
The Interplay between Autophagy and NLRP3 Inflammasome in Ischemia/Reperfusion Injury


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
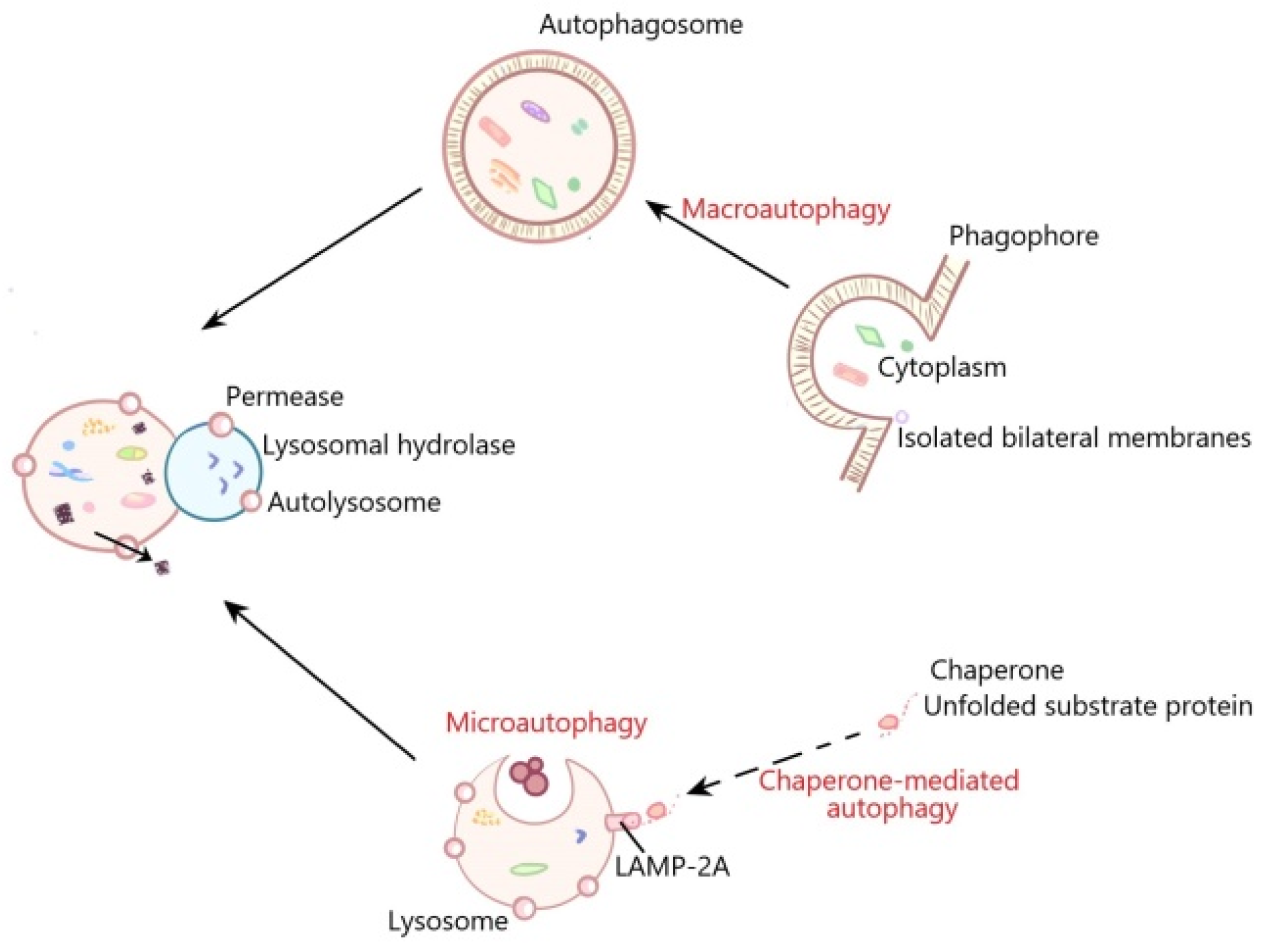
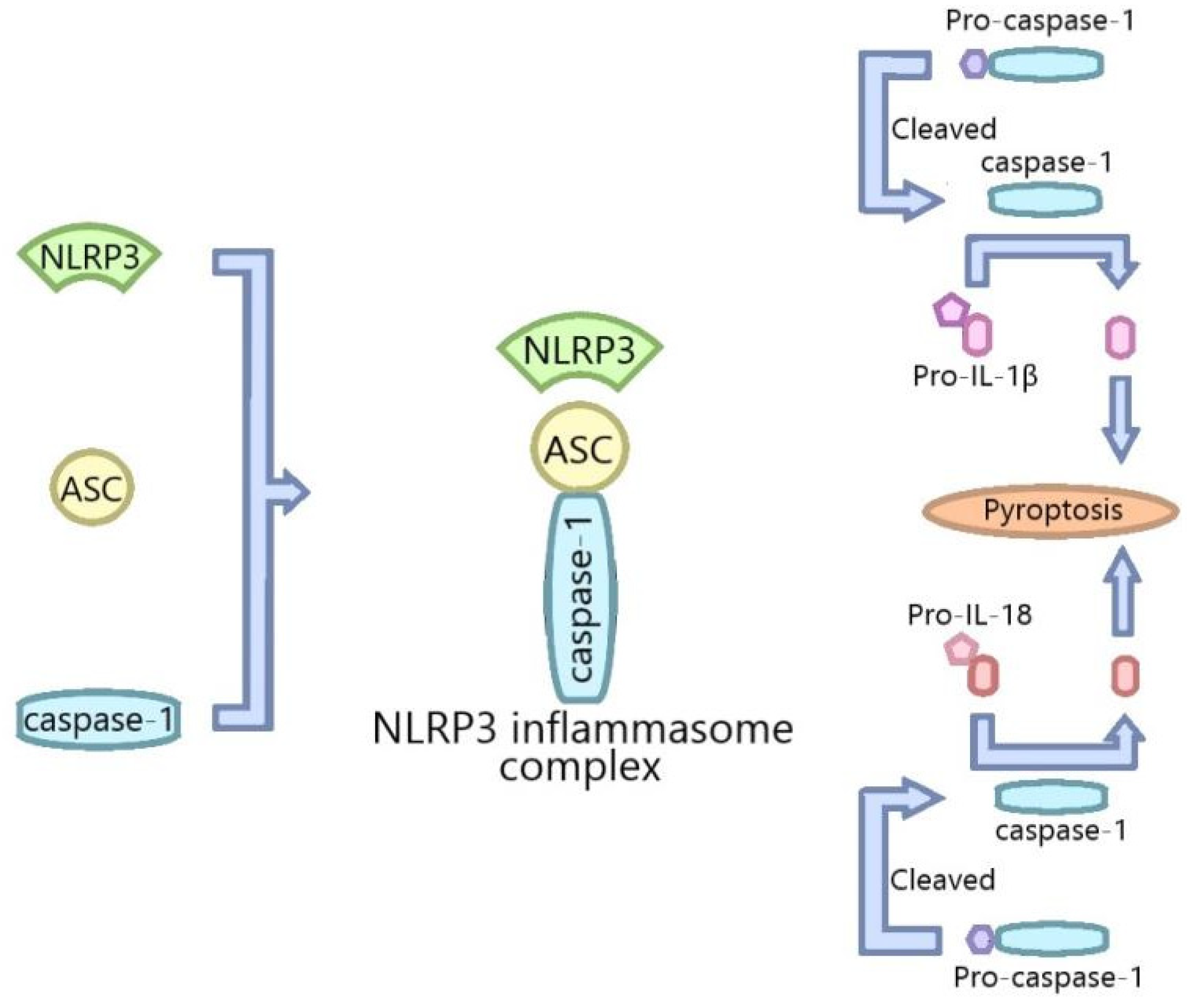
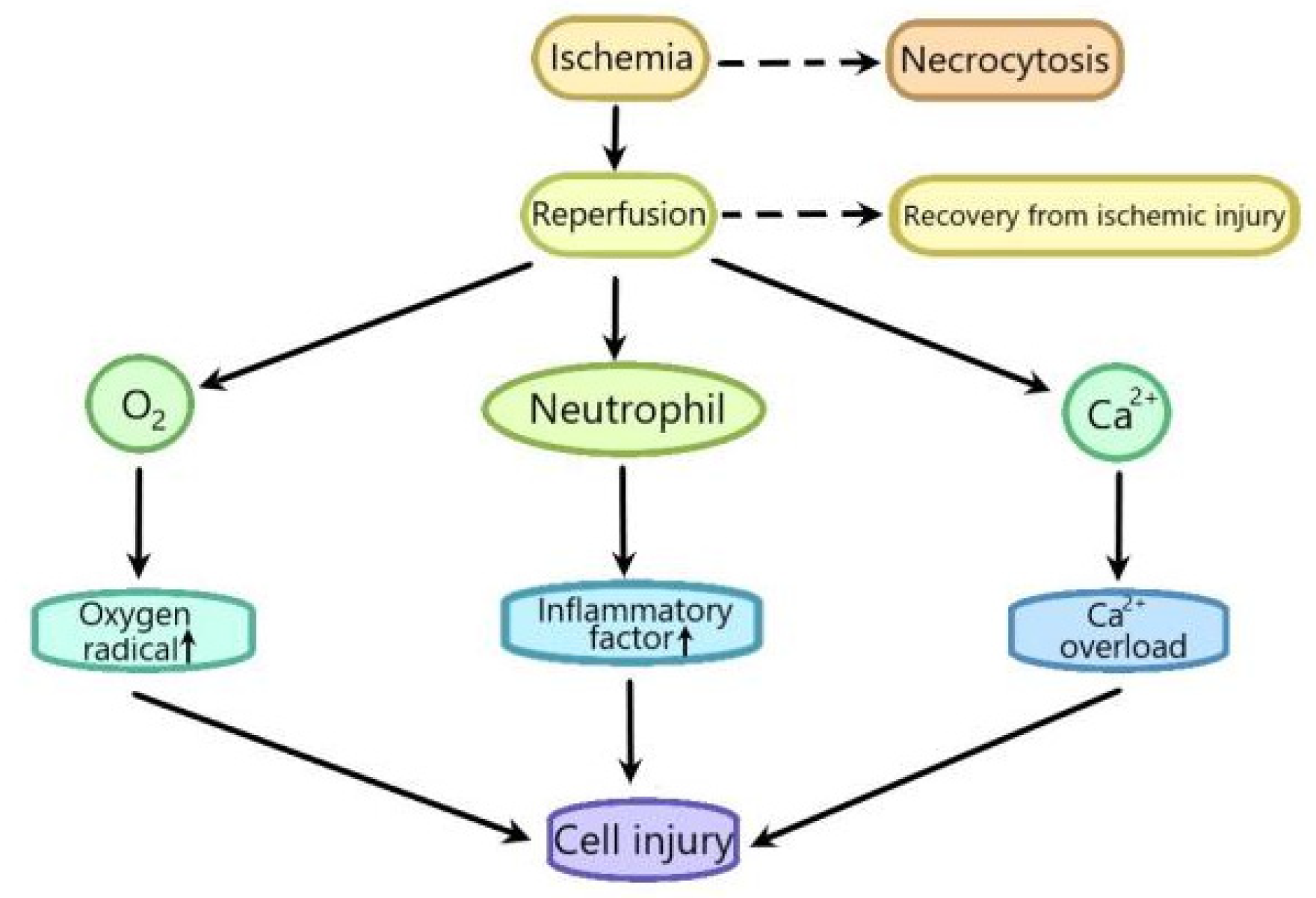
:1. Introduction
2. The Interplay between Autophagy and NLRP3 Inflammasome in Myocardial Ischemia/Reperfusion Injury
3. The Interplay between Autophagy and NLRP3 Inflammasome in Ischemia/Reperfusion Injury of the Nervous System
4. The Interplay between Autophagy and NLRP3 Inflammasome in Hepatic Ischemia/Reperfusion Injury
5. The Interplay between Autophagy and NLRP3 Inflammasome in Intestinal Ischemia/Reperfusion Injury
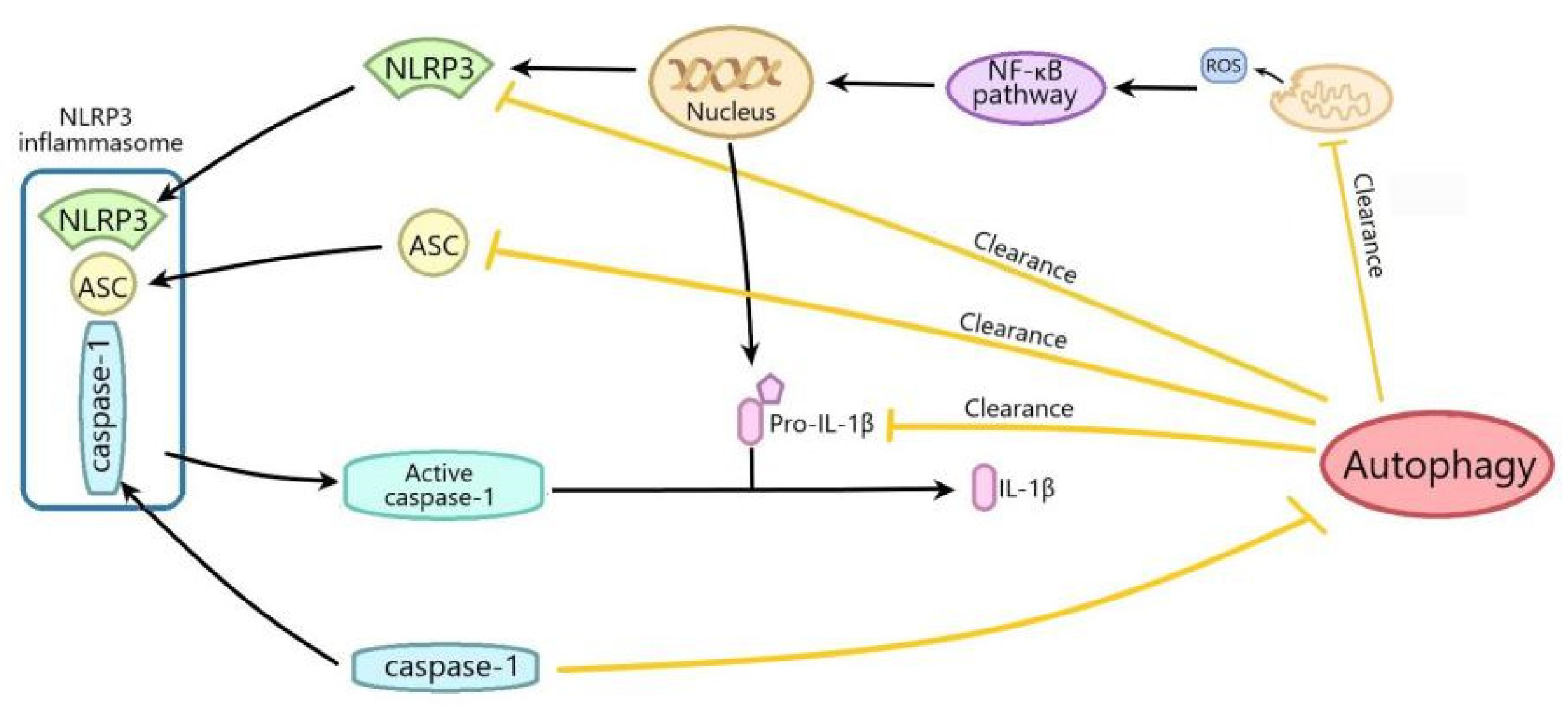
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ichimiya, T.; Yamakawa, T.; Hirano, T.; Yokoyama, Y.; Hayashi, Y.; Hirayama, D.; Wagatsuma, K.; Itoi, T.; Nakase, H. Autophagy and autophagy-related diseases: A review. Int. J. Mol. Sci. 2020, 21, 8274. [Google Scholar] [CrossRef]
- Zhou, B.; Liu, J.; Kang, R.; Klionsky, D.J.; Kroemer, G.; Tang, D. Ferroptosis is a type of autophagy-dependent cell death. Semin. Cancer Biol. 2020, 66, 89–100. [Google Scholar] [CrossRef]
- Klionsky, D.J. The molecular machinery of autophagy: Unanswered questions. J. Cell Sci. 2005, 118, 7–18. [Google Scholar] [CrossRef] [Green Version]
- Feng, Y.; He, D.; Yao, Z.; Klionsky, D.J. The machinery of macroautophagy. Cell Res. 2014, 24, 24–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuma, A.; Mizushima, N. Physiological role of autophagy as an intracellular recycling system: With an emphasis on nutrient metabolism. Semin. Cell Dev. Biol. 2010, 21, 683–690. [Google Scholar] [CrossRef] [PubMed]
- Yano, T.; Mita, S.; Ohmori, H.; Oshima, Y.; Fujimoto, Y.; Ueda, R.; Takada, H.; Goldman, W.E.; Fukase, K.; Silverman, N.; et al. Autophagic control of listeria through intracellular innate immune recognition in drosophila. Nat. Immunol. 2008, 9, 908–916. [Google Scholar] [CrossRef] [Green Version]
- Shelly, S.; Lukinova, N.; Bambina, S.; Berman, A.; Cherry, S. Autophagy is an essential component of Drosophila immunity against vesicular stomatitis virus. Immunity 2009, 30, 588–598. [Google Scholar] [CrossRef] [Green Version]
- Parzych, K.R.; Klionsky, D.J. An overview of autophagy: Morphology, mechanism, and regulation. Antioxid. Redox. Signal. 2014, 20, 460–473. [Google Scholar] [CrossRef] [Green Version]
- Oku, M.; Sakai, Y. Three distinct types of microautophagy based on membrane dynamics and molecular machineries. Bioessays 2018, 40, e1800008. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wu, D.; Wang, H. Hydrogen sulfide plays an important protective role by influencing autophagy in diseases. Physiol. Res. 2019, 68, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Mizushima, N.; Levine, B.; Cuervo, A.M.; Klionsky, D.J. Autophagy fights disease through cellular self-digestion. Nature 2008, 451, 1069–1075. [Google Scholar] [CrossRef] [Green Version]
- Mizushima, N.; Komatsu, M. Autophagy: Renovation of cells and tissues. Cell 2011, 147, 728–741. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mo, Y.; Sun, Y.; Liu, K.Y. Autophagy and inflammation in ischemic stroke. Neural. Regen. Res. 2020, 15, 1388–1396. [Google Scholar] [PubMed]
