jones, F.; Halliday, G.M. Intracellular and secreted forms of clusterin are elevated early in Alzheimer’s disease and associate with both Abeta and tau pathology. Neurobiol. Aging 2020, 89, 129–131. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Hayashi, I.; Wong, J.; Tugusheva, K.; Renger, J.J.; Zerbinatti, C. Intracellular clusterin interacts with brain isoforms of the bridging integrator 1 and with the microtubule-associated protein Tau in Alzheimer’s disease. PLoS ONE 2014, 9, e103187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, F.X.; Mayr, L.M.; Mucke, M.; Schonbrunner, E.R. Prolyl isomerases: Role in protein folding. Adv. Protein Chem. 1993, 44, 25–66. [Google Scholar] [PubMed]
- Morgan, A.A.; Rubenstein, E. Proline: The distribution, frequency, positioning, and common functional roles of proline and polyproline sequences in the human proteome. PLoS ONE 2013, 8, e53785. [Google Scholar] [CrossRef] [Green Version]
- Goode, B.L.; Denis, P.E.; Panda, D.; Radeke, M.J.; Miller, H.P.; Wilson, L.; Feinstein, S.C. Functional interactions between the proline-rich and repeat regions of tau enhance microtubule binding and assembly. Mol. Biol. Cell 1997, 8, 353–365. [Google Scholar] [CrossRef]
- Bielska, A.A.; Zondlo, N.J. Hyperphosphorylation of tau induces local polyproline II helix. Biochemistry 2006, 45, 5527–5537. [Google Scholar] [CrossRef]
- Koren, J.; Jinwal, U.K.; Davey, Z.; Kiray, J.; Arulselvam, K.; Dickey, C.A. Bending tau into shape: The emerging role of peptidyl-prolyl isomerases in tauopathies. Mol. Neurobiol. 2011, 44, 65–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chambraud, B.; Sardin, E.; Giustiniani, J.; Dounane, O.; Schumacher, M.; Goedert, M.; Baulieu, E.E. A role for FKBP52 in Tau protein function. Proc. Natl. Acad. Sci. USA 2010, 107, 2658–2663. [Google Scholar] [CrossRef] [Green Version]
- Lu, P.-J.; Wulf, G.; Zhou, X.Z.; Davies, P.; Lu, K.P. The prolyl isomerase Pin1 restores the function of Alzheimer-associated phosphorylated tau protein. Nature 1999, 399, 784–788. [Google Scholar] [CrossRef]
- Gruber, C.W.; Cemazar, M.; Heras, B.; Martin, J.L.; Craik, D.J. Protein disulfide isomerase: The structure of oxidative folding. Trends Biochem. Sci. 2006, 31, 455–464. [Google Scholar] [CrossRef]
- Xu, L.R.; Liu, X.L.; Chen, J.; Liang, Y. Protein disulfide isomerase interacts with tau protein and inhibits its fibrillization. PLoS ONE 2013, 8, e76657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wegmann, S.; Eftekharzadeh, B.; Tepper, K.; Zoltowska, K.M.; Bennett, R.E.; Dujardin, S.; Laskowski, P.R.; MacKenzie, D.; Kamath, T.; Commins, C.; et al. Tau protein liquid-liquid phase separation can initiate tau aggregation. EMBO J. 2018, 37, e98049. [Google Scholar] [CrossRef] [PubMed]
- Wang, K.; Liu, J.Q.; Zhong, T.; Liu, X.L.; Zeng, Y.; Qiao, X.; Xie, T.; Chen, Y.; Gao, Y.Y.; Tang, B.; et al. Phase Separation and Cytotoxicity of Tau are Modulated by Protein Disulfide Isomerase and S-nitrosylation of this Molecular Chaperone. J. Mol. Biol. 2020, 432, 2141–2163. [Google Scholar] [CrossRef] [PubMed]
- Uehara, T.; Nakamura, T.; Yao, D.; Shi, Z.Q.; Gu, Z.; Ma, Y.; Masliah, E.; Nomura, Y.; Lipton, S.A. S-nitrosylated protein-disulphide isomerase links protein misfolding to neurodegeneration. Nature 2006, 441, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Honjo, Y.; Horibe, T.; Torisawa, A.; Ito, H.; Nakanishi, A.; Mori, H.; Komiya, T.; Takahashi, R.; Kawakami, K. Protein disulfide isomerase P5-immunopositive inclusions in patients with Alzheimer’s disease. J. Alzheimers Dis. 2014, 38, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Blair, L.J.; Baker, J.D.; Sabbagh, J.J.; Dickey, C.A. The emerging role of peptidyl-prolyl isomerase chaperones in tau oligomerization, amyloid processing, and Alzheimer’s disease. J. Neurochem. 2015, 133, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Peak, S.L.; Gracia, L.; Lora, G.; Jinwal, U.K. Hsp90-interacting Co-chaperones and their Family Proteins in Tau Regulation: Introducing a Novel Role for Cdc37L1. Neuroscience 2021, 453, 312–323. [Google Scholar] [CrossRef]
- Amadoro, G.; Corsetti, V.; Atlante, A.; Florenzano, F.; Capsoni, S.; Bussani, R.; Mercanti, D.; Calissano, P. Interaction between NH2-tau fragment and Aβ in Alzheimer’s disease mitochondria contributes to the synaptic deterioration. Neurobiol. Aging 2012, 33, 833.e1–833.e25. [Google Scholar] [CrossRef]
- Elrod, J.W.; Molkentin, J.D. Physiologic functions of cyclophilin D and the mitochondrial permeability transition pore. Circ. J. 2013, 77, 1111–1122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schinzel, A.C.; Takeuchi, O.; Huang, Z.; Fisher, J.K.; Zhou, Z.; Rubens, J.; Hetz, C.; Danial, N.N.; Moskowitz, M.A.; Korsmeyer, S.J. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc. Natl. Acad. Sci. USA 2005, 102, 12005–12010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baines, C.P.; Kaiser, R.A.; Purcell, N.H.; Blair, N.S.; Osinska, H.; Hambleton, M.A.; Brunskill, E.W.; Sayen, M.R.; Gottlieb, R.A.; Dorn, G.W. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005, 434, 658–662. [Google Scholar] [CrossRef] [PubMed]
- Guo, C.; Sun, L.; Chen, X.; Zhang, D. Oxidative stress, mitochondrial damage and neurodegenerative diseases. Neural Regen. Res. 2013, 8, 2003. [Google Scholar] [PubMed]
- Berridge, M.J. Dysregulation of neural calcium signaling in Alzheimer disease, bipolar disorder and schizophrenia. Prion 2013, 7, 2–13. [Google Scholar] [CrossRef] [Green Version]
- Abeti, R.; Abramov, A.Y. Mitochondrial Ca2+ in neurodegenerative disorders. Pharmacol. Res. 2015, 99, 377–381. [Google Scholar] [CrossRef]
- Du, H.; Guo, L.; Fang, F.; Chen, D.; Sosunov, A.A.; McKhann, G.M.; Yan, Y.; Wang, C.; Zhang, H.; Molkentin, J.D. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer’s disease. Nat. Med. 2008, 14, 1097–1105. [Google Scholar] [CrossRef]
- Du, H.; Guo, L.; Zhang, W.; Rydzewska, M.; Yan, S. Cyclophilin D deficiency improves mitochondrial function and learning/memory in aging Alzheimer disease mouse model. Neurobiol. Aging 2011, 32, 398–406. [Google Scholar] [CrossRef] [Green Version]
- Jara, C.; Cerpa, W.; Tapia-Rojas, C.; Quintanilla, R.A. Tau deletion prevents cognitive impairment and mitochondrial dysfunction age-associated by a mechanism dependent on Cyclophilin-D (CypD). Front. Neurosci. 2020, 14, 1480. [Google Scholar]
- Pérez, M.J.; Jara, C.; Quintanilla, R.A. Contribution of tau pathology to mitochondrial impairment in neurodegeneration. Front. Neurosci. 2018, 12, 441. [Google Scholar] [CrossRef]
- Shimohama, S. Apoptosis in Alzheimer’s disease—An update. Apoptosis 2000, 5, 9–16. [Google Scholar] [CrossRef]
- Strasser, A.; O’Connor, L.; Dixit, V.M. Apoptosis signaling. Annu. Rev. Biochem. 2000, 69, 217–245. [Google Scholar] [CrossRef]
- Chung, C.-W.; Song, Y.-H.; Kim, I.-K.; Yoon, W.-J.; Ryu, B.-R.; Jo, D.-G.; Woo, H.-N.; Kwon, Y.-K.; Kim, H.-H.; Gwag, B.-J. Proapoptotic effects of tau cleavage product generated by caspase-3. Neurobiol. Dis. 2001, 8, 162–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cotman, C.W.; Poon, W.W.; Rissman, R.A.; Blurton-Jones, M. The role of caspase cleavage of tau in Alzheimer disease neuropathology. J. Neuropathol. Exp. Neurol. 2005, 64, 104–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zilka, N.; Kovacech, B.; Barath, P.; Kontsekova, E.; Novák, M. The Self-Perpetuating tau Truncation Circle; Portland Press Ltd.: London, UK, 2012. [Google Scholar]
- Miao, E.A.; Rajan, J.V.; Aderem, A. Caspase-1-induced pyroptotic cell death. Immunol. Rev. 2011, 243, 206–214. [Google Scholar] [CrossRef]
- Chu, J.; Lauretti, E.; Praticò, D. Caspase-3-dependent cleavage of Akt modulates tau phosphorylation via GSK3β kinase: Implications for Alzheimer’s disease. Mol. Psychiatry 2017, 22, 1002–1008. [Google Scholar] [CrossRef]
- Glushakova, O.Y.; Glushakov, A.O.; Borlongan, C.V.; Valadka, A.B.; Hayes, R.L.; Glushakov, A.V. Role of Caspase-3-Mediated Apoptosis in Chronic Caspase-3-Cleaved Tau Accumulation and Blood–Brain Barrier Damage in the Corpus Callosum after Traumatic Brain Injury in Rats. J. Neurotrauma 2018, 35, 157–173. [Google Scholar] [CrossRef]
- Horowitz, P.M.; Patterson, K.R.; Guillozet-Bongaarts, A.L.; Reynolds, M.R.; Carroll, C.A.; Weintraub, S.T.; Bennett, D.A.; Cryns, V.L.; Berry, R.W.; Binder, L.I. Early N-terminal changes and caspase-6 cleavage of tau in Alzheimer’s disease. J. Neurosci. 2004, 24, 7895–7902. [Google Scholar] [CrossRef]
- Guo, H.; Albrecht, S.; Bourdeau, M.; Petzke, T.; Bergeron, C.; LeBlanc, A.C. Active caspase-6 and caspase-6-cleaved tau in neuropil threads, neuritic plaques, and neurofibrillary tangles of Alzheimer’s disease. Am. J. Pathol. 2004, 165, 523–531. [Google Scholar] [CrossRef]
- Zhao, H.; Zhao, W.; Lok, K.; Wang, Z.; Yin, M. A Synergic Role of Caspase-6 and Caspase-3 in Tau Truncation at D421 Induced by H2O2. Cell. Mol. Neurobiol. 2014, 34, 369–378. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, S.; Bogdanovic, N.; Ghetti, B.; Winblad, B.; LeBlanc, A.C. Caspase-6 activation in familial Alzheimer disease brains carrying amyloid precursor protein or presenilin I or presenilin II mutations. J. Neuropathol. Exp. Neurol. 2009, 68, 1282–1293. [Google Scholar] [CrossRef]
- Wang, X.J.; Cao, Q.; Zhang, Y.; Su, X.D. Activation and regulation of caspase-6 and its role in neurodegenerative diseases. Annu. Rev. Pharm. Toxicol. 2015, 55, 553–572. [Google Scholar] [CrossRef] [Green Version]
- Gervais, F.G.; Xu, D.; Robertson, G.S.; Vaillancourt, J.P.; Zhu, Y.; Huang, J.; LeBlanc, A.; Smith, D.; Rigby, M.; Shearman, M.S.; et al. Involvement of caspases in proteolytic cleavage of Alzheimer’s amyloid-beta precursor protein and amyloidogenic A beta peptide formation. Cell 1999, 97, 395–406. [Google Scholar] [CrossRef] [Green Version]
- Pellegrini, L.; Passer, B.J.; Tabaton, M.; Ganjei, J.K.; D’Adamio, L. Alternative, non-secretase processing of Alzheimer’s beta-amyloid precursor protein during apoptosis by caspase-6 and -8. J. Biol. Chem. 1999, 274, 21011–21016. [Google Scholar] [CrossRef] [Green Version]
- Lu, D.C.; Rabizadeh, S.; Chandra, S.; Shayya, R.F.; Ellerby, L.M.; Ye, X.; Salvesen, G.S.; Koo, E.H.; Bredesen, D.E. A second cytotoxic proteolytic peptide derived from amyloid beta-protein precursor. Nat. Med. 2000, 6, 397–404. [Google Scholar] [CrossRef]
- Park, S.A.; Shaked, G.M.; Bredesen, D.E.; Koo, E.H. Mechanism of cytotoxicity mediated by the C31 fragment of the amyloid precursor protein. Biochem. Biophys. Res. Commun. 2009, 388, 450–455. [Google Scholar] [CrossRef] [Green Version]
- Gonçalves, A.V.; Margolis, S.R.; Quirino, G.F.; Mascarenhas, D.P.; Rauch, I.; Nichols, R.D.; Ansaldo, E.; Fontana, M.F.; Vance, R.E.; Zamboni, D.S. Gasdermin-D and Caspase-7 are the key Caspase-1/8 substrates downstream of the NAIP5/NLRC4 inflammasome required for restriction of Legionella pneumophila. PLoS Pathog. 2019, 15, e1007886. [Google Scholar] [CrossRef]
