
EphB4 as a Novel Target for the EGFR-Independent Suppressive Effects of Osimertinib on Cell Cycle Progression in Non-Small Cell Lung Cancer

 , , , , ,
, , , , ,
Abstract
:1. Introduction
2. Results
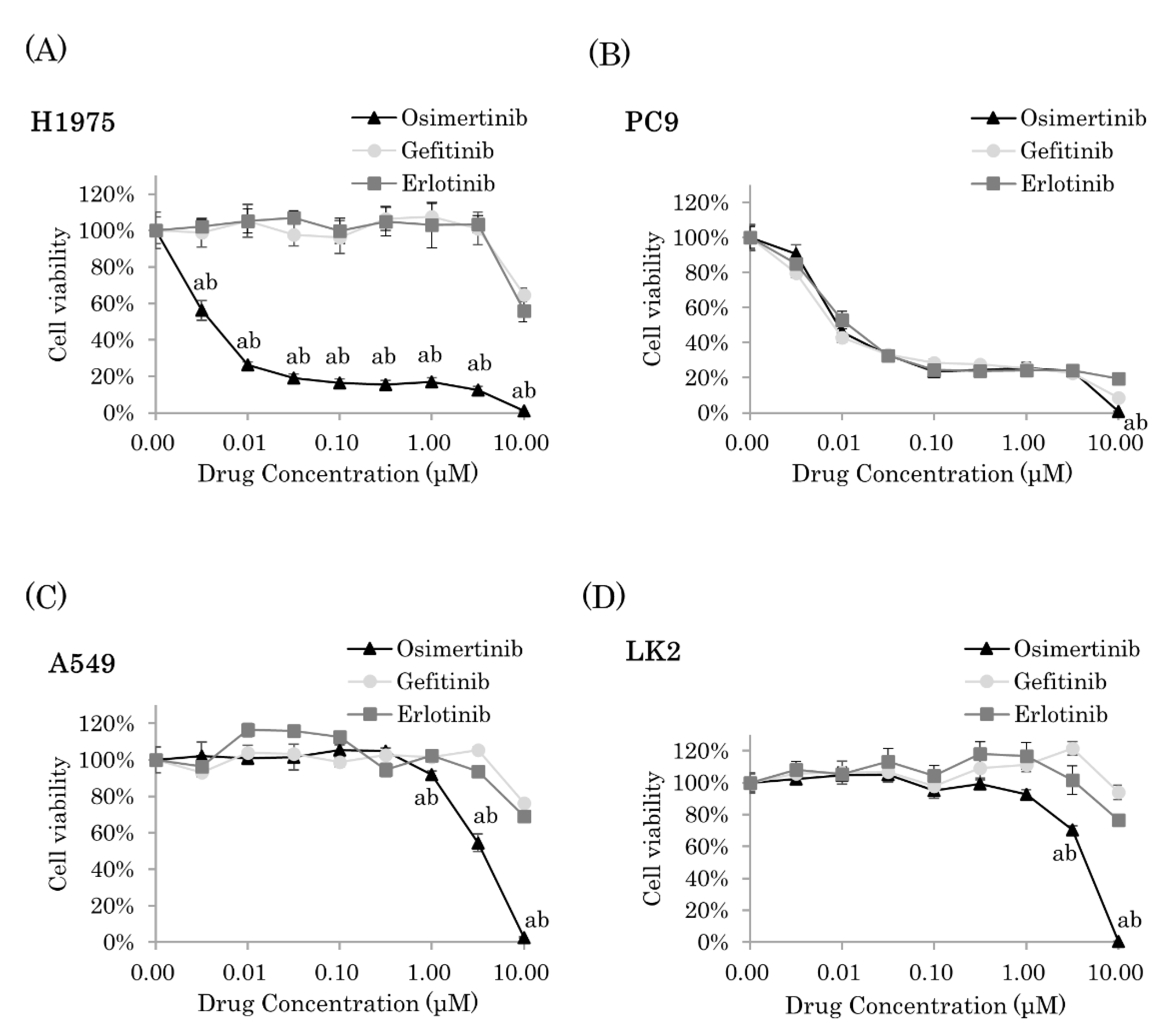
2.1. Osimertinib Significantly Inhibited Cell Proliferation of NSCLC Cells
2.2. Osimertinib Suppressed Cell Cycle Progression Independent of EGFR Pathways
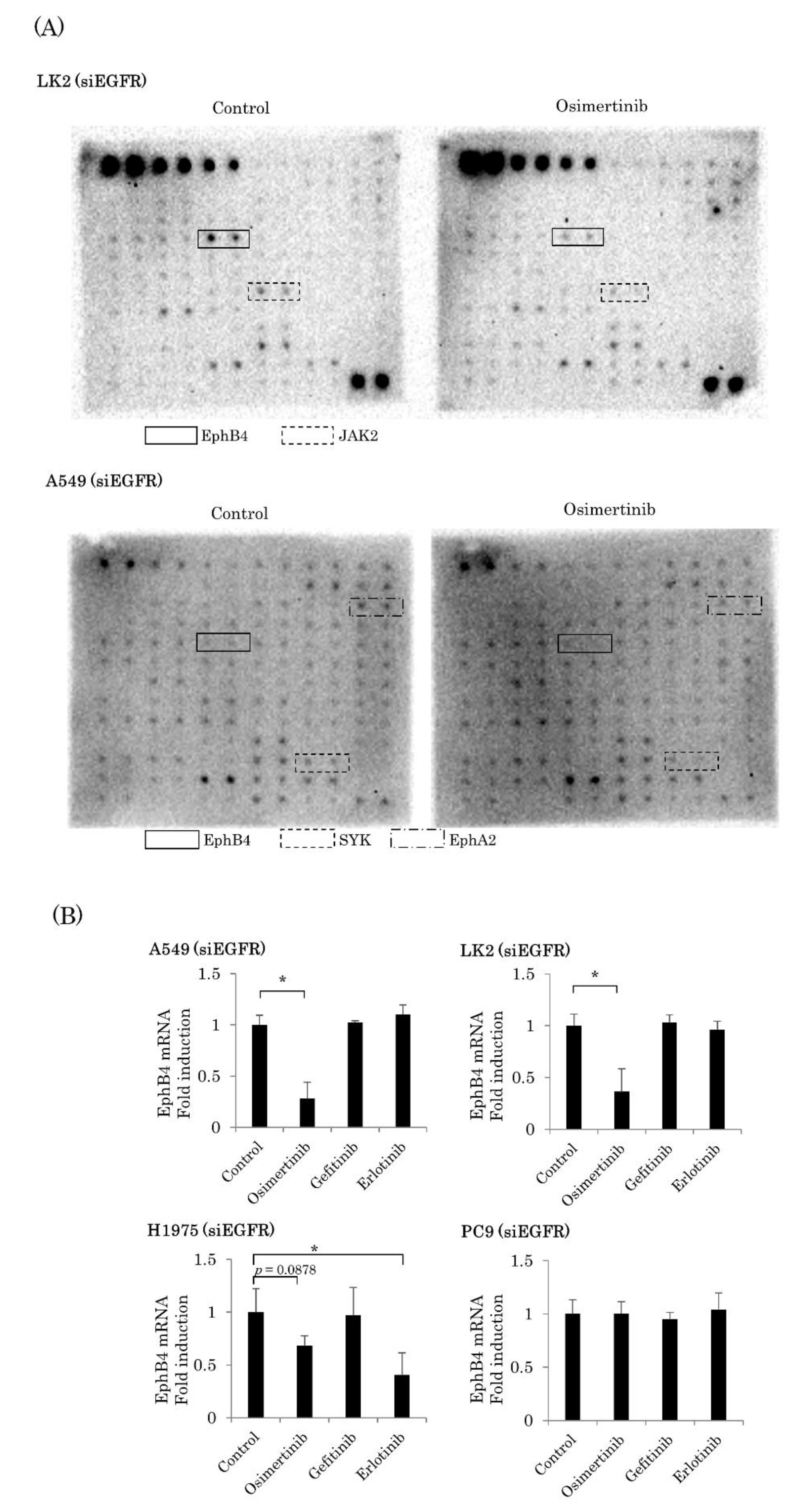
2.3. EGFR-Independent Effect of Osimertinib on EphB4 in NSCLC
2.4. Involvement of EphB4 in the EGFR-Independent Inhibitory Effects of Osimertinib
2.5. EphB4 Was a Poor Prognostic Factor in EGFR Mutation-Positive Lung Adenocarcinoma
3. Discussion
4. Material and Methods
4.1. Reagents and Antibodies
4.2. Cell Culture
4.3. Cell Viability Assays
4.4. Western Blotting
4.5. RNA Interference
4.6. Cell Cycle Analysis
4.7. Antibody Microarray Analysis of Phosphorylation of Cell Cycle Factors
4.8. Real Time RT-PCR
4.9. Human Receptor Tyrosine Kinase Phosphorylation Antibody Array
4.10. Patients and Tissue Preparation
4.11. Immunohistochemistry
4.12. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| ALK | anaplastic lymphoma kinase |
| CCN | Cyclin |
| CDK | Cyclin-dependent kinase |
| DMSO | Dimethyl sulfoxide |
| EGFR | epidermal growth factor receptor |
| EGFR-TKI | EGFR-tyrosine kinase inhibitor |
| Eph | ephrin receptor |
| FBS | fetal bovine serum |
| HER2 | Human EGFR-related 2 |
| JAK2 | Janus activating kinase 2 |
| LI | labelling index |
| NSCLC | non-small cell lung cancer |
| PARP | poly(ADP-ribose) polymerase |
| PBS | phosphate-buffered saline |
| RFS | Relapse-free survival |
| RPMI | Roswell Park Memorial Institute media |
| TKI | tyrosine kinase inhibitor |
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Y.; Au, J.S.-K.; Thongprasert, S.; Srinivasan, S.; Tsai, C.-M.; Khoa, M.T.; Heeroma, K.; Itoh, Y.; Cornelio, G.; Yang, P.-C. A Prospective, Molecular Epidemiology Study of EGFR Mutations in Asian Patients with Advanced Non–Small-Cell Lung Cancer of Adenocarcinoma Histology (PIONEER). J. Thorac. Oncol. 2014, 9, 154–162. [Google Scholar] [CrossRef] [Green Version]
- Mitsudomi, T.; Morita, S.; Yatabe, Y.; Negoro, S.; Okamoto, I.; Tsurutani, J.; Seto, T.; Satouchi, M.; Tada, H.; Hirashima, T.; et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): An open label, randomised phase 3 trial. Lancet Oncol. 2010, 11, 121–128. [Google Scholar] [CrossRef]