- Levine, B.; Kroemer, G. Biological functions of autophagy genes: A disease perspective. Cell 2019, 176, 11–42. [Google Scholar] [CrossRef] [Green Version]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol. Cell 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Yaribeygi, H.; Katsiki, N.; Butler, A.E.; Sahebkar, A. Effects of antidiabetic drugs on NLRP3 inflammasome activity, with a focus on diabetic kidneys. Drug Discov. Today 2019, 24, 256–262. [Google Scholar] [CrossRef]
- Ozaki, E.; Campbell, M.; Doyle, S.L. Targeting the NLRP3 inflammasome in chronic inflammatory diseases: Current perspectives. J. Inflamm. Res. 2015, 8, 15–27. [Google Scholar]
- Jo, E.K.; Kim, J.K.; Shin, D.M.; Sasakawa, C. Molecular mechanisms regulating NLRP3 inflammasome activation. Cell. Mol. Immunol. 2016, 13, 148–159. [Google Scholar] [CrossRef] [Green Version]
- Lv, S.; Li, X.; Wang, H. The role of the effects of endoplasmic reticulum stress on NLRP3 inflammasome in diabetes. Front. Cell Dev. Biol. 2021, 9, 663528. [Google Scholar] [CrossRef]
- Lu, A.; Magupalli, V.G.; Ruan, J.; Yin, Q.; Atianand, M.K.; Vos, M.R.; Schroder, G.; Fitzgerald, K.A.; Wu, H.; Egelman, E.H. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 2014, 156, 1193–1206. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Meng, C.; Zhang, J.; Wu, J.; Zhao, J. Inhibition of GSK-3β alleviates cerebral ischemia/reperfusion injury in rats by suppressing NLRP3 inflammasome activation through autophagy. Int. Immunopharmacol. 2019, 68, 234–241. [Google Scholar] [CrossRef]
- Lu, F.; Lan, Z.; Xin, Z.; He, C.; Guo, Z.; Xia, X.; Hu, T. Emerging insights into molecular mechanisms underlying pyroptosis and functions of inflammasomes in diseases. J. Cell. Physiol. 2020, 235, 3207–3221. [Google Scholar] [CrossRef]
- Martinon, F.; Petrilli, V.; Mayor, A.; Tardivel, A.; Tschopp, J. Gout-Associated uric acid crystals activate the NALP3 inflammasome. Nature 2006, 440, 237–241. [Google Scholar] [CrossRef] [Green Version]
- Duewell, P.; Kono, H.; Rayner, K.J.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nunez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halle, A.; Hornung, V.; Petzold, G.C.; Stewart, C.R.; Monks, B.G.; Reinheckel, T.; Fitzgerald, K.A.; Latz, E.; Moore, K.J.; Golenbock, D.T. The NALP3 inflammasome is involved in the innate immune response to amyloid-β. Nat. Immunol. 2008, 9, 857–865. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.M.; Kim, J.J.; Kim, H.J.; Shong, M.; Ku, B.J.; Jo, E.K. Upregulated NLRP3 inflammasome activation in patients with type 2 diabetes. Diabetes 2013, 62, 194–204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peralta, C.; Jimenez-Castro, M.B.; Gracia-Sancho, J. Hepatic ischemia and reperfusion injury: Effects on the liver sinusoidal milieu. J. Hepatol. 2013, 59, 1094–1106. [Google Scholar] [CrossRef] [Green Version]
- Soares, R.O.S.; Losada, D.M.; Jordani, M.C.; Evora, P.; Castro, E.S.O. Ischemia/Reperfusion Injury revisited: An Overview of the latest pharmacological strategies. Int. J. Mol. Sci. 2019, 20, 5034. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, H.F.; Tuo, Q.Z.; Yin, Q.Z.; Lei, P. The pathological role of ferroptosis in ischemia/reperfusion-related injury. Zool. Res. 2020, 41, 220–230. [Google Scholar] [CrossRef] [PubMed]