- Ayers, K.L.; Mirshahi, U.L.; Wardeh, A.H.; Murray, M.F.; Hao, K.; Glicksberg, B.S.; Li, S.; Carey, D.J.; Chen, R. A loss of function variant in CASP7 protects against Alzheimer’s disease in homozygous APOE epsilon4 allele carriers. BMC Genom. 2016, 17 (Suppl. 2), 445. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Zhu, C.; Beecham, G.; Vardarajan, B.N.; Ma, Y.; Lancour, D.; Farrell, J.J.; Chung, J.; Alzheimer’s Disease Sequencing, P.; Mayeux, R.; et al. A rare missense variant of CASP7 is associated with familial late-onset Alzheimer’s disease. Alzheimers Dement 2019, 15, 441–452. [Google Scholar] [CrossRef]
- Muzio, M.; Stockwell, B.R.; Stennicke, H.R.; Salvesen, G.S.; Dixit, V.M. An induced proximity model for caspase-8 activation. J. Biol. Chem. 1998, 273, 2926–2930. [Google Scholar] [CrossRef] [Green Version]
- Stennicke, H.R.; Jurgensmeier, J.M.; Shin, H.; Deveraux, Q.; Wolf, B.B.; Yang, X.; Zhou, Q.; Ellerby, H.M.; Ellerby, L.M.; Bredesen, D.; et al. Pro-caspase-3 is a major physiologic target of caspase-8. J. Biol. Chem. 1998, 273, 27084–27090. [Google Scholar] [CrossRef] [Green Version]
- Rohn, T.T.; Head, E.; Nesse, W.H.; Cotman, C.W.; Cribbs, D.H. Activation of caspase-8 in the Alzheimer’s disease brain. Neurobiol. Dis. 2001, 8, 1006–1016. [Google Scholar] [CrossRef] [Green Version]
- Rehker, J.; Rodhe, J.; Nesbitt, R.R.; Boyle, E.A.; Martin, B.K.; Lord, J.; Karaca, I.; Naj, A.; Jessen, F.; Helisalmi, S.; et al. Caspase-8, association with Alzheimer’s Disease and functional analysis of rare variants. PLoS ONE 2017, 12, e0185777. [Google Scholar]
- O’Brien, R.J.; Xu, D.; Petralia, R.S.; Steward, O.; Huganir, R.L.; Worley, P. Synaptic clustering of AMPA receptors by the extracellular immediate-early gene product Narp. Neuron 1999, 23, 309–323. [Google Scholar] [CrossRef] [Green Version]
- Allen, N.J.; Bennett, M.L.; Foo, L.C.; Wang, G.X.; Chakraborty, C.; Smith, S.J.; Barres, B.A. Astrocyte glypicans 4 and 6 promote formation of excitatory synapses via GluA1 AMPA receptors. Nature 2012, 486, 410–414. [Google Scholar] [CrossRef]
- Groc, L.; Gustafsson, B.; Hanse, E. AMPA signalling in nascent glutamatergic synapses: There and not there! Trends Neurosci. 2006, 29, 132–139. [Google Scholar] [CrossRef]
- Farhy-Tselnicker, I.; van Casteren, A.C.; Lee, A.; Chang, V.T.; Aricescu, A.R.; Allen, N.J. Astrocyte-secreted glypican 4 regulates release of neuronal pentraxin 1 from axons to induce functional synapse formation. Neuron 2017, 96, 428–445.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeGregorio-Rocasolano, N.; Gasull, T.; Trullas, R. Overexpression of neuronal pentraxin 1 is involved in neuronal death evoked by low K+ in cerebellar granule cells. J. Biol. Chem. 2001, 276, 796–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Enguita, M.; DeGregorio-Rocasolano, N.; Abad, A.; Trullas, R. Glycogen synthase kinase 3 activity mediates neuronal pentraxin 1 expression and cell death induced by potassium deprivation in cerebellar granule cells. Mol. Pharmacol. 2005, 67, 1237–1246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abad, M.A.; Enguita, M.; De Gregorio-Rocasolano, N.; Ferrer, I.; Trullas, R. Neuronal pentraxin 1 contributes to the neuronal damage evoked by amyloid-β and is overexpressed in dystrophic neurites in Alzheimer’s brain. J. Neurosci. 2006, 26, 12735–12747. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Vicente, M.; Sovak, G.; Cuervo, A.M. Protein degradation and aging. Exp. Gerontol. 2005, 40, 622–633. [Google Scholar] [CrossRef] [PubMed]
- Gafni, J.; Ellerby, L.M. Calpain activation in Huntington’s disease. J. Neurosci. 2002, 22, 4842–4849. [Google Scholar] [CrossRef]
- Gafni, J.; Hermel, E.; Young, J.E.; Wellington, C.L.; Hayden, M.R.; Ellerby, L.M. Inhibition of calpain cleavage of huntingtin reduces toxicity: Accumulation of calpain/caspase fragments in the nucleus. J. Biol. Chem. 2004, 279, 20211–20220. [Google Scholar] [CrossRef] [Green Version]
- Anderson, J.P.; Walker, D.E.; Goldstein, J.M.; De Laat, R.; Banducci, K.; Caccavello, R.J.; Barbour, R.; Huang, J.; Kling, K.; Lee, M. Phosphorylation of Ser-129 is the dominant pathological modification of α-synuclein in familial and sporadic Lewy body disease. J. Biol. Chem. 2006, 281, 29739–29752. [Google Scholar] [CrossRef] [Green Version]
- Berke, S.J.S.; Schmied, F.A.F.; Brunt, E.R.; Ellerby, L.M.; Paulson, H.L. Caspase-mediated proteolysis of the polyglutamine disease protein ataxin-3. J. Neurochem. 2004, 89, 908–918. [Google Scholar] [CrossRef]
- Goti, D.; Katzen, S.M.; Mez, J.; Kurtis, N.; Kiluk, J.; Ben-Haïem, L.; Jenkins, N.A.; Copeland, N.G.; Kakizuka, A.; Sharp, A.H. A mutant ataxin-3 putative-cleavage fragment in brains of Machado-Joseph disease patients and transgenic mice is cytotoxic above a critical concentration. J. Neurosci. 2004, 24, 10266–10279. [Google Scholar] [CrossRef]
- Forloni, G.; Angeretti, N.; Chiesa, R.; Monzani, E.; Salmona, M.; Bugiani, O.; Tagliavini, F. Neurotoxicity of a prion protein fragment. Nature 1993, 362, 543–546. [Google Scholar] [CrossRef] [PubMed]
- Soto, C. Unfolding the role of protein misfolding in neurodegenerative diseases. Nat. Rev. Neurosci. 2003, 4, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Novak, M.; Kabat, J.; Wischik, C.M. Molecular characterization of the minimal protease resistant tau unit of the Alzheimer’s disease paired helical filament. EMBO J. 1993, 12, 365–370. [Google Scholar] [CrossRef] [PubMed]
- Wischik, C.M.; Novak, M.; Edwards, P.C.; Klug, A.; Tichelaar, W.; Crowther, R.A. Structural characterization of the core of the paired helical filament of Alzheimer disease. Proc. Natl. Acad. Sci. USA 1988, 85, 4884–4888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wischik, C.M.; Novak, M.; Thogersen, H.C.; Edwards, P.C.; Runswick, M.J.; Jakes, R.; Walker, J.E.; Milstein, C.; Roth, M.; Klug, A. Isolation of a fragment of tau derived from the core of the paired helical filament of Alzheimer disease. Proc. Natl. Acad. Sci. USA 1988, 85, 4506–4510. [Google Scholar] [CrossRef] [Green Version]
- Quinn, J.P.; Corbett, N.J.; Kellett, K.A.; Hooper, N.M. Tau proteolysis in the pathogenesis of tauopathies: Neurotoxic fragments and novel biomarkers. J. Alzheimer’s Dis. 2018, 63, 13–33. [Google Scholar] [CrossRef] [Green Version]
- Zilka, N.; Filipcik, P.; Koson, P.; Fialova, L.; Skrabana, R.; Zilkova, M.; Rolkova, G.; Kontsekova, E.; Novak, M. Truncated tau from sporadic Alzheimer’s disease suffices to drive neurofibrillary degeneration in vivo. FEBS Lett. 2006, 580, 3582–3588. [Google Scholar] [CrossRef] [Green Version]
- Igaz, L.M.; Kwong, L.K.; Chen-Plotkin, A.; Winton, M.J.; Unger, T.L.; Xu, Y.; Neumann, M.; Trojanowski, J.Q.; Lee, V.M. Expression of TDP-43 C-terminal Fragments in Vitro Recapitulates Pathological Features of TDP-43 Proteinopathies. J. Biol. Chem. 2009, 284, 8516–8524. [Google Scholar] [CrossRef] [Green Version]
- Filipcik, P.; Zilka, N.; Bugos, O.; Kucerak, J.; Koson, P.; Novak, P.; Novak, M. First transgenic rat model developing progressive cortical neurofibrillary tangles. Neurobiol. Aging 2012, 33, 1448–1456. [Google Scholar] [CrossRef]
- Tennstaedt, A.; Popsel, S.; Truebestein, L.; Hauske, P.; Brockmann, A.; Schmidt, N.; Irle, I.; Sacca, B.; Niemeyer, C.M.; Brandt, R.; et al. Human high temperature requirement serine protease A1 (HTRA1) degrades tau protein aggregates. J. Biol. Chem. 2012, 287, 20931–20941. [Google Scholar] [CrossRef] [Green Version]
- Glading, A.; Bodnar, R.; Reynolds, I.; Shiraha, H.; Satish, L.; Potter, D.; Blair, H.; Wells, A. Epidermal growth factor activates m-calpain (calpain II), at least in part, by extracellular signal-regulated kinase-mediated phosphorylation. Mol. Cell. Biol. 2004, 24, 2499–2512. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.S.; Ksiezak-Reding, H. Calpain-induced proteolysis of normal human tau and tau associated with paired helical filaments. Eur. J. Biochem. 1995, 233, 9–17. [Google Scholar] [CrossRef]
- Baudry, M.; Bi, X. Calpain-1 and Calpain-2: The Yin and Yang of Synaptic Plasticity and Neurodegeneration. Trends Neurosci 2016, 39, 235–245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurbatskaya, K.; Phillips, E.C.; Croft, C.L.; Dentoni, G.; Hughes, M.M.; Wade, M.A.; Al-Sarraj, S.; Troakes, C.; O’Neill, M.J.; Perez-Nievas, B.G.; et al. Upregulation of calpain activity precedes tau phosphorylation and loss of synaptic proteins in Alzheimer’s disease brain. Acta Neuropathol. Commun. 2016, 4, 34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, S.Y.; Ferreira, A. The generation of a 17 kDa neurotoxic fragment: An alternative mechanism by which tau mediates beta-amyloid-induced neurodegeneration. J. Neurosci. 2005, 25, 5365–5375. [Google Scholar] [CrossRef] [PubMed]
- Garg, S.; Timm, T.; Mandelkow, E.M.; Mandelkow, E.; Wang, Y. Cleavage of Tau by calpain in Alzheimer’s disease: The quest for the toxic 17 kD fragment. Neurobiol. Aging 2011, 32, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, S.E.; Motoi, Y.; Ishiguro, K.; Tabira, T.; Kametani, F.; Hasegawa, M.; Hattori, N. The twenty-four KDa C-terminal tau fragment increases with aging in tauopathy mice: Implications of prion-like properties. Hum. Mol. Genet. 2015, 24, 6403–6416. [Google Scholar] [CrossRef] [Green Version]
- Cicognola, C.; Satir, T.M.; Brinkmalm, G.; Matecko-Burmann, I.; Agholme, L.; Bergstrom, P.; Becker, B.; Zetterberg, H.; Blennow, K.; Hoglund, K. Tauopathy-Associated Tau Fragment Ending at Amino Acid 224 Is Generated by Calpain-2 Cleavage. J. Alzheimers Dis. 2020, 74, 1143–1156. [Google Scholar] [CrossRef]
- Cicognola, C.; Brinkmalm, G.; Wahlgren, J.; Portelius, E.; Gobom, J.; Cullen, N.C.; Hansson, O.; Parnetti, L.; Constantinescu, R.; Wildsmith, K.; et al. Novel tau fragments in cerebrospinal fluid: Relation to tangle pathology and cognitive decline in Alzheimer’s disease. Acta Neuropathol. 2019, 137, 279–296. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Song, M.; Liu, X.; Kang, S.S.; Kwon, I.S.; Duong, D.M.; Seyfried, N.T.; Hu, W.T.; Liu, Z.; Wang, J.Z.; et al. Cleavage of tau by asparagine endopeptidase mediates the neurofibrillary pathology in Alzheimer’s disease. Nat. Med. 2014, 20, 1254–1262. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.-H.; Liu, P.; Liu, X.; Manfredsson, F.P.; Sandoval, I.M.; Yu, S.P.; Wang, J.-Z.; Ye, K. Delta-secretase phosphorylation by SRPK2 enhances its enzymatic activity, provoking pathogenesis in Alzheimer’s disease. Mol. Cell 2017, 67, 812–825.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, P.; Joberty, G.; Buist, A.; Vanoosthuyse, A.; Stancu, I.C.; Vasconcelos, B.; Pierrot, N.; Faelth-Savitski, M.; Kienlen-Campard, P.; Octave, J.N.; et al. Tau interactome mapping based identification of Otub1 as Tau deubiquitinase involved in accumulation of pathological Tau forms in vitro and in vivo. Acta Neuropathol. 2017, 133, 731–749. [Google Scholar] [CrossRef] [Green Version]
