
Effect of L- to D-Amino Acid Substitution on Stability and Activity of Antitumor Peptide RDP215 against Human Melanoma and Glioblastoma

Abstract
:1. Introduction
2. Results
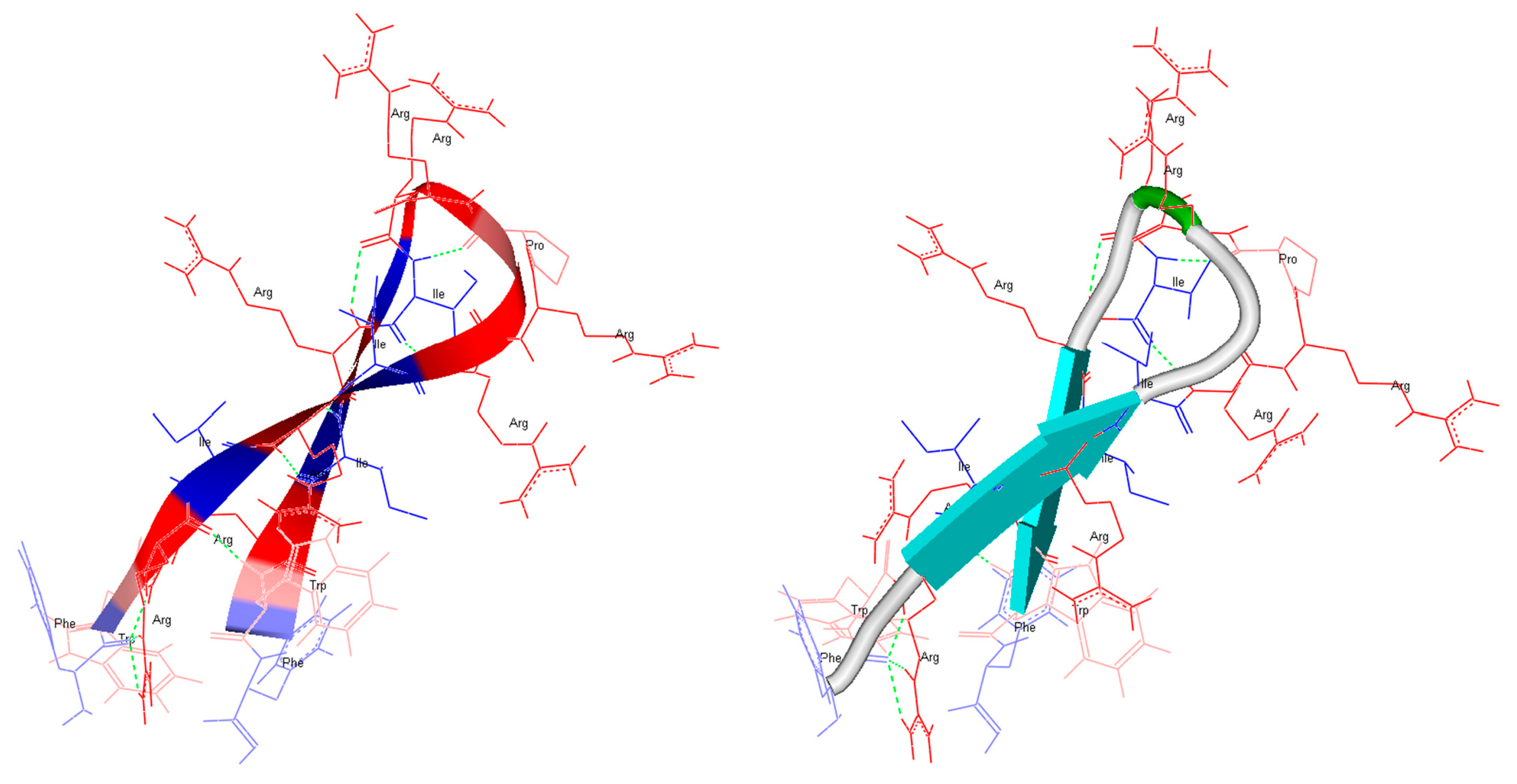
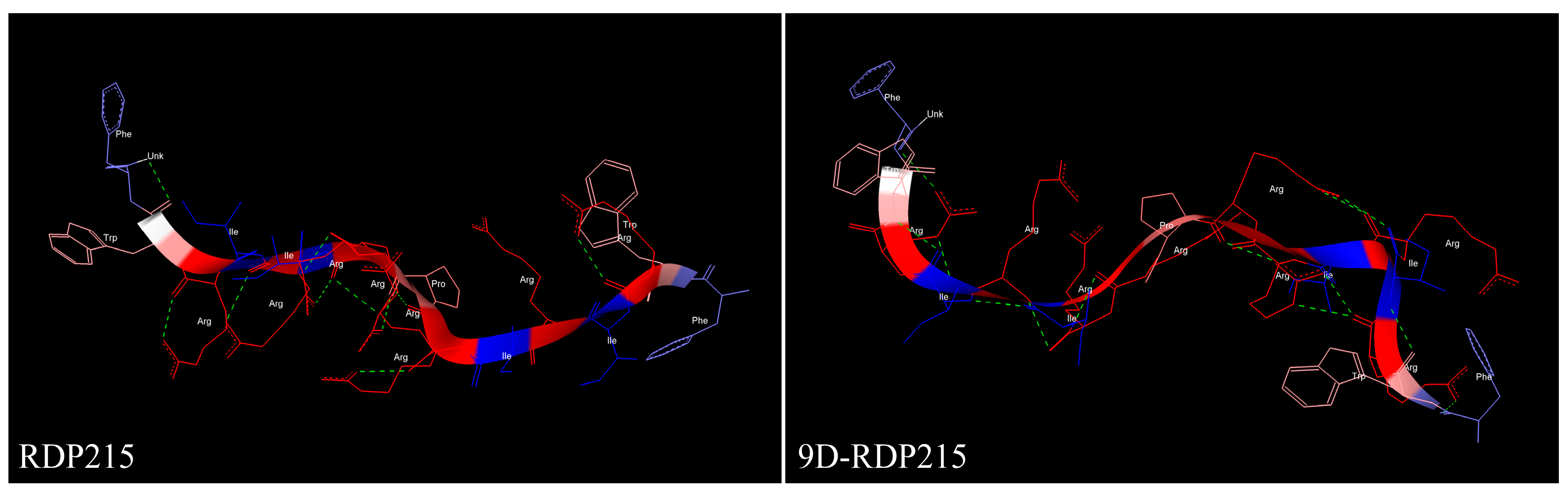
2.1. In Silico Structure Prediction of RDP215 and 9D-RDP215