- Hurst, S.; Gonnot, F.; Dia, M.; Crola Da Silva, C.; Gomez, L.; Sheu, S.S. Phosphorylation of cyclophilin D at serine 191 regulates mitochondrial permeability transition pore opening and cell death after ischemia-reperfusion. Cell Death Dis. 2020, 11, 661. [Google Scholar] [CrossRef] [PubMed]
- Webster, K.A. Mitochondrial membrane permeabilization and cell death during myocardial infarction: Roles of calcium and reactive oxygen species. Future Cardiol. 2012, 8, 863–884. [Google Scholar] [CrossRef] [Green Version]
- Corona, J.C.; Duchen, M.R. Impaired mitochondrial homeostasis and neurodegeneration: Towards new therapeutic targets? J. Bioenerg. Biomembr. 2015, 47, 89–99. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, S.; Wang, Y.; Chen, Y.; Cao, F. The role of the autophagy in myocardial ischemia/reperfusion injury. Biochim. Biophys. Acta 2015, 1852, 271–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xin, R.; Pan, Y.L.; Wang, Y.; Wang, S.Y.; Wang, R.; Xia, B.; Qin, R.N.; Fu, Y.; Wu, Y.H. Nickel-refining fumes induce NLRP3 activation dependent on mitochondrial damage and ROS production in Beas-2B cells. Arch. Biochem. Biophys. 2019, 676, 108148. [Google Scholar] [CrossRef]
- Lin, J.; Huang, H.F.; Yang, S.K.; Duan, J.; Qu, S.M.; Yuan, B.; Zeng, Z. The effect of Ginsenoside Rg1 in hepatic ischemia reperfusion (I/R) injury ameliorates ischemia-reperfusion-induced liver injury by inhibiting apoptosis. Biomed. Pharmacother. 2020, 129, 110398. [Google Scholar] [CrossRef]
- Deguchi, H.; Ikeda, M.; Ide, T.; Tadokoro, T.; Ikeda, S.; Okabe, K.; Ishikita, A.; Saku, K.; Matsushima, S.; Tsutsui, H. Roxadustat markedly reduces myocardial ischemia reperfusion injury in mice. Circ. J. 2020, 84, 1028–1033. [Google Scholar] [CrossRef] [Green Version]
- Zhong, L.; Wang, Z.F.; Xiao, B.J. Research on the antioxidant effect of Enshi banqiao radix codonopsis on brain ischemia/reperfusion (I/R)injury. Chin. J. Appl. Physiol. 2012, 28, 314–316. [Google Scholar]
- Hausenloy, D.J.; Yellon, D.M. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J. Clin. Investig. 2013, 123, 92–100. [Google Scholar] [CrossRef]
- Neri, M.; Riezzo, I.; Pascale, N.; Pomara, C.; Turillazzi, E. Ischemia/Reperfusion injury following acute myocardial infarction: A critical issue for clinicians and forensic pathologists. Mediat. Inflamm. 2017, 2017, 7018393. [Google Scholar] [CrossRef]
- Li, G.; Qian, W.; Zhao, C. Analyzing the anti-ischemia-reperfusion injury effects of ginsenoside Rb1 mediated through the inhibition of p38α MAPK. Can. J. Physiol. Pharmacol. 2016, 94, 97–103. [Google Scholar] [CrossRef]
- Wang, F.; Gao, Q.; Yang, J.; Wang, C.; Cao, J.; Sun, J.; Fan, Z.; Fu, L. Artemisinin suppresses myocardial ischemia-reperfusion injury via NLRP3 inflammasome mechanism. Mol. Cell. Biochem. 2020, 474, 171–180. [Google Scholar] [CrossRef]
- Zhang, J.; Huang, L.; Shi, X.; Yang, L.; Hua, F.; Ma, J.; Zhu, W.; Liu, X.; Xuan, R.; Shen, Y.; et al. Metformin protects against myocardial ischemia-reperfusion injury and cell pyroptosis via AMPK/NLRP3 inflammasome pathway. Aging 2020, 12, 24270–24287. [Google Scholar] [CrossRef]