- Dall, E.; Brandstetter, H. Structure and function of legumain in health and disease. Biochimie 2016, 122, 126–150. [Google Scholar] [CrossRef] [Green Version]
- Basurto-Islas, G.; Grundke-Iqbal, I.; Tung, Y.C.; Liu, F.; Iqbal, K. Activation of asparaginyl endopeptidase leads to Tau hyperphosphorylation in Alzheimer disease. J. Biol. Chem. 2013, 288, 17495–17507. [Google Scholar] [CrossRef] [Green Version]
- Harrigan, J.A.; Jacq, X.; Martin, N.M.; Jackson, S.P. Deubiquitylating enzymes and drug discovery: Emerging opportunities. Nat. Rev. Drug Discov. 2018, 17, 57–78. [Google Scholar] [CrossRef]
- Kontaxi, C.; Piccardo, P.; Gill, A.C. Lysine-Directed Post-translational Modifications of Tau Protein in Alzheimer’s Disease and Related Tauopathies. Front. Mol. Biosci. 2017, 4, 56. [Google Scholar] [CrossRef]
- Letronne, F.; Laumet, G.; Ayral, A.M.; Chapuis, J.; Demiautte, F.; Laga, M.; Vandenberghe, M.E.; Malmanche, N.; Leroux, F.; Eysert, F.; et al. ADAM30 Downregulates APP-Linked Defects Through Cathepsin D Activation in Alzheimer’s Disease. EBioMedicine 2016, 9, 278–292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khurana, V.; Elson-Schwab, I.; Fulga, T.A.; Sharp, K.A.; Loewen, C.A.; Mulkearns, E.; Tyynelä, J.; Scherzer, C.R.; Feany, M.B. Lysosomal dysfunction promotes cleavage and neurotoxicity of tau in vivo. PLoS Genet. 2010, 6, e1001026. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Egberts, F.; Heinrich, M.; Jensen, J.M.; Winoto-Morbach, S.; Pfeiffer, S.; Wickel, M.; Schunck, M.; Steude, J.; Saftig, P.; Proksch, E.; et al. Cathepsin D is involved in the regulation of transglutaminase 1 and epidermal differentiation. J. Cell Sci. 2004, 117 Pt 11, 2295–2307. [Google Scholar] [CrossRef] [Green Version]
- Banay-Schwartz, M.; Bracco, F.; DeGuzman, T.; Lajtha, A. Developmental changes in the breakdown of brain tubulin by cerebral cathepsin D. Neurochem. Res. 1983, 8, 51–61. [Google Scholar] [CrossRef] [PubMed]
- Banay-Schwartz, M.; Dahl, D.; Hui, K.S.; Lajtha, A. The breakdown of the individual neurofilament proteins by cathepsin D. Neurochem. Res. 1987, 12, 361–367. [Google Scholar] [CrossRef]
- Woessner, J.F., Jr.; Shamberger, R.J., Jr. Purification and properties of cathepsin D from bovine uterus. J. Biol. Chem. 1971, 246, 1951–1960. [Google Scholar] [CrossRef]
- Wolf, M.; Clark-Lewis, I.; Buri, C.; Langen, H.; Lis, M.; Mazzucchelli, L. Cathepsin D specifically cleaves the chemokines macrophage inflammatory protein-1 alpha, macrophage inflammatory protein-1 beta, and SLC that are expressed in human breast cancer. Am. J. Pathol. 2003, 162, 1183–1190. [Google Scholar] [CrossRef]
- Hiraiwa, M.; Martin, B.M.; Kishimoto, Y.; Conner, G.E.; Tsuji, S.; O’Brien, J.S. Lysosomal proteolysis of prosaposin, the precursor of saposins (sphingolipid activator proteins): Its mechanism and inhibition by ganglioside. Arch. Biochem. Biophys. 1997, 341, 17–24. [Google Scholar] [CrossRef] [PubMed]
- Kenessey, A.; Nacharaju, P.; Ko, L.W.; Yen, S.H. Degradation of tau by lysosomal enzyme cathepsin D: Implication for Alzheimer neurofibrillary degeneration. J. Neurochem. 1997, 69, 2026–2038. [Google Scholar] [CrossRef] [PubMed]
- Benuck, M.; Marks, N.; Hashim, G.A. Metabolic instability of myelin proteins. Breakdown of basic protein induced by brain cathepsin D. Eur. J. Biochem. 1975, 52, 615–621. [Google Scholar] [CrossRef]
- Sadik, G.; Kaji, H.; Takeda, K.; Yamagata, F.; Kameoka, Y.; Hashimoto, K.; Miyanaga, K.; Shinoda, T. In vitro processing of amyloid precursor protein by cathepsin D. Int. J. Biochem. Cell Biol. 1999, 31, 1327–1337. [Google Scholar] [CrossRef]
- Heinrich, M.; Neumeyer, J.; Jakob, M.; Hallas, C.; Tchikov, V.; Winoto-Morbach, S.; Wickel, M.; Schneider-Brachert, W.; Trauzold, A.; Hethke, A.; et al. Cathepsin D links TNF-induced acid sphingomyelinase to Bid-mediated caspase-9 and -3 activation. Cell Death Differ 2004, 11, 550–563. [Google Scholar] [CrossRef] [Green Version]
- Papassotiropoulos, A.; Bagli, M.; Kurz, A.; Kornhuber, J.; Forstl, H.; Maier, W.; Pauls, J.; Lautenschlager, N.; Heun, R. A genetic variation of cathepsin D is a major risk factor for Alzheimer’s disease. Ann. Neurol. 2000, 47, 399–403. [Google Scholar] [CrossRef]
- Poepsel, S.; Sprengel, A.; Sacca, B.; Kaschani, F.; Kaiser, M.; Gatsogiannis, C.; Raunser, S.; Clausen, T.; Ehrmann, M. Determinants of amyloid fibril degradation by the PDZ protease HTRA1. Nat. Chem. Biol. 2015, 11, 862–869. [Google Scholar] [CrossRef] [PubMed]
- Arai, T.; Guo, J.P.; McGeer, P.L. Proteolysis of non-phosphorylated and phosphorylated tau by thrombin. J. Biol. Chem. 2005, 280, 5145–5153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marshall, R.S.; Vierstra, R.D. Dynamic regulation of the 26S proteasome: From synthesis to degradation. Front. Mol. Biosci. 2019, 6, 40. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.Y.; Liu, S.J.; Li, H.L.; Wang, J.Z. Microtubule-associated protein tau is a substrate of ATP/Mg(2+)-dependent proteasome protease system. J. Neural. Transm. 2005, 112, 547–555. [Google Scholar] [CrossRef]
- Kanayama, H.O.; Tamura, T.; Ugai, S.; Kagawa, S.; Tanahashi, N.; Yoshimura, T.; Tanaka, K.; Ichihara, A. Demonstration that a human 26S proteolytic complex consists of a proteasome and multiple associated protein components and hydrolyzes ATP and ubiquitin-ligated proteins by closely linked mechanisms. Eur. J. Biochem. 1992, 206, 567–578. [Google Scholar] [CrossRef]
- Voges, D.; Zwickl, P.; Baumeister, W. The 26S proteasome: A molecular machine designed for controlled proteolysis. Annu. Rev. Biochem. 1999, 68, 1015–1068. [Google Scholar] [CrossRef]
- Bard, J.A.M.; Goodall, E.A.; Greene, E.R.; Jonsson, E.; Dong, K.C.; Martin, A. Structure and Function of the 26S Proteasome. Annu. Rev. Biochem. 2018, 87, 697–724. [Google Scholar] [CrossRef]
- Borodovsky, A.; Kessler, B.M.; Casagrande, R.; Overkleeft, H.S.; Wilkinson, K.D.; Ploegh, H.L. A novel active site-directed probe specific for deubiquitylating enzymes reveals proteasome association of USP14. EMBO J. 2001, 20, 5187–5196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, B.-H.; Lu, Y.; Prado, M.A.; Shi, Y.; Tian, G.; Sun, S.; Elsasser, S.; Gygi, S.P.; King, R.W.; Finley, D. USP14 deubiquitinates proteasome-bound substrates that are ubiquitinated at multiple sites. Nature 2016, 532, 398–401. [Google Scholar] [CrossRef] [PubMed]
- Keck, S.; Nitsch, R.; Grune, T.; Ullrich, O. Proteasome inhibition by paired helical filament-tau in brains of patients with Alzheimer’s disease. J. Neurochem. 2003, 85, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Myeku, N.; Clelland, C.L.; Emrani, S.; Kukushkin, N.V.; Yu, W.H.; Goldberg, A.L.; Duff, K.E. Tau-driven 26S proteasome impairment and cognitive dysfunction can be prevented early in disease by activating cAMP-PKA signaling. Nat. Med. 2016, 22, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Thibaudeau, T.A.; Anderson, R.T.; Smith, D.M. A common mechanism of proteasome impairment by neurodegenerative disease-associated oligomers. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef]
- Sengupta, S.; Horowitz, P.M.; Karsten, S.L.; Jackson, G.R.; Geschwind, D.H.; Fu, Y.; Berry, R.W.; Binder, L.I. Degradation of tau protein by puromycin-sensitive aminopeptidase in vitro. Biochemistry 2006, 45, 15111–15119. [Google Scholar] [CrossRef]
- Karsten, S.L.; Sang, T.K.; Gehman, L.T.; Chatterjee, S.; Liu, J.; Lawless, G.M.; Sengupta, S.; Berry, R.W.; Pomakian, J.; Oh, H.S.; et al. A genomic screen for modifiers of tauopathy identifies puromycin-sensitive aminopeptidase as an inhibitor of tau-induced neurodegeneration. Neuron 2006, 51, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Zhou, R.; Zhou, Q.; Guo, X.; Yan, C.; Ke, M.; Lei, J.; Shi, Y. Structural basis of Notch recognition by human gamma-secretase. Nature 2019, 565, 192–197. [Google Scholar] [CrossRef]
- Zhou, R.; Yang, G.; Guo, X.; Zhou, Q.; Lei, J.; Shi, Y. Recognition of the amyloid precursor protein by human gamma-secretase. Science 2019, 363, eaaw0930. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.; Cai, F.; Wu, Y.; Bozorgmehr, T.; Wang, Z.; Zhang, S.; Huang, D.; Guo, J.; Shen, L.; Rankin, C.; et al. A presenilin-1 mutation causes Alzheimer disease without affecting Notch signaling. Mol. Psychiatry 2020, 25, 603–613. [Google Scholar] [CrossRef] [PubMed]
- Russo, C.; Schettini, G.; Saido, T.C.; Hulette, C.; Lippa, C.; Lannfelt, L.; Ghetti, B.; Gambetti, P.; Tabaton, M.; Teller, J.K. Presenilin-1 mutations in Alzheimer’s disease. Nature 2000, 405, 531–532. [Google Scholar] [CrossRef] [PubMed]
- Takashima, A.; Murayama, M.; Murayama, O.; Kohno, T.; Honda, T.; Yasutake, K.; Nihonmatsu, N.; Mercken, M.; Yamaguchi, H.; Sugihara, S.; et al. Presenilin 1 associates with glycogen synthase kinase-3beta and its substrate tau. Proc. Natl. Acad. Sci. USA 1998, 95, 9637–9641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kelleher, R.J., 3rd; Shen, J. Presenilin-1 mutations and Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, 629–631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon-Weeks, P.R.; Fournier, A.E. Neuronal cytoskeleton in synaptic plasticity and regeneration. J. Neurochem. 2014, 129, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, A.Y.; Wang, X.; Schwarz, T.L. Axonal transport and the delivery of pre-synaptic components. Curr. Opin. Neurobiol. 2008, 18, 495–503. [Google Scholar] [CrossRef] [Green Version]
- Witte, H.; Bradke, F. The role of the cytoskeleton during neuronal polarization. Curr. Opin. Neurobiol. 2008, 18, 479–487. [Google Scholar] [CrossRef]
- Alonso, A.d.C.; Zaidi, T.; Novak, M.; Barra, H.S.; Grundke-Iqbal, I.; Iqbal, K. Interaction of tau isoforms with Alzheimer’s disease abnormally hyperphosphorylated tau and in VitroPhosphorylation into the disease-like protein. J. Biol. Chem. 2001, 276, 37967–37973. [Google Scholar] [CrossRef] [PubMed]
- Paonessa, F.; Evans, L.D.; Solanki, R.; Larrieu, D.; Wray, S.; Hardy, J.; Jackson, S.P.; Livesey, F.J. Microtubules deform the nuclear membrane and disrupt nucleocytoplasmic transport in tau-mediated frontotemporal dementia. Cell Rep. 2019, 26, 582–593.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seiberlich, V.; Goldbaum, O.; Zhukareva, V.; Richter-Landsberg, C. The small molecule inhibitor PR-619 of deubiquitinating enzymes affects the microtubule network and causes protein aggregate formation in neural cells: Implications for neurodegenerative diseases. Biochim. Biophys. Acta Mol. Cell Res. 2012, 1823, 2057–2068. [Google Scholar] [CrossRef] [Green Version]
- Souter, S.; Lee, G. Microtubule-associated protein tau in human prostate cancer cells: Isoforms, phosphorylation, and interactions. J. Cell. Biochem. 2009, 108, 555–564. [Google Scholar] [CrossRef] [Green Version]
- Lai, R.Y.; Harrington, C.R.; Wischik, C.M. Absence of a role for phosphorylation in the tau pathology of Alzheimer’s disease. Biomolecules 2016, 6, 19. [Google Scholar] [CrossRef] [Green Version]