- Maemondo, M.; Inoue, A.; Kobayashi, K.; Sugawara, S.; Oizumi, S.; Isobe, H.; Gemma, A.; Harada, M.; Yoshizawa, H.; Kinoshita, I.; et al. Gefitinib or Chemotherapy for Non–Small-Cell Lung Cancer with Mutated EGFR. N. Engl. J. Med. 2010, 362, 2380–2388. [Google Scholar] [CrossRef] [Green Version]
- Mok, T.S.; Wu, Y.-L.; Thongprasert, S.; Yang, C.-H.; Chu, D.-T.; Saijo, N.; Sunpaweravong, P.; Han, B.; Margono, B.; Ichinose, Y.; et al. Gefitinib or Carboplatin–Paclitaxel in Pulmonary Adenocarcinoma. N. Engl. J. Med. 2009, 361, 947–957. [Google Scholar] [CrossRef]
- Rosell, R.; Carcereny, E.; Gervais, R.; Vergnenegre, A.; Massuti, B.; Felip, E.; Palmero, R.; Garcia-Gomez, R.; Pallares, C.; Sanchez, J.M.; et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012, 13, 239–246. [Google Scholar] [CrossRef]
- Zhou, C.; Wu, Y.-L.; Chen, G.; Feng, J.; Liu, X.-Q.; Wang, C.; Zhang, S.; Wang, J.; Zhou, S.; Ren, S.; et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): A multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011, 12, 735–742. [Google Scholar] [CrossRef]
- Miller, V.A.; Hirsh, V.; Cadranel, J.; Chen, Y.-M.; Park, K.; Kim, S.-W.; Zhou, C.; Su, W.-C.; Wang, M.; Sun, Y.; et al. Afatinib versus placebo for patients with advanced, metastatic non-small-cell lung cancer after failure of erlotinib, gefitinib, or both, and one or two lines of chemotherapy (LUX-Lung 1): A phase 2b/3 randomised trial. Lancet Oncol. 2012, 13, 528–538. [Google Scholar] [CrossRef]
- Kobayashi, S.; Boggon, T.J.; Dayaram, T.; Jänne, P.A.; Kocher, O.; Meyerson, M.; Johnson, B.E.; Eck, M.J.; Tenen, D.G.; Halmos, B. EGFR mutation and resistance of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2005, 352, 786–792. [Google Scholar] [CrossRef] [PubMed]
- Sequist, L.V.; Waltman, B.A.; Dias-Santagata, D.; Digumarthy, S.; Turke, A.B.; Fidias, P.; Bergethon, K.; Shaw, A.T.; Gettinger, S.; Cosper, A.K.; et al. Genotypic and Histological Evolution of Lung Cancers Acquiring Resistance to EGFR Inhibitors. Sci. Transl. Med. 2011, 3, 75ra26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cross, D.A.E.; Ashton, S.E.; Ghiorghiu, S.; Eberlein, C.; Nebhan, C.A.; Spitzler, P.J.; Orme, J.P.; Finlay, M.R.V.; Ward, R.A.; Mellor, M.J.; et al. AZD9291, an Irreversible EGFR TKI, Overcomes T790M-Mediated Resistance to EGFR Inhibitors in Lung Cancer. Cancer Discov. 2014, 4, 1046–1061. [Google Scholar] [CrossRef] [Green Version]