2.2. Effect of RDP215 and 9D-RDP215 on Liposomal Model Systems Mimicking Cancer and Non-Cancer Membranes
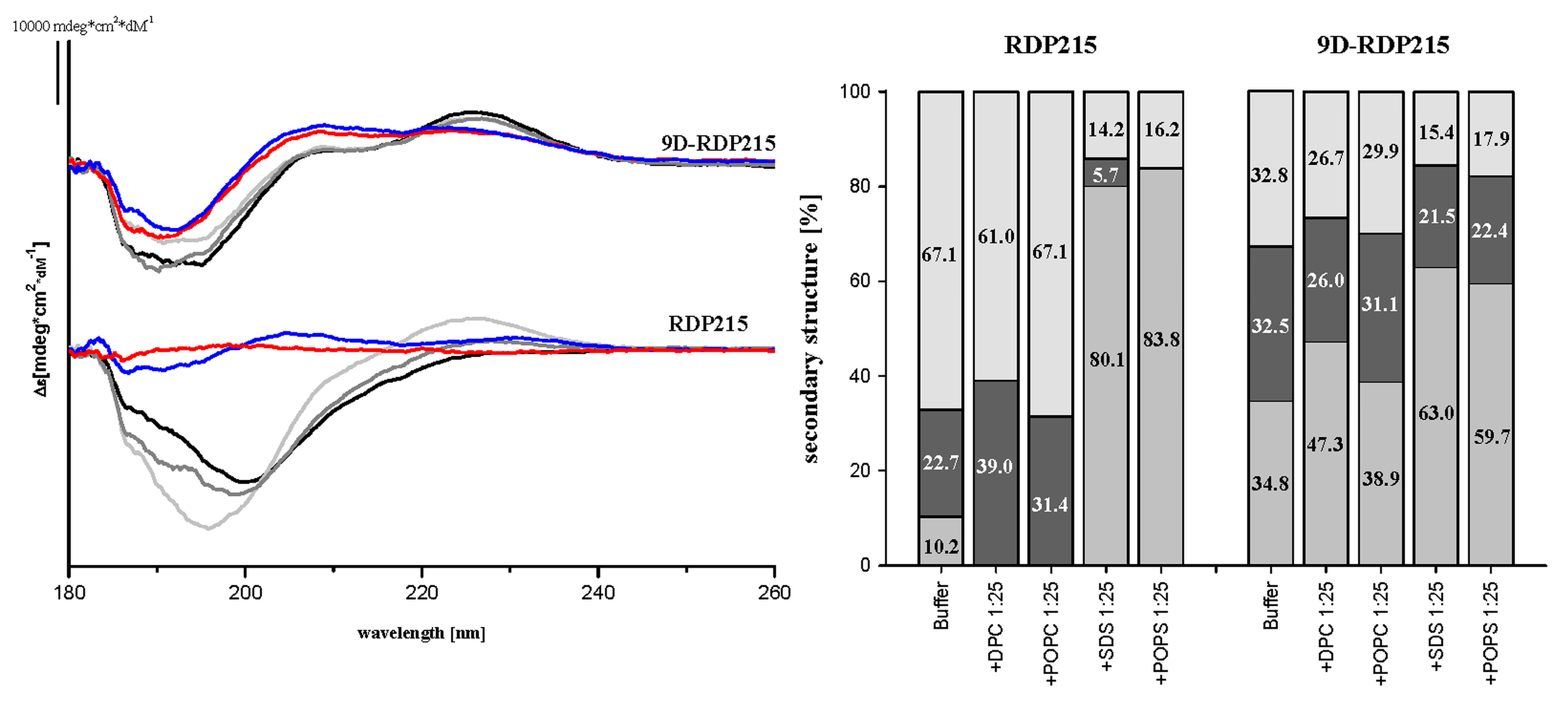
2.2.1. Membrane Induced Structure of RDP215 and 9D-RDP215