- Meng, Z.; Song, M.Y.; Li, C.F.; Zhao, J.Q. shRNA interference of NLRP3 inflammasome alleviate ischemia reperfusion-induced myocardial damage through autophagy activation. Biochem. Biophys. Res. Commun. 2017, 494, 728–735. [Google Scholar] [CrossRef] [PubMed]
- Hadi, N.R.; Al-Amran, F.; Yousif, M.; Zamil, S.T. Antiapoptotic effect of simvastatin ameliorates myocardial ischemia/reperfusion injury. ISRN Pharmacol. 2013, 2013, 815094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Q.Y.; Ma, J.Q.; Duan, Y.Y.; Sun, Y.; Yu, S.; Li, B.; Zhang, G.M. Carthamin yellow protects the heart against ischemia/reperfusion injury with reduced reactive oxygen species release and inflammatory response. J. Cardiovasc. Pharmacol. 2019, 74, 228–234. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, W.; Leng, Y.; Xiong, Y.; Xia, Z. Ferroptosis is involved in diabetes myocardial ischemia/reperfusion injury through endoplasmic reticulum stress. DNA Cell Biol. 2020, 39, 210–225. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Zhu, L.; Yuan, W.; Sun, L.; Xia, Z.; Zhang, Z.; Yao, W. Diabetes aggravates myocardial ischaemia reperfusion injury via activating Nox2-related programmed cell death in an AMPK-dependent manner. J. Cell. Mol. Med. 2020, 24, 6670–6679. [Google Scholar] [CrossRef]
- Qiu, Z.; He, Y.; Ming, H.; Lei, S.; Leng, Y.; Xia, Z.Y. Lipopolysaccharide (LPS) Aggravates High Glucose- and Hypoxia/Reoxygenation-Induced Injury through Activating ROS-Dependent NLRP3 Inflammasome-Mediated Pyroptosis in H9C2 Cardiomyocytes. J. Diabetes Res. 2019, 2019, 8151836. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.Y.; Dong, B.; Fang, Z.F.; Hu, X.Q.; Tang, L.; Zhou, S.H. Knockdown of lncRNA AK139328 alleviates myocardial ischaemia/reperfusion injury in diabetic mice via modulating miR-204-3p and inhibiting autophagy. J. Cell. Mol. Med. 2018, 22, 4886–4898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, D.; He, Y.; Ye, X.; Cai, Y.; Xu, J.; Zhang, L.; Li, M.; Liu, H.; Wang, S.; Xia, Z. Activation of autophagy inhibits nucleotide-binding oligomerization domain-like receptor protein 3 inflammasome activation and attenuates myocardial ischemia-reperfusion injury in diabetic rats. J. Diabetes Investig. 2020, 11, 1126–1136. [Google Scholar] [CrossRef]
- Ma, M.; Hui, J.; Zhang, Q.Y.; Zhu, Y.; He, Y.; Liu, X.J. Long non-coding RNA nuclear-enriched abundant transcript 1 inhibition blunts myocardial ischemia reperfusion injury via autophagic flux arrest and apoptosis in streptozotocin-induced diabetic rats. Atherosclerosis 2018, 277, 113–122. [Google Scholar] [CrossRef]
- Zhang, Y.M.; Qu, X.Y.; Tao, L.N.; Zhai, J.H.; Gao, H.; Song, Y.Q.; Zhang, S.X. XingNaoJing injection ameliorates cerebral ischaemia/reperfusion injury via SIRT1-mediated inflammatory response inhibition. Pharm. Biol. 2020, 58, 16–24. [Google Scholar] [CrossRef]
- Qiu, J.; Wang, M.; Zhang, J.; Cai, Q.; Lu, D.; Li, Y.; Dong, Y.; Zhao, T.; Chen, H. The neuroprotection of Sinomenine against ischemic stroke in mice by suppressing NLRP3 inflammasome via AMPK signaling. Int. Immunopharmacol. 2016, 40, 492–500. [Google Scholar] [CrossRef]