- Jänne, P.A.; Yang, J.C.-H.; Kim, D.-W.; Planchard, D.; Ohe, Y.; Ramalingam, S.S.; Ahn, M.-J.; Kim, S.-W.; Su, W.-C.; Horn, L.; et al. AZD9291 in EGFR Inhibitor–Resistant Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2015, 372, 1689–1699. [Google Scholar] [CrossRef] [PubMed]
- Mok, T.S.; Wu, Y.-L.; Ahn, M.-J.; Garassino, M.C.; Kim, H.R.; Ramalingam, S.S.; Shepherd, F.A.; He, Y.; Akamatsu, H.; Theelen, W.S.; et al. Osimertinib or Platinum-Pemetrexed in EGFR T790M-Positive Lung Cancer. N. Engl. J. Med. 2017, 376, 629–640. [Google Scholar] [CrossRef] [Green Version]
- Soria, J.-C.; Ohe, Y.; Vansteenkiste, J.; Reungwetwattana, T.; Chewaskulyong, B.; Lee, K.H.; Dechaphunkul, A.; Imamura, F.; Nogami, N.; Kurata, T.; et al. Osimertinib in UntreatedEGFR-Mutated Advanced Non–Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 378, 113–125. [Google Scholar] [CrossRef]
- Alvarez, J.V.; Greulich, H.; Sellers, W.R.; Meyerson, M.; Frank, D.A. Signal Transducer and Activator of Transcription 3 Is Required for the Oncogenic Effects of Non–Small-Cell Lung Cancer–Associated Mutations of the Epidermal Growth Factor Receptor. Cancer Res. 2006, 66, 3162–3168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karachaliou, N.; Codony-Servat, J.; Teixidó, C.; Pilotto, S.; Drozdowskyj, A.; Servat, C.C.; Gimenez-Capitan, A.; Molina-Vila, M.A.; Bertran-Alamillo, J.; Gervais, R.; et al. BIM and mTOR expression levels predict outcome to erlotinib in EGFR-mutant non-small-cell lung cancer. Sci. Rep. 2015, 5, 17499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sordella, R.; Bell, D.W.; Haber, D.A.; Settleman, J. Gefitinib-Sensitizing EGFR Mutations in Lung Cancer Activate Anti-Apoptotic Pathways. Science 2004, 305, 1163–1167. [Google Scholar] [CrossRef]
- Yadav, M.; Singh, A.K.; Kumar, H.; Rao, G.; Chakravarti, B.; Gurjar, A.; Dogra, S.; Kushwaha, S.; Vishwakarma, A.L.; Yadav, P.N.; et al. Epidermal growth factor receptor inhibitor cancer drug gefitinib modulates cell growth and differentiation of acute myeloid leukemia cells via histamine receptors. Biochim. Biophys. Acta (BBA) Gen. Subj. 2016, 1860, 2178–2190. [Google Scholar] [CrossRef]