- Alonso, A.d.C.; Grundke-Iqbal, I.; Barra, H.S.; Iqbal, K. Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration: Sequestration of microtubule-associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. Proc. Natl. Acad. Sci. USA 1997, 94, 298–303. [Google Scholar] [CrossRef] [Green Version]
- Harada, A.; Teng, J.; Takei, Y.; Oguchi, K.; Hirokawa, N. MAP2 is required for dendrite elongation, PKA anchoring in dendrites, and proper PKA signal transduction. J. Cell Biol. 2002, 158, 541–549. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Jang, Y.N.; Kim, J.Y.; Kim, N.; Noh, S.; Kim, H.; Queenan, B.N.; Bellmore, R.; Mun, J.Y.; Park, H. Microtubule-associated protein 2 mediates induction of long-term potentiation in hippocampal neurons. FASEB J. 2020, 34, 6965–6983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gumy, L.F.; Katrukha, E.A.; Grigoriev, I.; Jaarsma, D.; Kapitein, L.C.; Akhmanova, A.; Hoogenraad, C.C. MAP2 Defines a Pre-axonal Filtering Zone to Regulate KIF1- versus KIF5-Dependent Cargo Transport in Sensory Neurons. Neuron 2017, 94, 347–362.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brouhard, G.J.; Rice, L.M. Microtubule dynamics: An interplay of biochemistry and mechanics. Nat. Rev. Mol. Cell Biol. 2018, 19, 451–463. [Google Scholar] [CrossRef] [PubMed]
- Weisenberg, R.C.; Deery, W.J.; Dickinson, P.J. Tubulin-nucleotide interactions during the polymerization and depolymerization of microtubules. Biochemistry 1976, 15, 4248–4254. [Google Scholar] [CrossRef]
- Panda, D.; Goode, B.L.; Feinstein, S.C.; Wilson, L. Kinetic stabilization of microtubule dynamics at steady state by tau and microtubule-binding domains of tau. Biochemistry 1995, 34, 11117–11127. [Google Scholar] [CrossRef] [PubMed]
- Drechsel, D.N.; Hyman, A.; Cobb, M.H.; Kirschner, M. Modulation of the dynamic instability of tubulin assembly by the microtubule-associated protein tau. Mol. Biol. Cell 1992, 3, 1141–1154. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, I.; Wilde, A.; Mal, T.K.; Ikura, M. Structural basis for the activation of microtubule assembly by the EB1 and p150Glued complex. Mol. Cell 2005, 19, 449–460. [Google Scholar] [CrossRef]
- Go, C.D.; Knight, J.D.; Rajasekharan, A.; Rathod, B.; Hesketh, G.G.; Abe, K.T.; Youn, J.-Y.; Samavarchi-Tehrani, P.; Zhang, H.; Zhu, L.Y. A proximity-dependent biotinylation map of a human cell: An interactive web resource. bioRxiv 2021. [Google Scholar] [CrossRef] [Green Version]
- Askham, J.M.; Vaughan, K.T.; Goodson, H.V.; Morrison, E.E. Evidence that an interaction between EB1 and p150Glued is required for the formation and maintenance of a radial microtubule array anchored at the centrosome. Mol. Biol. Cell 2002, 13, 3627–3645. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Wu, J.; De Heus, C.; Grigoriev, I.; Liv, N.; Yao, Y.; Smal, I.; Meijering, E.; Klumperman, J.; Qi, R.Z. EB1 and EB3 regulate microtubule minus end organization and Golgi morphology. J. Cell Biol. 2017, 216, 3179–3198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez-Rios, S.; Denarier, E.; Prezel, E.; Vinit, A.; Stoppin-Mellet, V.; Devred, F.; Barbier, P.; Peyrot, V.; Sayas, C.L.; Avila, J. Tau antagonizes end-binding protein tracking at microtubule ends through a phosphorylation-dependent mechanism. Mol. Biol. Cell 2016, 27, 2924–2934. [Google Scholar] [CrossRef] [PubMed]
- Sayas, C.L.; Medina, M.; Cuadros, R.; Ollá, I.; García, E.; Pérez, M.; Ferrer, I.; Hernández, F.; Avila, J. Role of tau N-terminal motif in the secretion of human tau by End Binding proteins. PLoS ONE 2019, 14, e0210864. [Google Scholar] [CrossRef] [Green Version]
- Malki, I.; Cantrelle, F.X.; Sottejeau, Y.; Lippens, G.; Lambert, J.C.; Landrieu, I. Regulation of the interaction between the neuronal BIN 1 isoform 1 and Tau proteins–role of the SH 3 domain. FEBS J. 2017, 284, 3218–3229. [Google Scholar] [CrossRef] [Green Version]
- Chapuis, J.; Hansmannel, F.; Gistelinck, M.; Mounier, A.; Van Cauwenberghe, C.; Kolen, K.; Geller, F.; Sottejeau, Y.; Harold, D.; Dourlen, P. Increased expression of BIN1 mediates Alzheimer genetic risk by modulating tau pathology. Mol. Psychiatry 2013, 18, 1225–1234. [Google Scholar] [CrossRef] [Green Version]
- Wu, T.; Shi, Z.; Baumgart, T. Mutations in BIN1 associated with centronuclear myopathy disrupt membrane remodeling by affecting protein density and oligomerization. PLoS ONE 2014, 9, e93060. [Google Scholar] [CrossRef]
- Wechsler-Reya, R.; Sakamuro, D.; Zhang, J.; Duhadaway, J.; Prendergast, G.C. Structural analysis of the human BIN1 gene. Evidence for tissue-specific transcriptional regulation and alternate RNA splicing. J. Biol. Chem. 1997, 272, 31453–31458. [Google Scholar] [CrossRef] [Green Version]
- Seshadri, S.; Fitzpatrick, A.L.; Ikram, M.A.; DeStefano, A.L.; Gudnason, V.; Boada, M.; Bis, J.C.; Smith, A.V.; Carassquillo, M.M.; Lambert, J.C.; et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA 2010, 303, 1832–1840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kingwell, K. Alzheimer disease: BIN1 variant increases risk of Alzheimer disease through tau. Nat. Rev. Neurol. 2013, 9, 184. [Google Scholar] [CrossRef] [PubMed]
- Dharmalingam, E.; Haeckel, A.; Pinyol, R.; Schwintzer, L.; Koch, D.; Kessels, M.M.; Qualmann, B. F-BAR proteins of the syndapin family shape the plasma membrane and are crucial for neuromorphogenesis. J. Neurosci. 2009, 29, 13315–13327. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Navarro, M.V.; Peng, G.; Molinelli, E.; Goh, S.L.; Judson, B.L.; Rajashankar, K.R.; Sondermann, H. Molecular mechanism of membrane constriction and tubulation mediated by the F-BAR protein Pacsin/Syndapin. Proc. Natl. Acad. Sci. USA 2009, 106, 12700–12705. [Google Scholar] [CrossRef] [Green Version]
- Kessels, M.M.; Qualmann, B. Syndapins integrate N-WASP in receptor-mediated endocytosis. EMBO J. 2002, 21, 6083–6094. [Google Scholar] [CrossRef] [Green Version]
- Plomann, M.; Lange, R.; Vopper, G.; Cremer, H.; Heinlein, U.A.; Scheff, S.; Baldwin, S.A.; Leitges, M.; Cramer, M.; Paulsson, M.; et al. PACSIN, a brain protein that is upregulated upon differentiation into neuronal cells. Eur. J. Biochem. 1998, 256, 201–211. [Google Scholar] [CrossRef] [Green Version]
- Pérez-Otaño, I.; Luján, R.; Tavalin, S.J.; Plomann, M.; Modregger, J.; Liu, X.-B.; Jones, E.G.; Heinemann, S.F.; Lo, D.C.; Ehlers, M.D. Endocytosis and synaptic removal of NR3A-containing NMDA receptors by PACSIN1/syndapin1. Nat. Neurosci. 2006, 9, 611–621. [Google Scholar] [CrossRef] [Green Version]
- Carmignoto, G.; Vicini, S. Activity-dependent decrease in NMDA receptor responses during development of the visual cortex. Science 1992, 258, 1007–1011. [Google Scholar] [CrossRef] [PubMed]
- Hestrin, S. Developmental regulation of NMDA receptor-mediated synaptic currents at a central synapse. Nature 1992, 357, 686–689. [Google Scholar] [CrossRef] [PubMed]
- Sheng, M.; Cummings, J.; Roldan, L.A.; Jan, Y.N.; Jan, L.Y. Changing subunit composition of heteromeric NMDA receptors during development of rat cortex. Nature 1994, 368, 144–147. [Google Scholar] [CrossRef] [PubMed]
- Yamagishi, A.; Masuda, M.; Ohki, T.; Onishi, H.; Mochizuki, N. A novel actin bundling/filopodium-forming domain conserved in insulin receptor tyrosine kinase substrate p53 and missing in metastasis protein. J. Biol. Chem. 2004, 279, 14929–14936. [Google Scholar] [CrossRef] [Green Version]
- Scita, G.; Confalonieri, S.; Lappalainen, P.; Suetsugu, S. IRSp53: Crossing the road of membrane and actin dynamics in the formation of membrane protrusions. Trends Cell Biol. 2008, 18, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Kang, J.; Park, H.; Kim, E. IRSp53/BAIAP2 in dendritic spine development, NMDA receptor regulation, and psychiatric disorders. Neuropharmacology 2016, 100, 27–39. [Google Scholar] [CrossRef] [Green Version]
- Disanza, A.; Mantoani, S.; Hertzog, M.; Gerboth, S.; Frittoli, E.; Steffen, A.; Berhoerster, K.; Kreienkamp, H.-J.; Milanesi, F.; Di Fiore, P.P. Regulation of cell shape by Cdc42 is mediated by the synergic actin-bundling activity of the Eps8–IRSp53 complex. Nat. Cell Biol. 2006, 8, 1337–1347. [Google Scholar] [CrossRef]
- Okamura-Oho, Y.; Miyashita, T.; Yamada, M. Distinctive tissue distribution and phosphorylation of IRSp53 isoforms. Biochem. Biophys. Res. Commun. 2001, 289, 957–960. [Google Scholar] [CrossRef]
- Fujiwara, T.; Mammoto, A.; Kim, Y.; Takai, Y. Rho small G-protein-dependent binding of mDia to an Src homology 3 domain-containing IRSp53/BAIAP2. Biochem. Biophys. Res. Commun. 2000, 271, 626–629. [Google Scholar] [CrossRef]
- Choi, J.; Ko, J.; Racz, B.; Burette, A.; Lee, J.-R.; Kim, S.; Na, M.; Lee, H.W.; Kim, K.; Weinberg, R.J. Regulation of dendritic spine morphogenesis by insulin receptor substrate 53, a downstream effector of Rac1 and Cdc42 small GTPases. J. Neurosci. 2005, 25, 869–879. [Google Scholar] [CrossRef] [Green Version]
- Fromer, M.; Pocklington, A.J.; Kavanagh, D.H.; Williams, H.J.; Dwyer, S.; Gormley, P.; Georgieva, L.; Rees, E.; Palta, P.; Ruderfer, D.M. De novo mutations in schizophrenia implicate synaptic networks. Nature 2014, 506, 179–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Purcell, S.M.; Moran, J.L.; Fromer, M.; Ruderfer, D.; Solovieff, N.; Roussos, P.; O’dushlaine, C.; Chambert, K.; Bergen, S.E.; Kähler, A. A polygenic burden of rare disruptive mutations in schizophrenia. Nature 2014, 506, 185–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levy, D.; Ronemus, M.; Yamrom, B.; Lee, Y.-h.; Leotta, A.; Kendall, J.; Marks, S.; Lakshmi, B.; Pai, D.; Ye, K. Rare de novo and transmitted copy-number variation in autistic spectrum disorders. Neuron 2011, 70, 886–897. [Google Scholar] [CrossRef] [Green Version]
- Toma, C.; Hervás, A.; Balmaña, N.; Vilella, E.; Aguilera, F.; Cuscó, I.; del Campo, M.; Caballero, R.; De Diego-Otero, Y.; Ribasés, M. Association study of six candidate genes asymmetrically expressed in the two cerebral hemispheres suggests the involvement of BAIAP2 in autism. J. Psychiatr. Res. 2011, 45, 280–282. [Google Scholar] [CrossRef]
- Liu, L.; Sun, L.; Li, Z.-H.; Li, H.-M.; Wei, L.-P.; Wang, Y.-F.; Qian, Q.-J. BAIAP2 exhibits association to childhood ADHD especially predominantly inattentive subtype in Chinese Han subjects. Behav. Brain Funct. 2013, 9, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Herrmann, L.; Wiegmann, C.; Arsalan-Werner, A.; Hilbrich, I.; Jager, C.; Flach, K.; Suttkus, A.; Lachmann, I.; Arendt, T.; Holzer, M. Hook proteins: Association with Alzheimer pathology and regulatory role of hook3 in amyloid beta generation. PLoS ONE 2015, 10, e0119423. [Google Scholar] [CrossRef] [Green Version]
- Goitre, L.; Trapani, E.; Trabalzini, L.; Retta, S.F. The Ras superfamily of small GTPases: The unlocked secrets. Ras Signal. 2014, 1120, 1–18. [Google Scholar]