- Sun, X.; Liu, H.; Sun, Z.; Zhang, B.; Wang, X.; Liu, T.; Pan, T.; Gao, Y.; Jiang, X.; Li, H. Acupuncture protects against cerebral ischemia-reperfusion injury via suppressing endoplasmic reticulum stress-mediated autophagy and apoptosis. Mol. Med. 2020, 26, 105. [Google Scholar] [CrossRef]
- Burns, J.; Yokota, T.; Ashihara, H.; Lean, M.E.; Crozier, A. Plant foods and herbal sources of resveratrol. J. Agric. Food Chem. 2002, 50, 3337–3340. [Google Scholar] [CrossRef] [PubMed]
- Shrikanta, A.; Kumar, A.; Govindaswamy, V. Resveratrol content and antioxidant properties of underutilized fruits. J. Food Sci. Technol. 2015, 52, 383–390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.; Dong, J.; Zheng, D.; Li, X.; Ding, D.; Xu, H. Reperfusion combined with intraarterial administration of resveratrol-loaded nanoparticles improved cerebral ischemia-reperfusion injury in rats. Nanomedicine 2020, 28, 102208. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Li, Z.; Wang, Y.; Hou, Y.; Li, L.; Zhao, J. Resveratrol alleviates cerebral ischemia/reperfusion injury in rats by inhibiting NLRP3 inflammasome activation through Sirt1-dependent autophagy induction. Int. Immunopharmacol. 2017, 50, 208–215. [Google Scholar] [CrossRef]
- Wang, P.; Xu, T.Y.; Guan, Y.F.; Tian, W.W.; Viollet, B.; Rui, Y.C.; Zhai, Q.W.; Su, D.F.; Miao, C.Y. Nicotinamide phosphoribosyltransferase protects against ischemic stroke through SIRT1-dependent adenosine monophosphate-activated kinase pathway. Ann. Neurol. 2011, 69, 360–374. [Google Scholar] [CrossRef] [PubMed]
- Fu, C.; Zhang, X.; Lu, Y.; Wang, F.; Xu, Z.; Liu, S.; Zheng, H.; Liu, X. Geniposide inhibits NLRP3 inflammasome activation via autophagy in BV-2 microglial cells exposed to oxygen-glucose deprivation/reoxygenation. Int. Immunopharmacol. 2020, 84, 106547. [Google Scholar] [CrossRef]
- Chien, T.; Weng, Y.T.; Chang, S.Y.; Lai, H.L.; Chiu, F.L.; Kuo, H.C.; Chuang, D.M.; Chern, Y. GSK3beta negatively regulates TRAX, a scaffold protein implicated in mental disorders, for NHEJ-mediated DNA repair in neurons. Mol. Psychiatry 2018, 23, 2375–2390. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Liu, Y.; Zhu, J.; Lei, S.; Dong, Y.; Li, L.; Jiang, B.; Tan, L.; Wu, J.; Yu, S.; et al. GSK-3β downregulates Nrf2 in cultured cortical neurons and in a rat model of cerebral ischemia-reperfusion. Sci. Rep. 2016, 6, 20196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mancinelli, R.; Carpino, G.; Petrungaro, S.; Mammola, C.L.; Tomaipitinca, L.; Filippini, A.; Facchiano, A.; Ziparo, E.; Giampietri, C. Multifaceted roles of GSK-3 in cancer and autophagy-related diseases. Oxidative Med. Cell. Longev. 2017, 2017, 4629495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weikel, K.A.; Cacicedo, J.M.; Ruderman, N.B.; Ido, Y. Knockdown of GSK3β increases basal autophagy and AMPK signalling in nutrient-laden human aortic endothelial cells. Biosci. Rep. 2016, 36, e00382. [Google Scholar] [CrossRef]
- Liu, K.; Sun, Y.; Gu, Z.; Shi, N.; Zhang, T.; Sun, X. Mitophagy in ischaemia/reperfusion induced cerebral injury. Neurochem. Res. 2013, 38, 1295–1300. [Google Scholar] [CrossRef]