- Ko, J.-C.; Chiu, H.-C.; Wo, T.-Y.; Huang, Y.-J.; Tseng, S.-C.; Huang, Y.-C.; Chen, H.-J.; Syu, J.-J.; Chen, C.-Y.; Jian, Y.-T.; et al. Inhibition of p38 MAPK-dependent MutS homologue-2 (MSH2) expression by metformin enhances gefitinib-induced cytotoxicity in human squamous lung cancer cells. Lung Cancer 2013, 82, 397–406. [Google Scholar] [CrossRef]
- Sugita, S.; Ito, K.; Yamashiro, Y.; Moriya, S.; Che, X.-F.; Yokoyama, T.; Hiramoto, M.; Miyazawa, K. EGFR-independent autophagy induction with gefitinib and enhancement of its cytotoxic effect by targeting autophagy with clarithromycin in non-small cell lung cancer cells. Biochem. Biophys. Res. Commun. 2015, 461, 28–34. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Li, S.; Hai, J.; Wang, X.; Chen, T.; Quinn, M.M.; Gao, P.; Zhang, Y.; Ji, H.; Cross, D.A.; et al. Targeting HER2 Aberrations in Non–Small Cell Lung Cancer with Osimertinib. Clin. Cancer Res. 2018, 24, 2594–2604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.-R.; Suo, F.-Z.; Hu, B.; Guo, Y.-J.; Fu, D.-J.; Yu, B.; Zheng, Y.-C.; Liu, H.-M. Identification of osimertinib (AZD9291) as a lysine specific demethylase 1 inhibitor. Bioorganic Chem. 2019, 84, 164–169. [Google Scholar] [CrossRef] [PubMed]
- Tang, Z.-H.; Cao, W.-X.; Su, M.-X.; Chen, X.; Lu, J.-J. Osimertinib induces autophagy and apoptosis via reactive oxygen species generation in non-small cell lung cancer cells. Toxicol. Appl. Pharmacol. 2017, 321, 18–26. [Google Scholar] [CrossRef]
- Murai, K.K.; Pasquale, E.B. ‘Eph’ective signaling: Forward, reverse and crosstalk. J. Cell Sci. 2003, 116, 2823–2832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kania, A.; Klein, R. Mechanisms of ephrin–Eph signalling in development, physiology and disease. Nat. Rev. Mol. Cell Biol. 2016, 17, 240–256. [Google Scholar] [CrossRef] [PubMed]
- Holland, S.J.; Gale, N.W.; Gish, G.D.; Roth, R.A.; Songyang, Z.; Cantley, L.C.; Henkemeyer, M.; Yancopoulos, G.D.; Pawson, T. Juxtamembrane tyrosine residues couple the Eph family receptor EphB2/Nuk to specific SH2 domain proteins in neuronal cells. EMBO J. 1997, 16, 3877–3888. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pasquale, E.B. Eph-Ephrin Bidirectional Signaling in Physiology and Disease. Cell 2008, 133, 38–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noren, N.K.; Pasquale, E.B. Eph receptor–ephrin bidirectional signals that target Ras and Rho proteins. Cell. Signal. 2004, 16, 655–666. [Google Scholar] [CrossRef]