- Carazo-Salas, R.E.; Guarguaglini, G.; Gruss, O.J.; Segref, A.; Karsenti, E.; Mattaj, I.W. Generation of GTP-bound Ran by RCC1 is required for chromatin-induced mitotic spindle formation. Nature 1999, 400, 178–181. [Google Scholar] [CrossRef]
- Bao, X.; Liu, H.; Liu, X.; Ruan, K.; Zhang, Y.; Zhang, Z.; Hu, Q.; Liu, Y.; Akram, S.; Zhang, J. Mitosis-specific acetylation tunes Ran effector binding for chromosome segregation. J. Mol. Cell Biol. 2018, 10, 18–32. [Google Scholar] [CrossRef]
- Melchior, F.; Paschal, B.; Evans, J.; Gerace, L. Inhibition of nuclear protein import by nonhydrolyzable analogues of GTP and identification of the small GTPase Ran/TC4 as an essential transport factor. J. Cell Biol. 1993, 123, 1649–1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Görlich, D.; Pante, N.; Kutay, U.; Aebi, U.; Bischoff, F. Identification of different roles for RanGDP and RanGTP in nuclear protein import. EMBO J. 1996, 15, 5584–5594. [Google Scholar] [CrossRef] [Green Version]
- Rudack, T.; Jenrich, S.; Brucker, S.; Vetter, I.R.; Gerwert, K.; Kötting, C. Catalysis of GTP hydrolysis by small GTPases at atomic detail by integration of X-ray crystallography, experimental, and theoretical IR spectroscopy. J. Biol. Chem. 2015, 290, 24079–24090. [Google Scholar] [CrossRef] [Green Version]
- Fleming, L.M.; Weisgraber, K.H.; Strittmatter, W.J.; Troncoso, J.C.; Johnson, G.V. Differential binding of apolipoprotein E isoforms to tau and other cytoskeletal proteins. Exp. Neurol. 1996, 138, 252–260. [Google Scholar] [CrossRef]
- Yu, Y.; Kuang, Y.L.; Lei, D.; Zhai, X.; Zhang, M.; Krauss, R.M.; Ren, G. Polyhedral 3D structure of human plasma very low density lipoproteins by individual particle cryo-electron tomography1. J. Lipid Res. 2016, 57, 1879–1888. [Google Scholar] [CrossRef] [Green Version]
- Dawson, P.A.; Rudel, L.L. Intestinal cholesterol absorption. Curr Opin Lipidol 1999, 10, 315–320. [Google Scholar] [CrossRef]
- Otvos, J.D. Measurement of lipoprotein subclass profiles by nuclear magnetic resonance spectroscopy. Clin. Lab. 2002, 48, 171–180. [Google Scholar]
- Rall, S.C., Jr.; Weisgraber, K.H.; Mahley, R.W. Human apolipoprotein E. The complete amino acid sequence. J. Biol. Chem. 1982, 257, 4171–4178. [Google Scholar] [CrossRef]
- Heinsinger, N.M.; Gachechiladze, M.A.; Rebeck, G.W. Apolipoprotein E Genotype Affects Size of ApoE Complexes in Cerebrospinal Fluid. J. Neuropathol. Exp. Neurol. 2016, 75, 918–924. [Google Scholar] [CrossRef] [Green Version]
- Roses, A.D.; Saunders, A.M. APOE is a major susceptibility gene for Alzheimer’s disease. Curr. Opin. Biotechnol. 1994, 5, 663–667. [Google Scholar] [CrossRef]
- Strittmatter, W.J.; Saunders, A.M.; Goedert, M.; Weisgraber, K.H.; Dong, L.M.; Jakes, R.; Huang, D.Y.; Pericak-Vance, M.; Schmechel, D.; Roses, A.D. Isoform-specific interactions of apolipoprotein E with microtubule-associated protein tau: Implications for Alzheimer disease. Proc. Natl. Acad. Sci. USA 1994, 91, 11183–11186. [Google Scholar] [CrossRef] [Green Version]
- Bullido, M.J.; Aldudo, J.; Frank, A.; Coria, F.; Avila, J.; Valdivieso, F. A polymorphism in the tau gene associated with risk for Alzheimer’s disease. Neurosci. Lett. 2000, 278, 49–52. [Google Scholar] [CrossRef]
- Yu, L.; Boyle, P.A.; Leurgans, S.; Schneider, J.A.; Bennett, D.A. Disentangling the effects of age and APOE on neuropathology and late life cognitive decline. Neurobiol. Aging 2014, 35, 819–826. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhaenens, C.-M.; Van Brussel, E.; Schraen-Maschke, S.; Pasquier, F.; Delacourte, A.; Sablonnière, B. Association study of three polymorphisms of kinesin light-chain 1 gene with Alzheimer’s disease. Neurosci. Lett. 2004, 368, 290–292. [Google Scholar] [CrossRef] [PubMed]
- Rahman, A.; Friedman, D.S.; Goldstein, L.S. Two kinesin light chain genes in mice: Identification and characterization of the encoded proteins. J. Biol. Chem. 1998, 273, 15395–15403. [Google Scholar] [CrossRef] [Green Version]
- Konecna, A.; Frischknecht, R.; Kinter, J.; Ludwig, A.; Steuble, M.; Meskenaite, V.; Indermühle, M.; Engel, M.; Cen, C.; Mateos, J.-M. Calsyntenin-1 docks vesicular cargo to kinesin-1. Mol. Biol. Cell 2006, 17, 3651–3663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araki, Y.; Kawano, T.; Taru, H.; Saito, Y.; Wada, S.; Miyamoto, K.; Kobayashi, H.; Ishikawa, H.O.; Ohsugi, Y.; Yamamoto, T. The novel cargo Alcadein induces vesicle association of kinesin-1 motor components and activates axonal transport. EMBO J. 2007, 26, 1475–1486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vagnoni, A.; Perkinton, M.S.; Gray, E.H.; Francis, P.T.; Noble, W.; Miller, C.C. Calsyntenin-1 mediates axonal transport of the amyloid precursor protein and regulates Aβ production. Hum. Mol. Genet. 2012, 21, 2845–2854. [Google Scholar] [CrossRef] [Green Version]
- Vagnoni, A.; Rodriguez, L.; Manser, C.; De Vos, K.J.; Miller, C.C. Phosphorylation of kinesin light chain 1 at serine 460 modulates binding and trafficking of calsyntenin-1. J. Cell Sci. 2011, 124, 1032–1042. [Google Scholar] [CrossRef] [Green Version]
- Mórotz, G.M.; Glennon, E.B.; Greig, J.; Lau, D.H.; Bhembre, N.; Mattedi, F.; Muschalik, N.; Noble, W.; Vagnoni, A.; Miller, C.C. Kinesin light chain-1 serine-460 phosphorylation is altered in Alzheimer’s disease and regulates axonal transport and processing of the amyloid precursor protein. Acta Neuropathol. Commun. 2019, 7, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Morel, M.; Héraud, C.; Nicaise, C.; Suain, V.; Brion, J.-P. Levels of kinesin light chain and dynein intermediate chain are reduced in the frontal cortex in Alzheimer’s disease: Implications for axoplasmic transport. Acta Neuropathol. 2012, 123, 71–84. [Google Scholar] [CrossRef]
- Falzone, T.L.; Gunawardena, S.; McCleary, D.; Reis, G.F.; Goldstein, L.S. Kinesin-1 transport reductions enhance human tau hyperphosphorylation, aggregation and neurodegeneration in animal models of tauopathies. Hum. Mol. Genet. 2010, 19, 4399–4408. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroer, T.A. Dynactin. Annu. Rev. Cell Dev. Biol. 2004, 20, 759–779. [Google Scholar] [CrossRef] [PubMed]
- Ayloo, S.; Lazarus, J.E.; Dodda, A.; Tokito, M.; Ostap, E.M.; Holzbaur, E.L. Dynactin functions as both a dynamic tether and brake during dynein-driven motility. Nat. Commun. 2014, 5, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lill, R.; Nargang, F.E.; Neupert, W. Biogenesis of mitochondrial proteins. Curr. Opin. Cell Biol. 1996, 8, 505–512. [Google Scholar] [CrossRef] [Green Version]
- Chai, Y.L.; Xing, H.; Chong, J.R.; Francis, P.T.; Ballard, C.G.; Chen, C.P.; Lai, M.K. Mitochondrial translocase of the outer membrane alterations may underlie dysfunctional oxidative phosphorylation in Alzheimer’s disease. J. Alzheimer’s Dis. 2018, 61, 793–801. [Google Scholar] [CrossRef]
- Neupert, W.; Herrmann, J.M. Translocation of proteins into mitochondria. Annu. Rev. Biochem. 2007, 76, 723–749. [Google Scholar] [CrossRef] [Green Version]
- Petersen, C.A.H.; Alikhani, N.; Behbahani, H.; Wiehager, B.; Pavlov, P.F.; Alafuzoff, I.; Leinonen, V.; Ito, A.; Winblad, B.; Glaser, E. The amyloid β-peptide is imported into mitochondria via the TOM import machinery and localized to mitochondrial cristae. Proc. Natl. Acad. Sci. USA 2008, 105, 13145–13150. [Google Scholar] [CrossRef] [Green Version]
- Gawaz, M.; Douglas, M.; Klingenberg, M. Structure-function studies of adenine nucleotide transport in mitochondria. II. Biochemical analysis of distinct AAC1 and AAC2 proteins in yeast. J. Biol. Chem. 1990, 265, 14202–14208. [Google Scholar] [CrossRef]
- Krüger, J.; Hinttala, R.; Majamaa, K.; Remes, A.M. Mitochondrial DNA haplogroups in early-onset Alzheimer’s disease and frontotemporal lobar degeneration. Mol. Neurodegener. 2010, 5, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Copeland, W.C. Inherited mitochondrial diseases of DNA replication. Annu. Rev. Med. 2008, 59, 131–146. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, K.; Shimura, H.; Itaya, M.; Tanaka, R.; Mori, H.; Mizuno, Y.; Kosik, K.S.; Tanaka, S.; Hattori, N. Excitatory amino acid transporter 2 associates with phosphorylated tau and is localized in neurofibrillary tangles of tauopathic brains. FEBS Lett. 2009, 583, 2194–2200. [Google Scholar] [CrossRef] [Green Version]
- Benussi, L.; Ghidoni, R.; Paterlini, A.; Nicosia, F.; Alberici, A.C.; Signorini, S.; Barbiero, L.; Binetti, G. Interaction between tau and alpha-synuclein proteins is impaired in the presence of P301L tau mutation. Exp. Cell Res. 2005, 308, 78–84. [Google Scholar] [CrossRef]
- Cheng, F.; Vivacqua, G.; Yu, S. The role of alpha-synuclein in neurotransmission and synaptic plasticity. J. Chem. Neuroanat. 2011, 42, 242–248. [Google Scholar] [CrossRef]
- Pranke, I.M.; Morello, V.; Bigay, J.; Gibson, K.; Verbavatz, J.-M.; Antonny, B.; Jackson, C.L. α-Synuclein and ALPS motifs are membrane curvature sensors whose contrasting chemistry mediates selective vesicle binding. J. Cell Biol. 2011, 194, 89–103. [Google Scholar] [CrossRef] [Green Version]
- Jin, H.; Kanthasamy, A.; Ghosh, A.; Yang, Y.; Anantharam, V.; Kanthasamy, A.G. α-Synuclein negatively regulates protein kinase Cδ expression to suppress apoptosis in dopaminergic neurons by reducing p300 histone acetyltransferase activity. J. Neurosci. 2011, 31, 2035–2051. [Google Scholar] [CrossRef] [Green Version]
- Clayton, D.F.; George, J.M. Synucleins in synaptic plasticity and neurodegenerative disorders. J. Neurosci. Res. 1999, 58, 120–129. [Google Scholar] [CrossRef]
- Burré, J.; Sharma, M.; Tsetsenis, T.; Buchman, V.; Etherton, M.R.; Südhof, T.C. α-Synuclein promotes SNARE-complex assembly in vivo and in vitro. Science 2010, 329, 1663–1667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamilton, R.L. Lewy bodies in Alzheimer’s disease: A neuropathological review of 145 cases using α-synuclein immunohistochemistry. Brain Pathol. 2000, 10, 378–384. [Google Scholar] [CrossRef] [PubMed]
- Arai, Y.; Yamazaki, M.; Mori, O.; Muramatsu, H.; Asano, G.; Katayama, Y. α-Synuclein-positive structures in cases with sporadic Alzheimer’s disease: Morphology and its relationship to tau aggregation. Brain Res. 2001, 888, 287–296. [Google Scholar] [CrossRef]
- Giasson, B.I.; Forman, M.S.; Higuchi, M.; Golbe, L.I.; Graves, C.L.; Kotzbauer, P.T.; Trojanowski, J.Q.; Lee, V.M.-Y. Initiation and synergistic fibrillization of tau and alpha-synuclein. Science 2003, 300, 636–640. [Google Scholar] [CrossRef] [Green Version]
- Oikawa, T.; Nonaka, T.; Terada, M.; Tamaoka, A.; Hisanaga, S.-i.; Hasegawa, M. α-Synuclein fibrils exhibit gain of toxic function, promoting tau aggregation and inhibiting microtubule assembly. J. Biol. Chem. 2016, 291, 15046–15056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duka, T.; Duka, V.; Joyce, J.N.; Sidhu, A. α-Synuclein contributes to GSK-3β-catalyzed Tau phosphorylation in Parkinson’s disease models. FASEB J. 2009, 23, 2820–2830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doetsch, F.; Caille, I.; Lim, D.A.; Garcia-Verdugo, J.M.; Alvarez-Buylla, A. Subventricular zone astrocytes are neural stem cells in the adult mammalian brain. Cell 1999, 97, 703–716. [Google Scholar] [CrossRef] [Green Version]