- Tang, Y.C.; Tian, H.X.; Yi, T.; Chen, H.B. The critical roles of mitophagy in cerebral ischemia. Protein Cell 2016, 7, 699–713. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.L.; Wang, X.X.; Chen, L.; Tang, J.; Xia, Y.F.; Qian, K.; Qin, Z.H.; Waeber, C.; Sheng, R. A sphingosine kinase 2-mimicking TAT-peptide protects neurons against ischemia-reperfusion injury by activating BNIP3-mediated mitophagy. Neuropharmacology 2020, 181, 108326. [Google Scholar] [CrossRef]
- Seabright, A.P.; Fine, N.H.F.; Barlow, J.P.; Lord, S.O.; Musa, I.; Gray, A.; Bryant, J.A.; Banzhaf, M.; Lavery, G.G.; Hardie, D.G.; et al. AMPK activation induces mitophagy and promotes mitochondrial fission while activating TBK1 in a PINK1-Parkin independent manner. FASEB J. 2020, 34, 6284–6301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Yuan, Y.; Jiang, L.; Zhang, J.; Gao, J.; Shen, Z.; Zheng, Y.; Deng, T.; Yan, H.; Li, W.; et al. Endoplasmic reticulum stress induced by tunicamycin and thapsigargin protects against transient ischemic brain injury: Involvement of PARK2-dependent mitophagy. Autophagy 2014, 10, 1801–1813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, Q.; Li, Z.; Meng, C.; Wu, J.; Zhao, Y.; Zhao, J. Parkin-Dependent mitophagy is required for the inhibition of ATF4 on NLRP3 Inflammasome activation in cerebral ischemia-reperfusion injury in rats. Cells 2019, 8, 897. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Wang, X.C.; Gong, P.T.; Zhang, N.; Zhang, X.; Li, S.; Li, X.; Liu, S.X.; Zhang, X.X.; Li, W.; et al. ROS-mediated NLRP3 inflammasome activation participates in the response against Neospora caninum infection. Parasit. Vectors 2020, 13, 449. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhu, Q.; Song, W.; Gao, B. Exercise training and dietary restriction affect PINK1/Parkin and Bnip3/Nix-mediated cardiac mitophagy in mice. Gen. Physiol. Biophys. 2018, 37, 657–666. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Zhang, S.; Li, Q.; Liu, Z.; Mai, W.; Chen, W.; Lei, J.; Hu, H. miR-219a-5p ameliorates hepatic ischemia/reperfusion injury via impairing TP53BP2. Dig. Dis. Sci. 2019, 64, 2177–2186. [Google Scholar] [CrossRef] [PubMed]
- Hua, S.; Ma, M.; Fei, X.; Zhang, Y.; Gong, F.; Fang, M. Glycyrrhizin attenuates hepatic ischemia-reperfusion injury by suppressing HMGB1-dependent GSDMD-mediated kupffer cells pyroptosis. Int. Immunopharmacol. 2019, 68, 145–155. [Google Scholar] [CrossRef]
- Xu, Y.; Yao, J.; Zou, C.; Zhang, H.; Zhang, S.; Liu, J.; Ma, G.; Jiang, P.; Zhang, W. Asiatic acid protects against hepatic ischemia/reperfusion injury by inactivation of Kupffer cells via PPARgamma/NLRP3 inflammasome signaling pathway. Oncotarget 2017, 8, 86339–86355. [Google Scholar] [CrossRef]
- Lin, Q.; Li, S.; Jiang, N.; Shao, X.; Zhang, M.; Jin, H.; Zhang, Z.; Shen, J.; Zhou, Y.; Zhou, W.; et al. PINK1-parkin pathway of mitophagy protects against contrast-induced acute kidney injury via decreasing mitochondrial ROS and NLRP3 inflammasome activation. Redox Biol. 2019, 26, 101254. [Google Scholar] [CrossRef]