- Noren, N.K.; Foos, G.; Hauser, C.A.; Pasquale, E.B. The EphB4 receptor suppresses breast cancer cell tumorigenicity through an Abl–Crk pathway. Nat. Cell Biol. 2006, 8, 815–825. [Google Scholar] [CrossRef]
- Tu, Y.; He, S.; Fu, J.; Li, G.; Xu, R.; Lu, H.; Deng, J. Expression of EphrinB2 and EphB4 in glioma tissues correlated to the progression of glioma and the prognosis of glioblastoma patients. Clin. Transl. Oncol. 2012, 14, 214–220. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.R.; Singh, J.; Xia, G.; Krasnoperov, V.; Hassanieh, L.; Ley, E.J.; Scehnet, J.; Kumar, N.G.; Hawes, D.; Press, M.F.; et al. Receptor Tyrosine Kinase EphB4 Is a Survival Factor in Breast Cancer. Am. J. Pathol. 2006, 169, 279–293. [Google Scholar] [CrossRef] [Green Version]
- Castellano, G.; Reid, J.; Alberti, P.; Carcangiu, M.L.; Tomassetti, A.; Canevari, S. New Potential Ligand-Receptor Signaling Loops in Ovarian Cancer Identified in Multiple Gene Expression Studies. Cancer Res. 2006, 66, 10709–10719. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Choi, W.W.; Yan, R.; Yu, H.; Krasnoperov, V.; Kumar, S.R.; Schuckman, A.; Klumpp, D.J.; Pan, C.-X.; Quinn, D.; et al. The Differential Expression of EphB2 and EphB4 Receptor Kinases in Normal Bladder and in Transitional Cell Carcinoma of the Bladder. PLoS ONE 2014, 9, e105326. [Google Scholar] [CrossRef]
- Merchant, A.A.; Jorapur, A.; McManus, A.; Liu, R.; Krasnoperov, V.; Chaudhry, P.; Singh, M.; Harton, L.; Agajanian, M.; Kim, M.; et al. EPHB4 is a therapeutic target in AML and promotes leukemia cell survival via AKT. Blood Adv. 2017, 1, 1635–1644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oda, K.; Arakawa, H.; Tanaka, T.; Matsuda, K.; Tanikawa, C.; Mori, T.; Nishimori, H.; Tamai, K.; Tokino, T.; Nakamura, Y.; et al. p53AIP1, a Potential Mediator of p53-Dependent Apoptosis, and Its Regulation by Ser-46-Phosphorylated p53. Cell 2000, 102, 849–862. [Google Scholar] [CrossRef] [Green Version]
- Taira, N.; Nihira, K.; Yamaguchi, T.; Miki, Y.; Yoshida, K. DYRK2 Is Targeted to the Nucleus and Controls p53 via Ser46 Phosphorylation in the Apoptotic Response to DNA Damage. Mol. Cell 2007, 25, 725–738. [Google Scholar] [CrossRef]