- Seward, M.E.; Swanson, E.; Norambuena, A.; Reimann, A.; Cochran, J.N.; Li, R.; Roberson, E.D.; Bloom, G.S. Amyloid-β signals through tau to drive ectopic neuronal cell cycle re-entry in Alzheimer’s disease. J. Cell Sci. 2013, 126, 1278–1286. [Google Scholar] [CrossRef] [Green Version]
- Frost, B.; Hemberg, M.; Lewis, J.; Feany, M.B. Tau promotes neurodegeneration through global chromatin relaxation. Nat. Neurosci. 2014, 17, 357–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernández-Ortega, K.; Garcia-Esparcia, P.; Gil, L.; Lucas, J.J.; Ferrer, I. Altered machinery of protein synthesis in Alzheimer’s: From the nucleolus to the ribosome. Brain Pathol. 2016, 26, 593–605. [Google Scholar] [CrossRef]
- Barbato, C.; Corbi, N.; Canu, N.; Fanciulli, M.; Serafino, A.; Ciotti, M.; Libri, V.; Bruno, T.; Amadoro, G.; De Angelis, R.; et al. Rb binding protein Che-1 interacts with Tau in cerebellar granule neurons. Modulation during neuronal apoptosis. Mol. Cell Neurosci 2003, 24, 1038–1050. [Google Scholar] [CrossRef] [PubMed]
- Ueberham, U.; Rohn, S.; Ueberham, E.; Wodischeck, S.; Hilbrich, I.; Holzer, M.; Bruckner, M.K.; Gruschka, H.; Arendt, T. Pin1 promotes degradation of Smad proteins and their interaction with phosphorylated tau in Alzheimer’s disease. Neuropathol. Appl. Neurobiol. 2014, 40, 815–832. [Google Scholar] [CrossRef]
- Bruno, T.; De Angelis, R.; De Nicola, F.; Barbato, C.; Di Padova, M.; Corbi, N.; Libri, V.; Benassi, B.; Mattei, E.; Chersi, A.; et al. Che-1 affects cell growth by interfering with the recruitment of HDAC1 by Rb. Cancer Cell 2002, 2, 387–399. [Google Scholar] [CrossRef] [Green Version]
- Wrighton, K.H.; Lin, X.; Feng, X.H. Phospho-control of TGF-beta superfamily signaling. Cell Res. 2009, 19, 8–20. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.Y.; Sun, J.; Ji, C.M.; Shen, L.; Chen, Z.J.; Xie, P.; Sun, Y.Z.; Yu, R.T. Selective deletion of apolipoprotein E in astrocytes ameliorates the spatial learning and memory deficits in Alzheimer’s disease (APP/PS1) mice by inhibiting TGF-beta/Smad2/STAT3 signaling. Neurobiol. Aging 2017, 54, 112–132. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; He, W.; Gayen, M.; Benoit, M.R.; Luo, X.; Hu, X.; Yan, R. Activated CX3CL1/Smad2 Signals Prevent Neuronal Loss and Alzheimer’s Tau Pathology-Mediated Cognitive Dysfunction. J. Neurosci. 2020, 40, 1133–1144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baig, S.; van Helmond, Z.; Love, S. Tau hyperphosphorylation affects Smad 2/3 translocation. Neuroscience 2009, 163, 561–570. [Google Scholar] [CrossRef] [PubMed]
- Anderson, P.; Kedersha, N. Stress granules: The Tao of RNA triage. Trends Biochem. Sci. 2008, 33, 141–150. [Google Scholar] [CrossRef]
- Liu-Yesucevitz, L.; Bassell, G.J.; Gitler, A.D.; Hart, A.C.; Klann, E.; Richter, J.D.; Warren, S.T.; Wolozin, B. Local RNA translation at the synapse and in disease. J. Neurosci. 2011, 31, 16086–16093. [Google Scholar] [CrossRef] [Green Version]
- Bai, B.; Hales, C.M.; Chen, P.C.; Gozal, Y.; Dammer, E.B.; Fritz, J.J.; Wang, X.; Xia, Q.; Duong, D.M.; Street, C.; et al. U1 small nuclear ribonucleoprotein complex and RNA splicing alterations in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2013, 110, 16562–16567. [Google Scholar] [CrossRef] [Green Version]
- Berson, A.; Barbash, S.; Shaltiel, G.; Goll, Y.; Hanin, G.; Greenberg, D.S.; Ketzef, M.; Becker, A.J.; Friedman, A.; Soreq, H. Cholinergic-associated loss of hnRNP-A/B in Alzheimer’s disease impairs cortical splicing and cognitive function in mice. EMBO Mol. Med. 2012, 4, 730–742. [Google Scholar] [CrossRef]
- Bishof, I.; Dammer, E.B.; Duong, D.M.; Kundinger, S.R.; Gearing, M.; Lah, J.J.; Levey, A.I.; Seyfried, N.T. RNA-binding proteins with basic-acidic dipeptide (BAD) domains self-assemble and aggregate in Alzheimer’s disease. J. Biol. Chem. 2018, 293, 11047–11066. [Google Scholar] [CrossRef] [Green Version]
- Vanderweyde, T.; Apicco, D.J.; Youmans-Kidder, K.; Ash, P.E.A.; Cook, C.; Lummertz da Rocha, E.; Jansen-West, K.; Frame, A.A.; Citro, A.; Leszyk, J.D.; et al. Interaction of tau with the RNA-Binding Protein TIA1 Regulates tau Pathophysiology and Toxicity. Cell Rep. 2016, 15, 1455–1466. [Google Scholar] [CrossRef] [Green Version]
- Pomeranz Krummel, D.A.; Oubridge, C.; Leung, A.K.; Li, J.; Nagai, K. Crystal structure of human spliceosomal U1 snRNP at 5.5 A resolution. Nature 2009, 458, 475–480. [Google Scholar] [CrossRef]
- Bai, B.; Chen, P.C.; Hales, C.M.; Wu, Z.; Pagala, V.; High, A.A.; Levey, A.I.; Lah, J.J.; Peng, J. Integrated approaches for analyzing U1-70K cleavage in Alzheimer’s disease. J. Proteome Res. 2014, 13, 4526–4534. [Google Scholar] [CrossRef] [PubMed]
- Diner, I.; Hales, C.M.; Bishof, I.; Rabenold, L.; Duong, D.M.; Yi, H.; Laur, O.; Gearing, M.; Troncoso, J.; Thambisetty, M.; et al. Aggregation properties of the small nuclear ribonucleoprotein U1-70K in Alzheimer disease. J. Biol. Chem. 2014, 289, 35296–35313. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raj, T.; Li, Y.I.; Wong, G.; Humphrey, J.; Wang, M.; Ramdhani, S.; Wang, Y.C.; Ng, B.; Gupta, I.; Haroutunian, V.; et al. Integrative transcriptome analyses of the aging brain implicate altered splicing in Alzheimer’s disease susceptibility. Nat. Genet. 2018, 50, 1584–1592. [Google Scholar] [CrossRef] [PubMed]
- Del Gatto-Konczak, F.; Bourgeois, C.F.; Le Guiner, C.; Kister, L.; Gesnel, M.-C.; Stévenin, J.; Breathnach, R. The RNA-binding protein TIA-1 is a novel mammalian splicing regulator acting through intron sequences adjacent to a 5′ splice site. Mol. Cell. Biol. 2000, 20, 6287–6299. [Google Scholar] [CrossRef] [PubMed]
- Gilks, N.; Kedersha, N.; Ayodele, M.; Shen, L.; Stoecklin, G.; Dember, L.M.; Anderson, P. Stress granule assembly is mediated by prion-like aggregation of TIA-1. Mol. Biol. Cell 2004, 15, 5383–5398. [Google Scholar] [CrossRef] [Green Version]
- Vanderweyde, T.; Yu, H.; Varnum, M.; Liu-Yesucevitz, L.; Citro, A.; Ikezu, T.; Duff, K.; Wolozin, B. Contrasting pathology of the stress granule proteins TIA-1 and G3BP in tauopathies. J. Neurosci. 2012, 32, 8270–8283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, L.; Ash, P.E.A.; Maziuk, B.F.; Ballance, H.I.; Boudeau, S.; Abdullatif, A.A.; Orlando, M.; Petrucelli, L.; Ikezu, T.; Wolozin, B. TIA1 regulates the generation and response to toxic tau oligomers. Acta Neuropathol. 2019, 137, 259–277. [Google Scholar] [CrossRef] [Green Version]
- Apicco, D.J.; Ash, P.E.A.; Maziuk, B.; LeBlang, C.; Medalla, M.; Al Abdullatif, A.; Ferragud, A.; Botelho, E.; Ballance, H.I.; Dhawan, U.; et al. Reducing the RNA binding protein TIA1 protects against tau-mediated neurodegeneration in vivo. Nat. Neurosci. 2018, 21, 72–80. [Google Scholar] [CrossRef] [Green Version]
- McKerracher, L.; Chamoux, M.; Arregui, C.O. Role of laminin and integrin interactions in growth cone guidance. Mol. Neurobiol. 1996, 12, 95–116. [Google Scholar] [CrossRef]
- Teng, F.Y.; Tang, B.L. Axonal regeneration in adult CNS neurons-signaling molecules and pathways. J. Neurochem. 2006, 96, 1501–1508. [Google Scholar] [CrossRef]
- McKenna, N.J.; O’Malley, B.W. Combinatorial control of gene expression by nuclear receptors and coregulators. Cell 2002, 108, 465–474. [Google Scholar] [CrossRef] [Green Version]
- Ochsner, S.A.; Abraham, D.; Martin, K.; Ding, W.; McOwiti, A.; Kankanamge, W.; Wang, Z.; Andreano, K.; Hamilton, R.A.; Chen, Y.; et al. The Signaling Pathways Project, an integrated ‘omics knowledgebase for mammalian cellular signaling pathways. Sci. Data 2019, 6, 252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mielke, K.; Herdegen, T. JNK and p38 stresskinases—Degenerative effectors of signal-transduction-cascades in the nervous system. Prog. Neurobiol. 2000, 61, 45–60. [Google Scholar] [CrossRef]
- Berridge, M.J. Calcium hypothesis of Alzheimer’s disease. Pflug. Arch. 2010, 459, 441–449. [Google Scholar] [CrossRef]
- Cau, Y.; Valensin, D.; Mori, M.; Draghi, S.; Botta, M. Structure, function, involvement in diseases and targeting of 14-3-3 proteins: An update. Curr. Med. Chem. 2018, 25, 5–21. [Google Scholar] [CrossRef] [PubMed]
- Cornell, B.; Toyo-Oka, K. 14-3-3 proteins in brain development: Neurogenesis, neuronal migration and neuromorphogenesis. Front. Mol. Neurosci. 2017, 10, 318. [Google Scholar] [CrossRef] [Green Version]
- Foote, M.; Zhou, Y. 14-3-3 proteins in neurological disorders. Int. J. Biochem. Mol. Biol. 2012, 3, 152. [Google Scholar]
- Agarwal-Mawal, A.; Qureshi, H.Y.; Cafferty, P.W.; Yuan, Z.; Han, D.; Lin, R.; Paudel, H.K. 14-3-3 connects glycogen synthase kinase-3 beta to tau within a brain microtubule-associated tau phosphorylation complex. J. Biol. Chem. 2003, 278, 12722–12728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadik, G.; Tanaka, T.; Kato, K.; Yamamori, H.; Nessa, B.N.; Morihara, T.; Takeda, M. Phosphorylation of tau at Ser214 mediates its interaction with 14-3-3 protein: Implications for the mechanism of tau aggregation. J. Neurochem. 2009, 108, 33–43. [Google Scholar] [CrossRef]
- Rosenquist, M. 14-3-3 proteins in apoptosis. Braz. J. Med. Biol. Res. 2003, 36, 403–408. [Google Scholar] [CrossRef]
- Jang, S.W.; Liu, X.; Fu, H.; Rees, H.; Yepes, M.; Levey, A.; Ye, K. Interaction of Akt-phosphorylated SRPK2 with 14-3-3 mediates cell cycle and cell death in neurons. J. Biol. Chem. 2009, 284, 24512–24525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Chen, X.; Yao, Z.; Shi, Y.; Xiong, J.; Zhou, J.; Su, Z.; Huang, Y. 14-3-3/Tau interaction and Tau amyloidogenesis. J. Mol. Neurosci. 2019, 68, 620–630. [Google Scholar] [CrossRef]
- McShea, A.; Zelasko, D.A.; Gerst, J.L.; Smith, M.A. Signal transduction abnormalities in Alzheimer’s disease: Evidence of a pathogenic stimuli. Brain Res. 1999, 815, 237–242. [Google Scholar] [CrossRef]
- Venezia, V.; Russo, C.; Repetto, E.; Salis, S.; Dolcini, V.; Genova, F.; Nizzari, M.; Mueller, U.; Schettini, G. Apoptotic cell death influences the signaling activity of the amyloid precursor protein through ShcA and Grb2 adaptor proteins in neuroblastoma SH-SY5Y cells. J. Neurochem. 2004, 90, 1359–1370. [Google Scholar] [CrossRef]
- Roder, H.M.; Eden, P.A.; Ingram, V.M. Brain protein kinase PK40erk converts TAU into a PHF-like form as found in Alzheimer’s disease. Biochem. Biophys. Res. Commun. 1993, 193, 639–647. [Google Scholar] [CrossRef]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A. Tissue-based map of the human proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef]