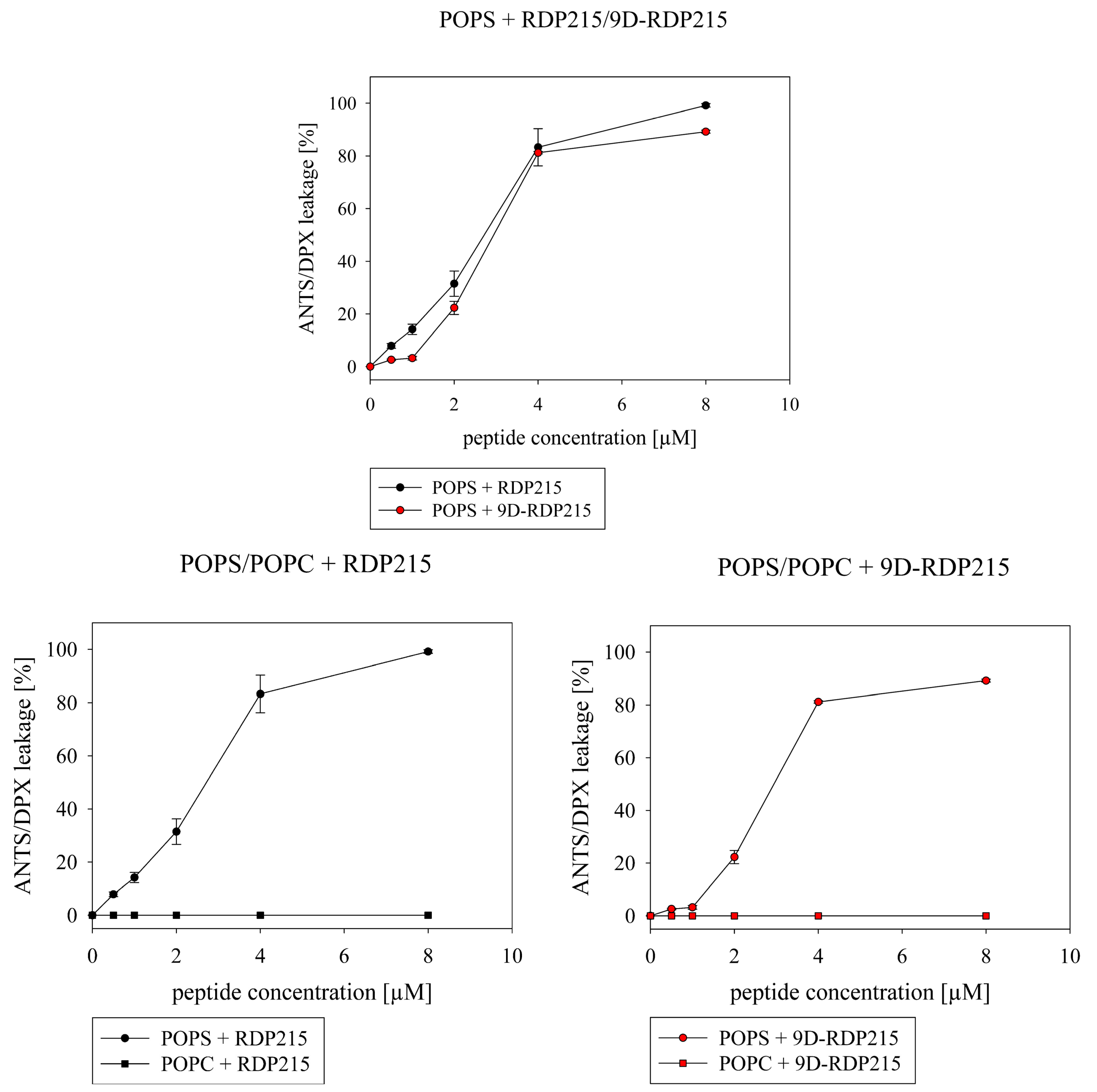
2.2.2. Effect of D-Amino Acids on Membrane Permeabilization
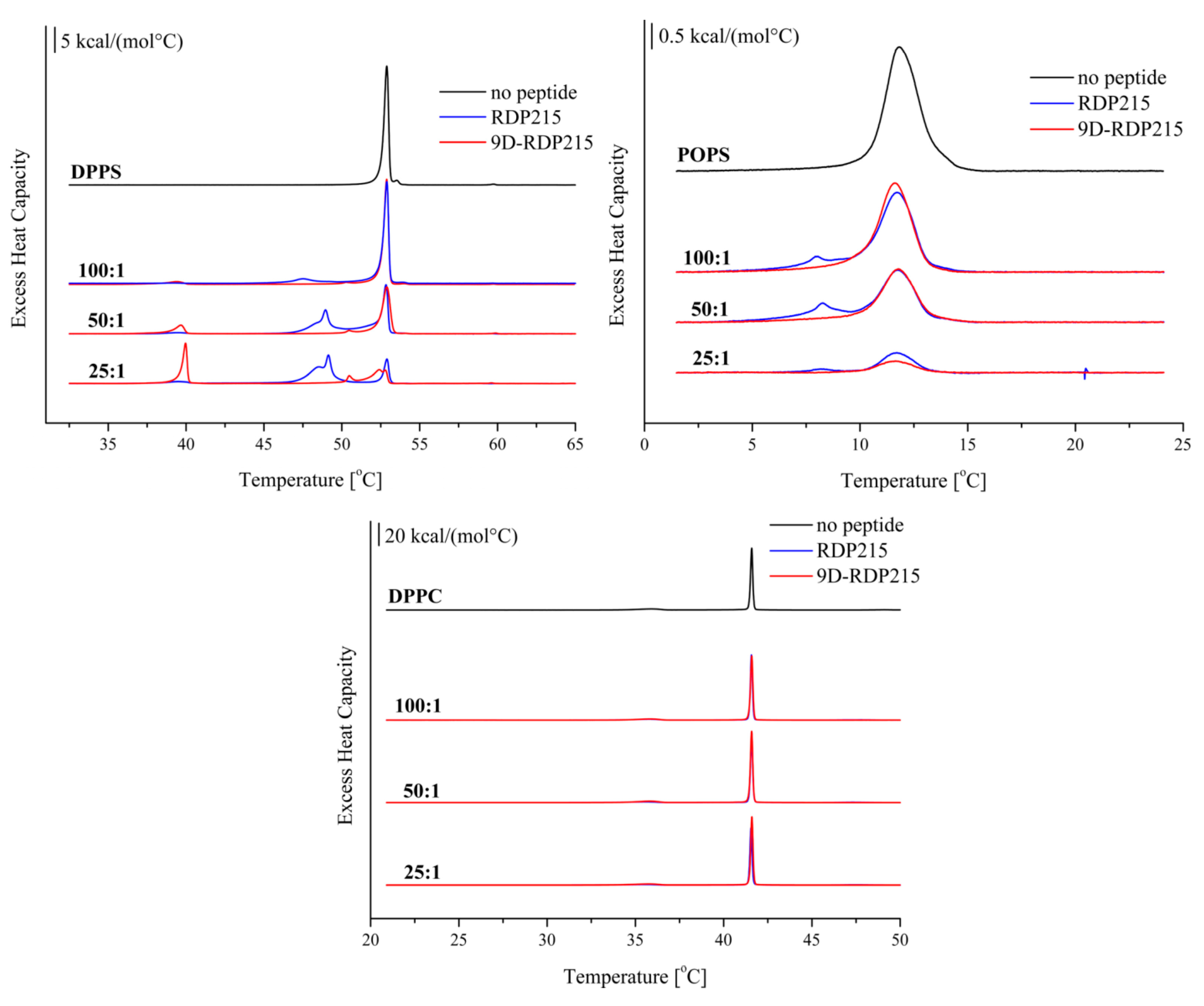