- Lin, Q.; Li, S.; Jiang, N.; Jin, H.; Shao, X.; Zhu, X.; Wu, J.; Zhang, M.; Zhang, Z.; Shen, J.; et al. Inhibiting NLRP3 inflammasome attenuates apoptosis in contrast-induced acute kidney injury through the upregulation of HIF1A and BNIP3-mediated mitophagy. Autophagy 2020, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Barazzuol, L.; Giamogante, F.; Brini, M.; Cali, T. PINK1/Parkin mediated mitophagy, Ca2+ signaling and ER-mitochondria contacts in Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 1772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Tang, Y.; Lu, J.; Zhang, W.; Zhu, Y.; Zhang, S.; Ma, G.; Jiang, P.; Zhang, W. PINK1-mediated mitophagy protects against hepatic ischemia/reperfusion injury by restraining NLRP3 inflammasome activation. Free Radic. Biol. Med. 2020, 160, 871–886. [Google Scholar] [CrossRef]
- Yu, J.; Nagasu, H.; Murakami, T.; Hoang, H.; Broderick, L.; Hoffman, H.M.; Horng, T. Inflammasome activation leads to Caspase-1-dependent mitochondrial damage and block of mitophagy. Proc. Natl. Acad. Sci. USA 2014, 111, 15514–15519. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Chen, Y.; Gao, J.; Chen, Y.; Zhou, C.; Lin, X.; Liu, C.; Zhao, M.; Xu, Y.; Ji, L.; et al. Eva1a ameliorates atherosclerosis by promoting re-endothelialization of injured arteries via Rac1/Cdc42/Arpc1b. Cardiovasc. Res. 2021, 117, 450–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Lu, G.; Hu, J.; Shen, X.; Ju, J.; Gao, Y.; Qu, L.; Xia, Y.; Chen, Y.; Bai, Y. EVA1A/TMEM166 Regulates embryonic neurogenesis by autophagy. Stem Cell Rep. 2016, 6, 396–410. [Google Scholar] [CrossRef] [Green Version]
- Shen, X.; Kan, S.; Liu, Z.; Lu, G.; Zhang, X.; Chen, Y.; Bai, Y. EVA1A inhibits GBM cell proliferation by inducing autophagy and apoptosis. Exp. Cell Res. 2017, 352, 130–138. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Han, S.; Chen, X.; Li, X.; Xia, N.; Pu, L. Eva1a inhibits NLRP3 activation to reduce liver ischemia-reperfusion injury via inducing autophagy in Kupffer cells. Mol. Immunol. 2021, 132, 82–92. [Google Scholar] [CrossRef]
- Xue, R.; Qiu, J.; Wei, S.; Liu, M.; Wang, Q.; Wang, P.; Sha, B.; Wang, H.; Shi, Y.; Zhou, J.; et al. Lycopene alleviates hepatic ischemia reperfusion injury via the Nrf2/HO-1 pathway mediated NLRP3 inflammasome inhibition in Kupffer cells. Ann. Transl. Med. 2021, 9, 631. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Liu, N.; Xie, X.; Bi, G.; Ba, H.; Li, L.; Zhang, J.; Deng, X.; Yao, Y.; Tang, Z.; et al. The macrophage-specific V-ATPase subunit ATP6V0D2 restricts inflammasome activation and bacterial infection by facilitating autophagosome-lysosome fusion. Autophagy 2019, 15, 960–975. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Wang, H.; Chen, X.; Han, S.; Zhu, Y.; Wang, H.; Cheng, F.; Pu, L. Inhibiting ATP6V0D2 Aggravates liver ischemia-reperfusion injury by promoting NLRP3 Activation via impairing autophagic flux independent of Notch1/Hes1. J. Immunol. Res. 2021, 2021, 6670495. [Google Scholar] [CrossRef]
- Schofield, Z.V.; Wu, M.C.L.; Hansbro, P.M.; Cooper, M.A.; Woodruff, T.M. Acetate protects against intestinal ischemia-reperfusion injury independent of its cognate free fatty acid 2 receptor. FASEB J. 2020, 34, 10418–10430. [Google Scholar] [CrossRef]
- Wang, Z.; Sun, R.; Wang, G.; Chen, Z.; Li, Y.; Zhao, Y.; Liu, D.; Zhao, H.; Zhang, F.; Yao, J.; et al. SIRT3-mediated deacetylation of PRDX3 alleviates mitochondrial oxidative damage and apoptosis induced by intestinal ischemia/reperfusion injury. Redox Biol. 2020, 28, 101343. [Google Scholar] [CrossRef] [PubMed]