- O’Brien, J.S.; Carson, G.S.; Seo, H.-C.; Hiraiwa, M.; Kishimoto, Y. Identification of prosaposin as a neurotrophic factor. Proc. Natl. Acad. Sci. USA 1994, 91, 9593–9596. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, J.L.; Ngo, T.; Schmidt, J.; Mrad, N.; Liew, C.K.; Jones, N.M.; Graham, R.M.; Smith, N.J. Metalloprotease cleavage of the N terminus of the orphan G protein–coupled receptor GPR37L1 reduces its constitutive activity. Sci. Signal. 2016, 9, ra36. [Google Scholar] [CrossRef]
- Vaz-Silva, J.; Gomes, P.; Jin, Q.; Zhu, M.; Zhuravleva, V.; Quintremil, S.; Meira, T.; Silva, J.; Dioli, C.; Soares-Cunha, C.; et al. Endolysosomal degradation of Tau and its role in glucocorticoid-driven hippocampal malfunction. EMBO J. 2018, 37, e99084. [Google Scholar] [CrossRef] [PubMed]
- Raiborg, C.; Stenmark, H. The ESCRT machinery in endosomal sorting of ubiquitylated membrane proteins. Nature 2009, 458, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Sheehan, P.; Zhu, M.; Beskow, A.; Vollmer, C.; Waites, C.L. Activity-Dependent Degradation of Synaptic Vesicle Proteins Requires Rab35 and the ESCRT Pathway. J. Neurosci. 2016, 36, 8668–8686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nixon, R.A.; Yang, D.-S. Autophagy failure in Alzheimer’s disease—Locating the primary defect. Neurobiol. Dis. 2011, 43, 38–45. [Google Scholar] [CrossRef] [Green Version]
- Green, K.N.; Billings, L.M.; Roozendaal, B.; McGaugh, J.L.; LaFerla, F.M. Glucocorticoids increase amyloid-β and tau pathology in a mouse model of Alzheimer’s disease. J. Neurosci. 2006, 26, 9047–9056. [Google Scholar] [CrossRef] [Green Version]
- Gireud-Goss, M.; Reyes, S.; Tewari, R.; Patrizz, A.; Howe, M.D.; Kofler, J.; Waxham, M.N.; McCullough, L.D.; Bean, A.J. The ubiquitin ligase UBE4B regulates amyloid precursor protein ubiquitination, endosomal trafficking, and amyloid β42 generation and secretion. Mol. Cell. Neurosci. 2020, 108, 103542. [Google Scholar] [CrossRef]
- Ittner, L.M.; Ke, Y.D.; Gotz, J. Phosphorylated Tau interacts with c-Jun N-terminal kinase-interacting protein 1 (JIP1) in Alzheimer disease. J. Biol. Chem. 2009, 284, 20909–20916. [Google Scholar] [CrossRef] [Green Version]
- Ozdemir, A.Y.; Rom, I.; Kovalevich, J.; Yen, W.; Adiga, R.; Dave, R.S.; Langford, D. PINCH in the cellular stress response to tau-hyperphosphorylation. PLoS ONE 2013, 8, e58232. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Bordoy, R.; Stanchi, F.; Moser, M.; Braun, A.; Kudlacek, O.; Wewer, U.M.; Yurchenco, P.D.; Fassler, R. PINCH1 regulates cell-matrix and cell-cell adhesions, cell polarity and cell survival during the peri-implantation stage. J. Cell Sci. 2005, 118 Pt 13, 2913–2921. [Google Scholar] [CrossRef] [Green Version]
- Rearden, A.; Hurford, R.; Luu, N.; Kieu, E.; Sandoval, M.; Perez-Liz, G.; Del Valle, L.; Powell, H.; Langford, T.D. Novel expression of PINCH in the central nervous system and its potential as a biomarker for human immunodeficiency virus-associated neurodegeneration. J. Neurosci. Res. 2008, 86, 2535–2542. [Google Scholar] [CrossRef]
- Jang, H.J.; Yang, Y.R.; Kim, J.K.; Choi, J.H.; Seo, Y.K.; Lee, Y.H.; Lee, J.E.; Ryu, S.H.; Suh, P.G. Phospholipase C-gamma1 involved in brain disorders. Adv. Biol. Regu.l 2013, 53, 51–62. [Google Scholar] [CrossRef] [PubMed]
- Kang, D.-S.; Kim, I.S.; Baik, J.-H.; Kim, D.; Cocco, L.; Suh, P.-G. The function of PLCγ1 in developing mouse mDA system. Adv. Biol. Regul. 2020, 75, 100654. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, H.; Campenot, R.B.; Vance, D.E.; Vance, J.E. Protection of neurons from apoptosis by apolipoprotein E-containing lipoproteins does not require lipoprotein uptake and involves activation of phospholipase Cgamma1 and inhibition of calcineurin. J. Biol. Chem. 2009, 284, 29605–29613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shimohama, S.; Matsushima, H.; Fujimoto, S.; Takenawa, T.; Taniguchi, T.; Kameyama, M.; Kimura, J. Differential involvement of phospholipase C isozymes in Alzheimer’s disease. Gerontology 1995, 41 (Suppl. 1), 13–19. [Google Scholar] [CrossRef]
- Hampel, H.; Ewers, M.; Bürger, K.; Annas, P.; Mörtberg, A.; Bogstedt, A.; Frölich, L.; Schröder, J.; Schönknecht, P.; Riepe, M.W. Lithium trial in Alzheimer’s disease: A randomized, single-blind, placebo-controlled, multicenter 10-week study. J. Clin. Psychiatry 2009, 70, 922–931. [Google Scholar] [CrossRef]
- Pagan, F.; Hebron, M.; Valadez, E.H.; Torres-Yaghi, Y.; Huang, X.; Mills, R.R.; Wilmarth, B.M.; Howard, H.; Dunn, C.; Carlson, A. Nilotinib effects in Parkinson’s disease and dementia with Lewy bodies. J. Parkinson’s Dis. 2016, 6, 503–517. [Google Scholar] [CrossRef] [Green Version]
- Nygaard, H.B.; Wagner, A.F.; Bowen, G.S.; Good, S.P.; MacAvoy, M.G.; Strittmatter, K.A.; Kaufman, A.C.; Rosenberg, B.J.; Sekine-Konno, T.; Varma, P. A phase Ib multiple ascending dose study of the safety, tolerability, and central nervous system availability of AZD0530 (saracatinib) in Alzheimer’s disease. Alzheimer’s Res. Ther. 2015, 7, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Dyck, C.H.; Nygaard, H.B.; Chen, K.; Donohue, M.C.; Raman, R.; Rissman, R.A.; Brewer, J.B.; Koeppe, R.A.; Chow, T.W.; Rafii, M.S. Effect of AZD0530 on cerebral metabolic decline in Alzheimer disease: A randomized clinical trial. JAMA Neurol. 2019, 76, 1219–1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Ser, T.; Steinwachs, K.C.; Gertz, H.J.; Andres, M.V.; Gomez-Carrillo, B.; Medina, M.; Vericat, J.A.; Redondo, P.; Fleet, D.; Leon, T. Treatment of Alzheimer’s disease with the GSK-3 inhibitor tideglusib: A pilot study. J. Alzheimer’s Dis. 2013, 33, 205–215. [Google Scholar] [CrossRef]
- Lovestone, S.; Boada, M.; Dubois, B.; Hüll, M.; Rinne, J.O.; Huppertz, H.-J.; Calero, M.; Andres, M.V.; Gómez-Carrillo, B.; Leon, T. A phase II trial of tideglusib in Alzheimer’s disease. J. Alzheimer’s Dis. 2015, 45, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Tolosa, E.; Litvan, I.; Höglinger, G.U.; Burn, D.; Lees, A.; Andrés, M.V.; Gómez-Carrillo, B.; León, T.; Del Ser, T.; Investigators, T. A phase 2 trial of the GSK-3 inhibitor tideglusib in progressive supranuclear palsy. Mov. Disord. 2014, 29, 470–478. [Google Scholar] [CrossRef] [PubMed]
- Höglinger, G.U.; Huppertz, H.J.; Wagenpfeil, S.; Andrés, M.V.; Belloch, V.; León, T.; Del Ser, T.; Investigators, T.M.; Gmez, J.; Tijero, B. Tideglusib reduces progression of brain atrophy in progressive supranuclear palsy in a randomized trial. Mov. Disord. 2014, 29, 479–487. [Google Scholar] [CrossRef] [PubMed]
- Tariot, P.N.; Schneider, L.S.; Cummings, J.; Thomas, R.G.; Raman, R.; Jakimovich, L.J.; Loy, R.; Bartocci, B.; Fleisher, A.; Ismail, M.S. Chronic divalproex sodium to attenuate agitation and clinical progression of Alzheimer disease. Arch. Gen. Psychiatry 2011, 68, 853–861. [Google Scholar] [CrossRef] [Green Version]
- Leclair-Visonneau, L.; Rouaud, T.; Debilly, B.; Durif, F.; Houeto, J.-L.; Kreisler, A.; Defebvre, L.; Lamy, E.; Volteau, C.; Nguyen, J.-M. Randomized placebo-controlled trial of sodium valproate in progressive supranuclear palsy. Clin. Neurol. Neurosurg. 2016, 146, 35–39. [Google Scholar] [CrossRef]
- Fleisher, A.; Truran, D.; Mai, J.; Langbaum, J.; Aisen, P.; Cummings, J.; Jack, C.; Weiner, M.; Thomas, R.; Schneider, L. Chronic divalproex sodium use and brain atrophy in Alzheimer disease. Neurology 2011, 77, 1263–1271. [Google Scholar] [CrossRef] [Green Version]
- Huisa, B.N.; Thomas, R.G.; Jin, S.; Oltersdorf, T.; Taylor, C.; Feldman, H.H. Memantine and acetylcholinesterase inhibitor use in Alzheimer’s disease clinical trials: Potential for confounding by indication. J. Alzheimer’s Dis. 2019, 67, 707–713. [Google Scholar] [CrossRef]
- Malpas, C.B.; Vivash, L.; Genc, S.; Saling, M.M.; Desmond, P.; Steward, C.; Hicks, R.J.; Callahan, J.; Brodtmann, A.; Collins, S. A phase IIa randomized control trial of VEL015 (Sodium Selenate) in mild-moderate Alzheimer’s disease. J. Alzheimer’s Dis. 2016, 54, 223–232. [Google Scholar] [CrossRef]
- VandeVrede, L.; Dale, M.L.; Fields, S.; Frank, M.; Hare, E.; Heuer, H.W.; Keith, K.; Koestler, M.; Ljubenkov, P.A.; McDermott, D. Open-Label Phase 1 Futility Studies of Salsalate and Young Plasma in Progressive Supranuclear Palsy. Mov. Disord. Clin. Pract. 2020, 7, 440–447. [Google Scholar] [CrossRef]
- Peng, Y.; Hu, Y.; Xu, S.; Li, P.; Li, J.; Lu, L.; Yang, H.; Feng, N.; Wang, L.; Wang, X. L-3-n-butylphthalide reduces tau phosphorylation and improves cognitive deficits in AβPP/PS1-Alzheimer’s transgenic mice. J. Alzheimer’s Dis. 2012, 29, 379–391. [Google Scholar] [CrossRef]
- Rohn, T.T.; Kokoulina, P.; Eaton, C.R.; Poon, W.W. Caspase activation in transgenic mice with Alzheimer-like pathology: Results from a pilot study utilizing the caspase inhibitor, Q-VD-OPh. Int. J. Clin. Exp. Med. 2009, 2, 300. [Google Scholar]
- Flores, J.; Noël, A.; Foveau, B.; Beauchet, O.; LeBlanc, A.C. Pre-symptomatic Caspase-1 inhibitor delays cognitive decline in a mouse model of Alzheimer disease and aging. Nat. Commun. 2020, 11, 1–14. [Google Scholar] [CrossRef]
- Flores, J.; Noël, A.; Foveau, B.; Lynham, J.; Lecrux, C.; LeBlanc, A.C. Caspase-1 inhibition alleviates cognitive impairment and neuropathology in an Alzheimer’s disease mouse model. Nat. Commun. 2018, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Selenica, M.-L.; Benner, L.; Housley, S.B.; Manchec, B.; Lee, D.C.; Nash, K.R.; Kalin, J.; Bergman, J.A.; Kozikowski, A.; Gordon, M.N. Histone deacetylase 6 inhibition improves memory and reduces total tau levels in a mouse model of tau deposition. Alzheimer’s Res. Ther. 2014, 6, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fan, S.-J.; Huang, F.-I.; Liou, J.-P.; Yang, C.-R. The novel histone de acetylase 6 inhibitor, MPT0G211, ameliorates tau phosphorylation and cognitive deficits in an Alzheimer’s disease model. Cell Death Dis. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-Y.; Fan, S.-J.; Huang, F.-I.; Chao, H.-Y.; Hsu, K.-C.; Lin, T.E.; Yeh, T.-K.; Lai, M.-J.; Li, Y.-H.; Huang, H.-L. 5-Aroylindoles act as selective histone deacetylase 6 inhibitors ameliorating Alzheimer’s disease phenotypes. J. Med. Chem. 2018, 61, 7087–7102. [Google Scholar] [CrossRef] [PubMed]
- Onishi, T.; Maeda, R.; Terada, M.; Sato, S.; Fujii, T.; Ito, M.; Hashikami, K.; Kawamoto, T.; Tanaka, M. A novel orally active HDAC6 inhibitor T-518 shows a therapeutic potential for Alzheimer’s disease and tauopathy in mice. Sci. Rep. 2021, 11, 1–11. [Google Scholar] [CrossRef]
- Dickey, C.A.; Kamal, A.; Lundgren, K.; Klosak, N.; Bailey, R.M.; Dunmore, J.; Ash, P.; Shoraka, S.; Zlatkovic, J.; Eckman, C.B. The high-affinity HSP90-CHIP complex recognizes and selectively degrades phosphorylated tau client proteins. J. Clin. Investig. 2007, 117, 648–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miyata, Y.; Li, X.; Lee, H.-F.; Jinwal, U.K.; Srinivasan, S.R.; Seguin, S.P.; Young, Z.T.; Brodsky, J.L.; Dickey, C.A.; Sun, D. Synthesis and initial evaluation of YM-08, a blood-brain barrier permeable derivative of the heat shock protein 70 (Hsp70) inhibitor MKT-077, which reduces tau levels. ACS Chem. Neurosci. 2013, 4, 930–939. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abisambra, J.; Jinwal, U.K.; Miyata, Y.; Rogers, J.; Blair, L.; Li, X.; Seguin, S.P.; Wang, L.; Jin, Y.; Bacon, J. Allosteric heat shock protein 70 inhibitors rapidly rescue synaptic plasticity deficits by reducing aberrant tau. Biol. Psychiatry 2013, 74, 367–374. [Google Scholar] [CrossRef] [Green Version]