- Li, J.; Sun, Y.; Wang, X.; Wang, J.; Zhu, Y. The expressions of EphB4 and ephrinB2 in lung adenocarcinomas: A high level of the EphB4 protein is associated with lymph node metastasis. Int. J. Clin. Exp. Pathol. 2019, 12, 3447–3452. [Google Scholar] [PubMed]
- Zheng, M.-F.; Ye, S.-G.; Chen, J.Y. EphB4 gene polymorphism and protein expression in non-small-cell lung cancer. Mol. Med. Rep. 2012, 6, 405–408. [Google Scholar] [CrossRef]
- Ferguson, B.D.; Liu, R.; Rolle, C.E.; Tan, Y.-H.C.; Krasnoperov, V.; Kanteti, R.; Tretiakova, M.S.; Cervantes, G.M.; Hasina, R.; Hseu, R.D.; et al. The EphB4 Receptor Tyrosine Kinase Promotes Lung Cancer Growth: A Potential Novel Therapeutic Target. PLoS ONE 2013, 8, e67668. [Google Scholar] [CrossRef] [Green Version]
- Koch, H.; Busto, M.D.C.; Kramer, K.; Médard, G.; Kuster, B. Chemical Proteomics Uncovers EPHA2 as a Mechanism of Acquired Resistance to Small Molecule EGFR Kinase Inhibition. J. Proteome Res. 2015, 14, 2617–2625. [Google Scholar] [CrossRef]
- Keum, J.S.; Kong, G.; Yang, S.C.; Shin, D.H.; Park, S.S.; Lee, J.H.; Lee, J.D. Cyclin D1 overexpression is an indicator of poor prognosis in resectable non-small cell lung cancer. Br. J. Cancer 1999, 81, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Gautschi, O.; Ratschiller, D.; Gugger, M.; Betticher, D.C.; Heighway, J. Cyclin D1 in non-small cell lung cancer: A key driver of malignant transformation. Lung Cancer 2007, 55, 1–14. [Google Scholar] [CrossRef]
- Willis, C.; Fiander, M.; Tran, D.; Korytowsky, B.; Thomas, J.-M.; Calderon, F.; Zyczynski, T.M.; Brixner, D.; Stenehjem, D. Tumor mutational burden in lung cancer: A systematic literature review. Oncotarget 2019, 10, 6604–6622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stallaert, W.; Brüggemann, Y.; Sabet, O.; Baak, L.; Gattiglio, M.; Bastiaens, P.I.H. Contact inhibitory Eph signaling suppresses EGF-promoted cell migration by decoupling EGFR activity from vesicular recycling. Sci. Signal. 2018, 11, eaat0114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xia, G.; Kumar, S.R.; Stein, J.P.; Singh, J.; Krasnoperov, V.; Zhu, S.; Hassanieh, L.; Smith, D.L.; Buscarini, M.; Broek, D.; et al. EphB4 receptor tyrosine kinase is expressed in bladder cancer and provides signals for cell survival. Oncogene 2005, 25, 769–780. [Google Scholar] [CrossRef] [Green Version]