- Jia, Y.; Cui, R.; Wang, C.; Feng, Y.; Li, Z.; Tong, Y.; Qu, K.; Liu, C.; Zhang, J. Metformin protects against intestinal ischemia-reperfusion injury and cell pyroptosis via TXNIP-NLRP3-GSDMD pathway. Redox Biol. 2020, 32, 101534. [Google Scholar] [CrossRef]
- Tan, Y.; Zuo, W.; Huang, L.; Zhou, B.; Liang, H.; Zheng, S.; Jia, W.; Chen, S.; Liu, J.; Yang, X.; et al. Nervilifordin F alleviates intestinal ischemia/reperfusion-induced acute lung injury via inhibiting inflammasome and mTOR pathway. Int. Immunopharmacol. 2020, 89, 107014. [Google Scholar] [CrossRef]
- Wang, Z.; Li, Z.; Feng, D.; Zu, G.; Li, Y.; Zhao, Y.; Wang, G.; Ning, S.; Zhu, J.; Zhang, F.; et al. Autophagy induction ameliorates inflammatory responses in intestinal ischemia-reperfusion through inhibiting NLRP3 inflammasome activation. Shock 2019, 52, 387–395. [Google Scholar] [CrossRef]
- Biasizzo, M.; Kopitar-Jerala, N. Interplay between NLRP3 Inflammasome and autophagy. Front. Immunol. 2020, 11, 591803. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Wang, Y.; Long, Z.; He, G. Interaction between autophagy and the NLRP3 inflammasome. Acta Biochim. Biophys. Sin. 2019, 51, 1087–1095. [Google Scholar] [CrossRef] [PubMed]
- Shi, B.; Ma, M.; Zheng, Y.; Pan, Y.; Lin, X. mTOR and Beclin1: Two key autophagy-related molecules and their roles in myocardial ischemia/reperfusion injury. J. Cell. Physiol. 2019, 234, 12562–12568. [Google Scholar] [CrossRef] [PubMed]
- Jia, J.; Abudu, Y.P.; Claude-Taupin, A.; Gu, Y.; Kumar, S.; Choi, S.W.; Peters, R.; Mudd, M.H.; Allers, L.; Salemi, M.; et al. Galectins control MTOR and AMPK in response to lysosomal damage to induce autophagy. Autophagy 2019, 15, 169–171. [Google Scholar] [CrossRef] [Green Version]
- Cosin-Roger, J.; Simmen, S.; Melhem, H.; Atrott, K.; Frey-Wagner, I.; Hausmann, M.; de Valliere, C.; Spalinger, M.R.; Spielmann, P.; Wenger, R.H.; et al. Hypoxia ameliorates intestinal inflammation through NLRP3/mTOR downregulation and autophagy activation. Nat. Commun. 2017, 8, 98. [Google Scholar] [CrossRef] [Green Version]
- Ye, J.; Lu, S.; Wang, M.; Ge, W.; Liu, H.; Qi, Y.; Fu, J.; Zhang, Q.; Zhang, B.; Sun, G.; et al. Hydroxysafflor Yellow A Protects Against Myocardial Ischemia/Reperfusion Injury via Suppressing NLRP3 Inflammasome and Activating Autophagy. Front. Pharmacol. 2020, 11, 1170. [Google Scholar] [CrossRef]
- Xu, L.J.; Chen, R.C.; Ma, X.Y.; Zhu, Y.; Sun, G.B.; Sun, X.B. Scutellarin protects against myocardial ischemia-reperfusion injury by suppressing NLRP3 inflammasome activation. Phytomedicine 2020, 68, 153169. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lv, S.; Liu, H.; Wang, H. The Interplay between Autophagy and NLRP3 Inflammasome in Ischemia/Reperfusion Injury. Int. J. Mol. Sci. 2021, 22, 8773. https://doi.org/10.3390/ijms22168773
Lv S, Liu H, Wang H. The Interplay between Autophagy and NLRP3 Inflammasome in Ischemia/Reperfusion Injury. International Journal of Molecular Sciences. 2021; 22(16):8773. https://doi.org/10.3390/ijms22168773
Chicago/Turabian StyleLv, Shuangyu, Huiyang Liu, and Honggang Wang. 2021. "The Interplay between Autophagy and NLRP3 Inflammasome in Ischemia/Reperfusion Injury" International Journal of Molecular Sciences 22, no. 16: 8773. https://doi.org/10.3390/ijms22168773