- Hardy, J.A.; Higgins, G.A. Alzheimer’s disease: The amyloid cascade hypothesis. Science 1992, 256, 184–186. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- van der Kant, R.; Langness, V.F.; Herrera, C.M.; Williams, D.A.; Fong, L.K.; Leestemaker, Y.; Steenvoorden, E.; Rynearson, K.D.; Brouwers, J.F.; Helms, J.B. Cholesterol metabolism is a druggable axis that independently regulates tau and amyloid-β in iPSC-derived Alzheimer’s disease neurons. Cell Stem Cell 2019, 24, 363–375.e9. [Google Scholar] [CrossRef] [Green Version]
- Young, J.E.; Fong, L.K.; Frankowski, H.; Petsko, G.A.; Small, S.A.; Goldstein, L.S. Stabilizing the retromer complex in a human stem cell model of Alzheimer’s disease reduces TAU phosphorylation independently of amyloid precursor protein. Stem Cell Rep. 2018, 10, 1046–1058. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhaskar, K.; Konerth, M.; Kokiko-Cochran, O.N.; Cardona, A.; Ransohoff, R.M.; Lamb, B.T. Regulation of tau pathology by the microglial fractalkine receptor. Neuron 2010, 68, 19–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, K.A.; Schultz, A.; Betensky, R.A.; Becker, J.A.; Sepulcre, J.; Rentz, D.; Mormino, E.; Chhatwal, J.; Amariglio, R.; Papp, K. Tau positron emission tomographic imaging in aging and early A lzheimer disease. Ann. Neurol. 2016, 79, 110–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schöll, M.; Lockhart, S.N.; Schonhaut, D.R.; O’Neil, J.P.; Janabi, M.; Ossenkoppele, R.; Baker, S.L.; Vogel, J.W.; Faria, J.; Schwimmer, H.D. PET imaging of tau deposition in the aging human brain. Neuron 2016, 89, 971–982. [Google Scholar] [CrossRef] [Green Version]
- Grothe, M.J.; Barthel, H.; Sepulcre, J.; Dyrba, M.; Sabri, O.; Teipel, S.J. Alzheimer’s Disease Neuroimaging Initiative. In vivo staging of regional amyloid deposition. Neurology 2017, 89, 2031–2038. [Google Scholar] [CrossRef] [Green Version]
- Jansen, W.J.; Ossenkoppele, R.; Knol, D.L.; Tijms, B.M.; Scheltens, P.; Verhey, F.R.; Visser, P.J.; Aalten, P.; Aarsland, D.; Alcolea, D. Prevalence of cerebral amyloid pathology in persons without dementia: A meta-analysis. JAMA 2015, 313, 1924–1938. [Google Scholar] [CrossRef]
- Gonzalez, M.W.; Kann, M.G. Chapter 4: Protein interactions and disease. PLoS Comput. Biol. 2012, 8, e1002819. [Google Scholar] [CrossRef] [Green Version]
- Morris, M.; Maeda, S.; Vossel, K.; Mucke, L. The many faces of tau. Neuron 2011, 70, 410–426. [Google Scholar] [CrossRef] [Green Version]





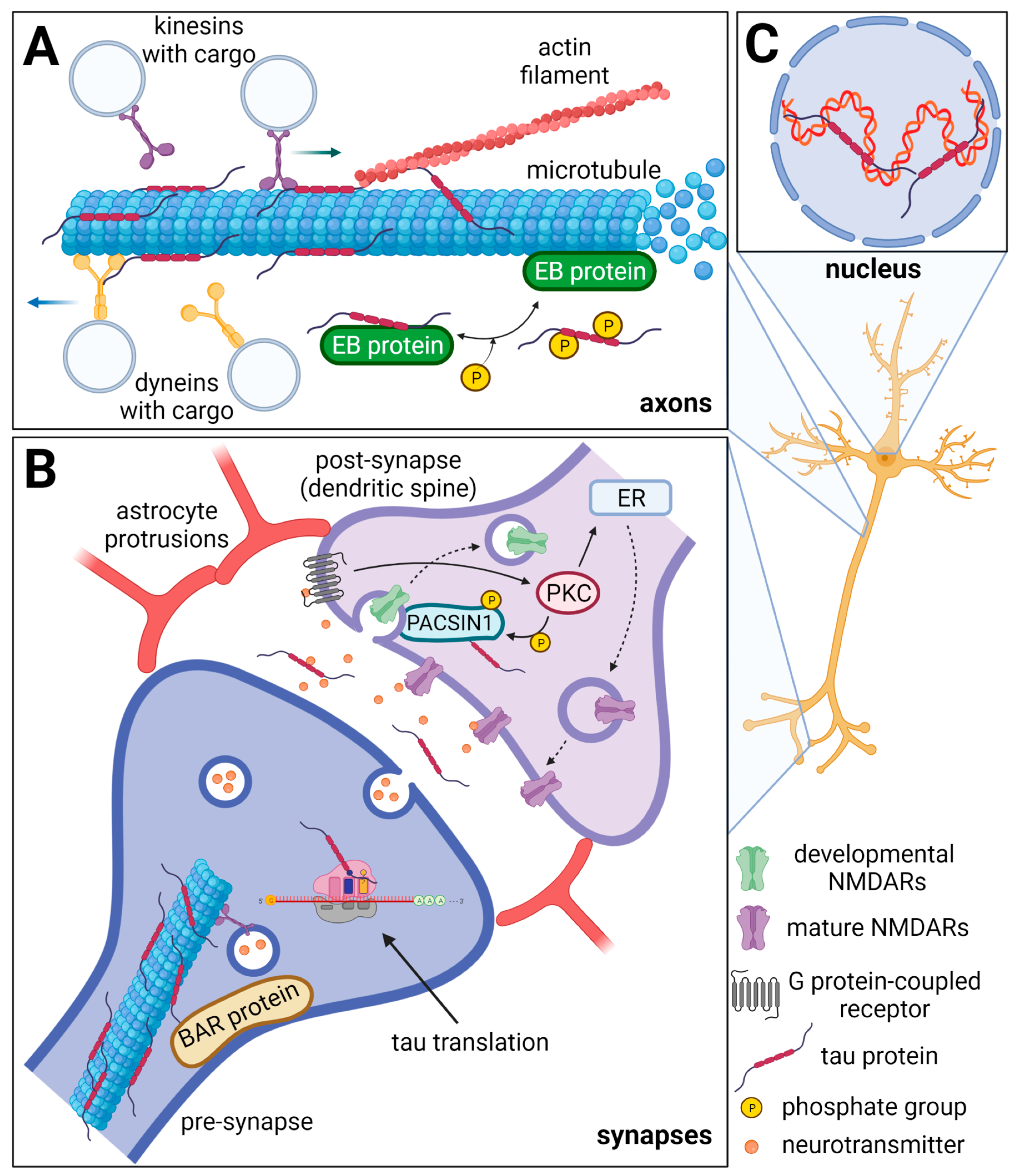{kind=link}
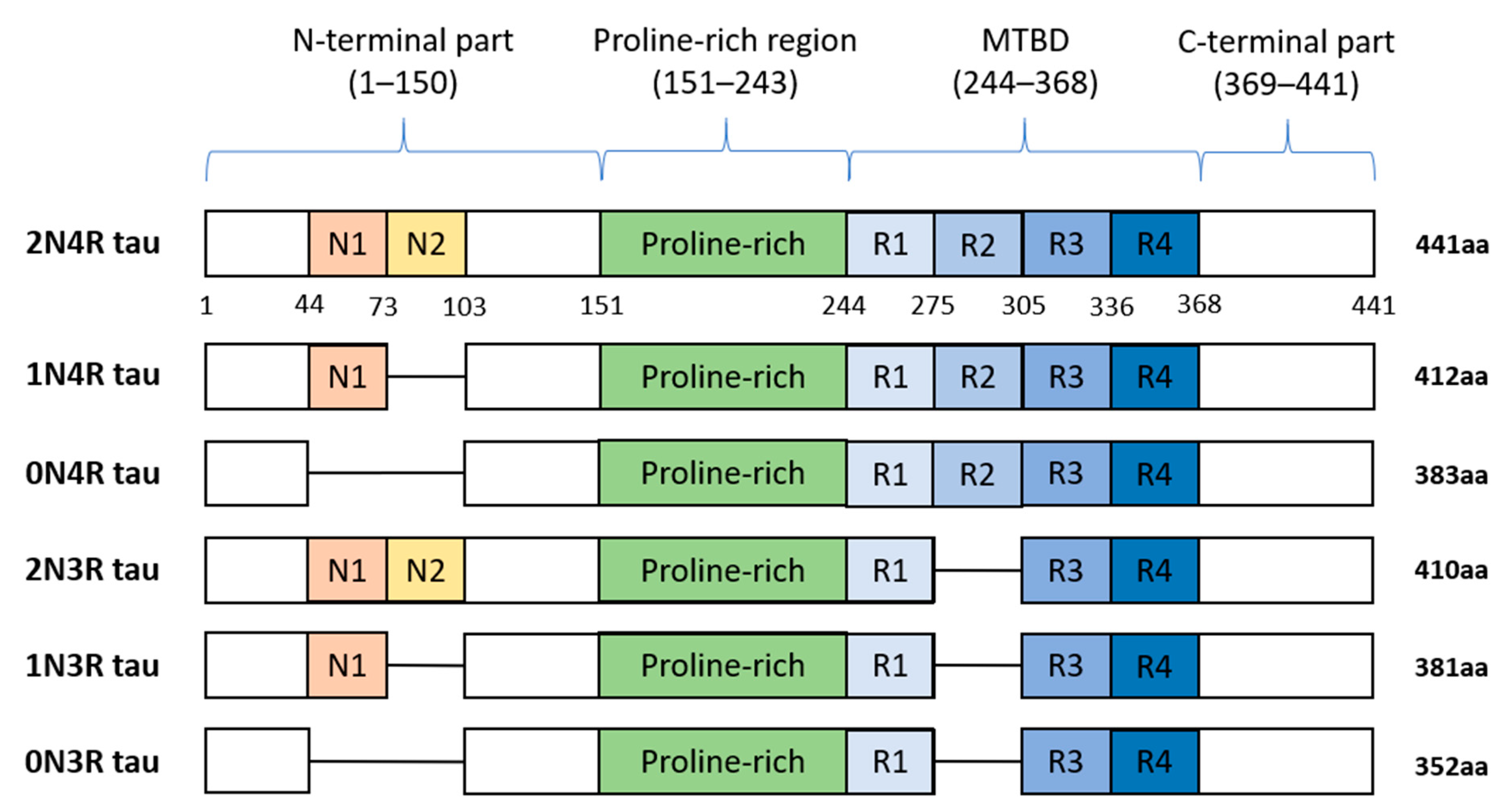{kind=link}
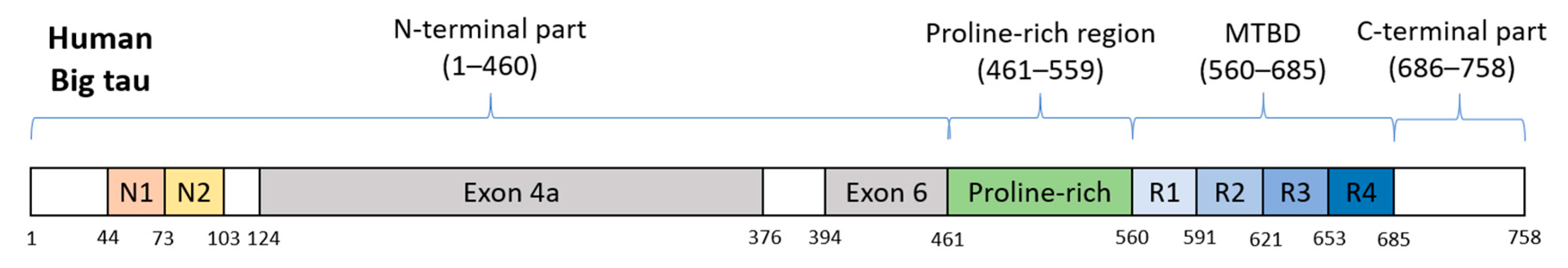{kind=link}
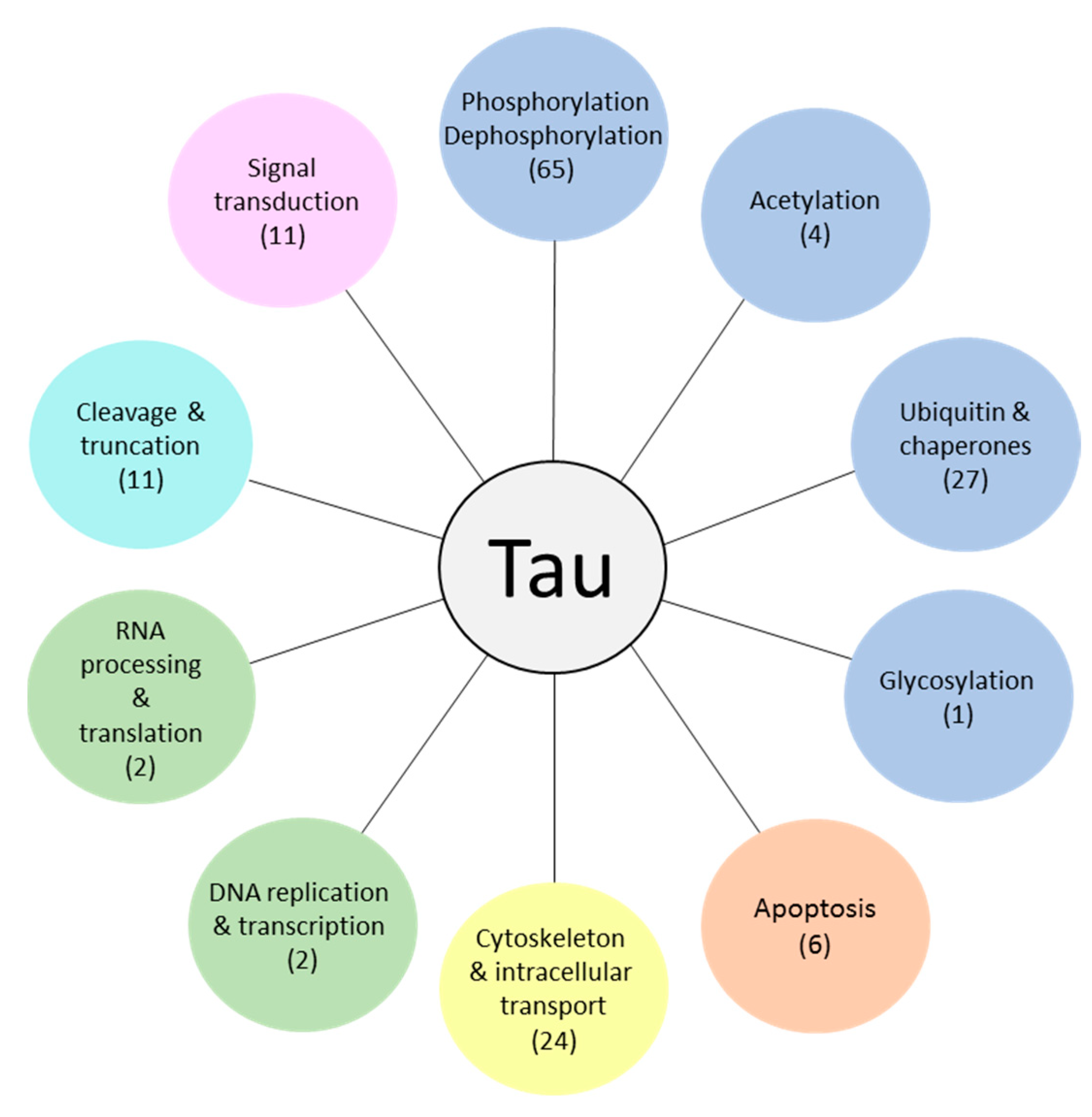{kind=link}
| Category | Proteins/Substances | Reference |
|---|---|---|
| Tubulin interacting proteins | microtubule-associated protein 2 | [122] |
| microtubule-associated protein 1B | [123,124] | |
| neurofilament proteins, vimentin | [125] | |
| heparansulfate proteoglycans | [126,127] | |
| amyloid precursor protein | [128,129] | |
| Kinases and other cytosol enzymes | casein kinase II | [130] |
| extracellular signal-related kinase-2 | [131] | |
| glycogen synthase kinase-3 | [132] | |
| phospholipase C-δ | [133] | |
| Stress molecules | advanced glycation end products | [134,135,136] |
| malondialdehyde | [137] | |
| heme oxygenase-1 | [136] | |
| Amyloid and amyloid-binding proteins | complement proteins (Clq, C3, C4d and C5b-9) | [138,139] |
| vitronectin | [140] | |
| apolipoprotein-E | [141,142] | |
| Others | C-series of gangliosides | [124] |
| ubiquitin and ubiquitin ligases | [143,144,145] | |
| synaptophysin | [146] | |
| anti-thrombin III | [147] | |
| lactotransferrin | [148] |
| Protein | Roles in Tau Pathology | References |
|---|---|---|
| ABL1 | involved in formation of apoptotic complex | [172] |
| AKT1 | neuroprotective activity; inactivates pro-apoptotic proteins and promotes cell survival | [173] |
| BRSK (1, 2) | unknown role in tau pathology | - |
| CAMK2A | involved in hyperphosphorylation of tau in AD | [174,175] |
| CDK (1, 5) | accelerate tau hyperphosphorylation and formation of NFT; act neuroprotective by apoptosis inhibition | [176,177] |
| CDK2 | inhibits tau-mediated tubulin polymerization by tau phosphorylation | [178] |
| CSNK1 (A1, D) | involved in formation of Aβ; colocalize with NFT, suggesting a role in tau aggregation | [179,180] |
| DYRK1A | overexpressed in AD; facilitates further tau phosphorylation by GSK3B | [181,182] |
| DYRK2 | overexpressed in AD; involved in activation of apoptosis | [181,183] |
| FYN | facilitates formation of NFT by tau hyperphosphorylation; activates PTK2B; potentiates Aβ-induced synapse damage | [184,185,186] |
| GSK3 (A, B) | responsible for hyperphosphorylation of tau; involved in activation of apoptosis; exacerbate tau pathology when activated by Aβ | [187,188,189] |
| CHEK (1, 2) | unknown role in tau pathology | - |
| LCK | it is downregulated in AD; unknown role in tau pathology | [190,191] |
| LRRK2 | contradictory reports: enhances abnormal tau phosphorylation and promote tauopathy or, no impact on tau pathology was shown | [192,193,194,195] |
| MAP2K7 | colocalize with hyperphosphorylated tau; contribute to Aβ accumulation | [196,197] |
| MAPK (1, 3, 8–14) | overactivated in AD; involved in abnormal tau phosphorylation | [198,199] |
| MARK (1–3) | phosphorylate tau aggregates; colocalizes with NFT | [200] |
| MARK4 | phosphorylate tau aggregates; mutation causes tau hyperphosphorylation, aggregation and tau-mediated neurodegeneration; colocalizes with NFT | [200,201] |
| PHKG1 | unknown role in tau pathology | - |
| PKN1 | regulates tau phosphorylation; colocalize with NFT | [202,203] |
| PRKACA | abnormally phosphorylate α-Synuclein-bound tau; facilitates further phosphorylation of tau by GSK3 kinase; colocalize with NFT; important for regulation of alternative splicing of exon 10 in MAPT | [204,205,206,207] |
| PRKC (A, B, E, G, I, Z) | neuroprotective role through inhibition of GSK3 kinase and reduced tau phosphorylation | [208,209] |
| PTK2B | hyperphosphorylates tau; is regulated by FYN kinase; colocalize with pathological tau proteins in brains of AD patients | [185,210] |
| RPS6K (A1, A3, A5, B1) | their attenuation is neuroprotective; possibly upregulate tau translation; contribute to tau aggregation and synapse damage; inhibit apoptosis | [211,212,213] |
| SGK1 | upregulated in AD; enhances tau hyperphosphorylation by GSK3B activation | [214] |
| SIK1 | unknown role in tau pathology | - |
| SRC | unknown role in tau pathology | - |
| SRPK (1, 2) | involved in regulation of alternative splicing of exon 10 in MAPT, where impaired splicing of exon 10 can lead to tauopathy | [215,216] |
| SYK | accumulation of pathological tau proteins cause overactivation of SYK, which further hyperphosphorylates tau and exacerbate pathology; contributes to the neuroinflammation; inhibits autophagic tau degradation; contributes to neuronal loss | [217,218,219] |
| TTBK1 | enhances tau phosphorylation and aggregation; involved in neurodegeneration | [220,221] |
| Protein | Roles in Tau Pathology | References |
|---|---|---|
| PPP1C (A, B, C) | downregulated in AD; neuroprotective function by reducing hyperphosphorylated tau levels; | [170,222] |
| PPP2C (A, B) | downregulated in AD what induces tau hyperphosphorylation; neuroprotective; regulate autophagic degradation of proteins | [170,222,223,224] |
| PPP5C | downregulated in AD what contributes to increased levels of hyperphosphorylated tau; dephosphorylates AD phospho-tau; protects against Aβ toxicity | [225,226,227] |
| PTPN11 | upregulated in AD; unknown role in tau pathology | [228] |
| Protein | Roles in Tau Pathology | References |
|---|---|---|
| PTPA | regulatory subunit of PPP2C (A, B); decreased expression in AD; enhances tau dephosphorylation; neuroprotective; regulate autophagic degradation of proteins | [223,224,229,230] |
| CDC37 | regulates tau phosphorylation, enhances phospho-tau stability at HSP90 scaffold, preventing its degradation, preserves CDK5 and AKT kinases | [231] |
| PIK3R1 | key unit of PI3K, part of the PI3K/AKT1/GSK3B signaling pathway, which is downregulated in AD, plays a role in insulin signaling pathway where it might exacerbate AD pathology | [232,233,234,235] |
| S100B | inhibits tau phosphorylation; enhances tau dephosphorylation by PPP5C; overexpression of S100B causes apoptosis, tau hyperphosphorylation, and inflammation | [236,237,238,239,240] |
| Name | Function | Phase (Disease) | Efficacy | Source |
|---|---|---|---|---|
| Dasatinib | Ab1 and Src kinase inhibitor | Phase I and II | Recruiting | NCT04063124 |
| Epigallocatechin gallate | DYRK1A kinase inhibitor | Phase II (AD) | No results posted | NCT00951834 |
| Lithium chloride | GSK-3 kinase inhibitor | Phase II (AD/PSP) | No effect | NCT00088387 [605] |
| Nilotinib | Ab1 kinase inhibitor | Phase II (AD/PD) | Unknown | NCT02947893 [606] |
| Saracatinib | Ab1, Src kinase inhibitor | Phase II (AD) | No effect | NCT02167256 [607,608] |
| Tideglusib | GSK-3 kinase inhibitor | Phase II (AD/PSP) | No effect | NCT01350362 [609,610,611,612] |
| Valproate | GSK-3 kinase inhibitor | Phase III (AD) | No effect | NCT00071721 [613,614,615] |
| Memantine | PPP2CA activator | Phase IV (AD) | No effect | NCT00469456 [616] |
| Sodium selenate | PPP2CA activator | Phase II (AD) | Small positive effects | [617] |
| Salsalate | Acetylation inhibitor | Phase I (AD/PSP) | Ongoing | NCT03277573 [618] |
| Minocycline | CDK5 kinase/caspase-3 inhibitor | Phase II (AD) | No effect | NCT01463384 |
| Nicotinamide | HDAC inhibitor | Phase II | Recruiting | NCT03061474 |
| Phenylbutyrate | HDAC inhibitor | Phase II | Ongoing | NCT03533257 |
| Vorinostat | HDAC inhibitor | Phase I | Recruiting | NCT03056495 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sinsky, J.; Pichlerova, K.; Hanes, J. Tau Protein Interaction Partners and Their Roles in Alzheimer’s Disease and Other Tauopathies. Int. J. Mol. Sci. 2021, 22, 9207. https://doi.org/10.3390/ijms22179207
Sinsky J, Pichlerova K, Hanes J. Tau Protein Interaction Partners and Their Roles in Alzheimer’s Disease and Other Tauopathies. International Journal of Molecular Sciences. 2021; 22(17):9207. https://doi.org/10.3390/ijms22179207
Chicago/Turabian StyleSinsky, Jakub, Karoline Pichlerova, and Jozef Hanes. 2021. "Tau Protein Interaction Partners and Their Roles in Alzheimer’s Disease and Other Tauopathies" International Journal of Molecular Sciences 22, no. 17: 9207. https://doi.org/10.3390/ijms22179207