- Xia, G.; Kumar, S.R.; Masood, R.; Zhu, S.; Reddy, R.; Krasnoperov, V.; Quinn, D.I.; Henshall, S.M.; Sutherland, R.L.; Pinski, J.K.; et al. EphB4 Expression and Biological Significance in Prostate Cancer. Cancer Res. 2005, 65, 4623–4632. [Google Scholar] [CrossRef] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EphB4 Immunoreactivity | |||
|---|---|---|---|
| Low (n = 43) | High (n = 41) | p Value | |
| Age | 67.5 ± 9.55 | 65.3 ± 9.41 | 0.2920 |
| Sex | 0.2639 | ||
| Male | 21 | 25 | |
| Female | 22 | 16 | |
| Smoking Index | 371.8 ± 521.7 | 482.8 ± 597.2 | 0.3665 |
| pStage | 0.6505 | ||
| I | 30 | 25 | |
| II | 4 | 6 | |
| III | 9 | 10 | |
| pT | 0.0135 * | ||
| 1 | 28 | 14 | |
| 2 | 14 | 23 | |
| 3 | 1 | 4 | |
| pN | 0.7048 | ||
| 0 | 34 | 31 | |
| 1–3 | 9 | 10 | |
| Tumor size | 23.14± 11.93 | 31.27± 12.13 | 0.0027 * |
| Ki-67 Labeling index (%) | 4.58± 5.61 | 13.14± 13.27 | 0.0004 * |
| EGFR mutation | 0.0827 | ||
| Positive | 27 (Ex19Del: 14, L858R: 12, Ex19Del & L858R: 1) | 18 (Ex19Del: 8, L858R: 7, Ex19Del & L858R: 2, G719X: 1) | |
| Negative | 16 | 23 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nanamiya, R.; Saito-Koyama, R.; Miki, Y.; Inoue, C.; Asavasupreechar, T.; Abe, J.; Sato, I.; Sasano, H. EphB4 as a Novel Target for the EGFR-Independent Suppressive Effects of Osimertinib on Cell Cycle Progression in Non-Small Cell Lung Cancer. Int. J. Mol. Sci. 2021, 22, 8522. https://doi.org/10.3390/ijms22168522
Nanamiya R, Saito-Koyama R, Miki Y, Inoue C, Asavasupreechar T, Abe J, Sato I, Sasano H. EphB4 as a Novel Target for the EGFR-Independent Suppressive Effects of Osimertinib on Cell Cycle Progression in Non-Small Cell Lung Cancer. International Journal of Molecular Sciences. 2021; 22(16):8522. https://doi.org/10.3390/ijms22168522
Chicago/Turabian StyleNanamiya, Ren, Ryoko Saito-Koyama, Yasuhiro Miki, Chihiro Inoue, Teeranut Asavasupreechar, Jiro Abe, Ikuro Sato, and Hironobu Sasano. 2021. "EphB4 as a Novel Target for the EGFR-Independent Suppressive Effects of Osimertinib on Cell Cycle Progression in Non-Small Cell Lung Cancer" International Journal of Molecular Sciences 22, no. 16: 8522. https://doi.org/10.3390/ijms22168522