2.2.3. Effect of D-Amino Acids on Membrane Perturbation and Destabilization
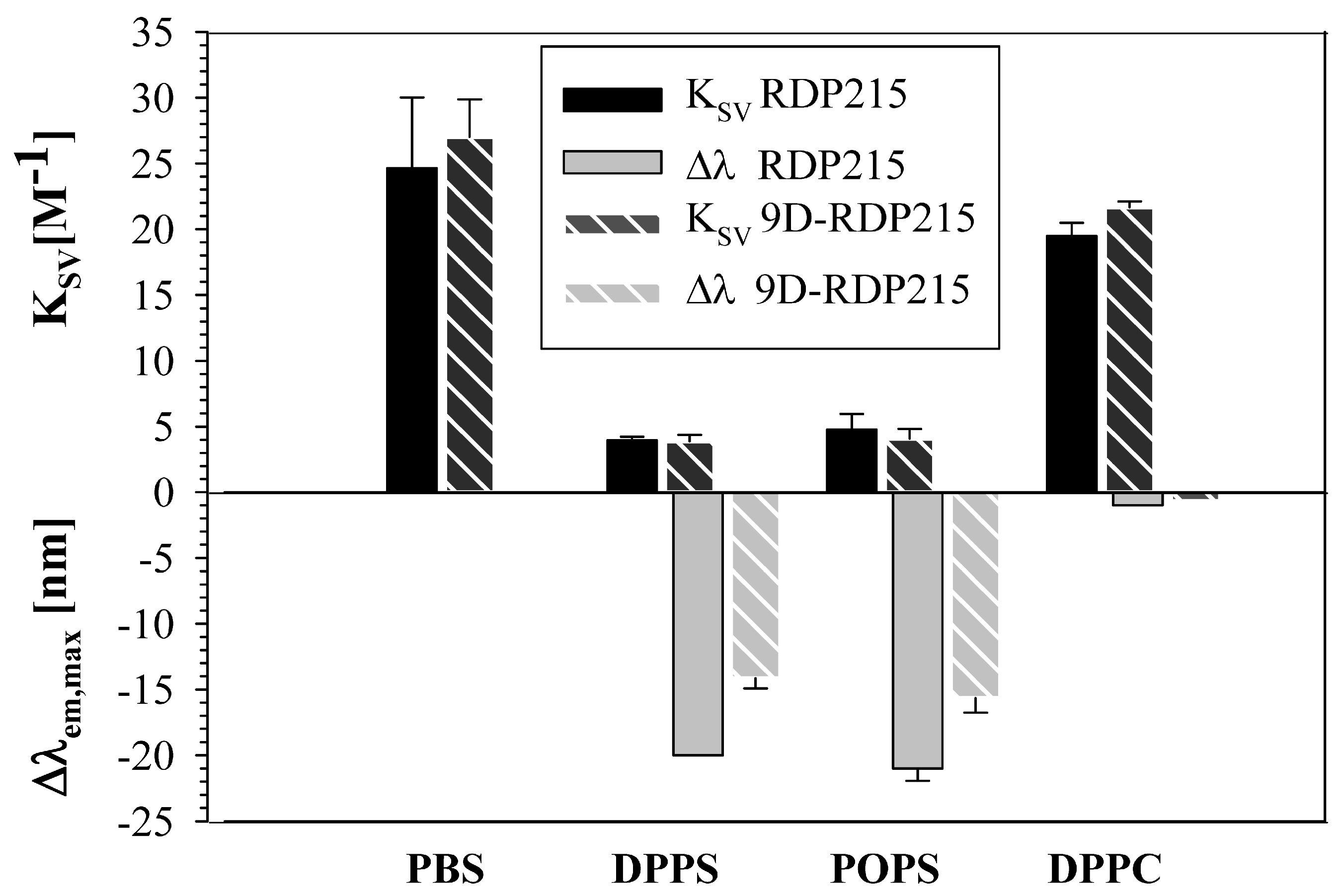
2.2.4. Membrane Interaction and Penetration by RDP215 and 9D-RDP215
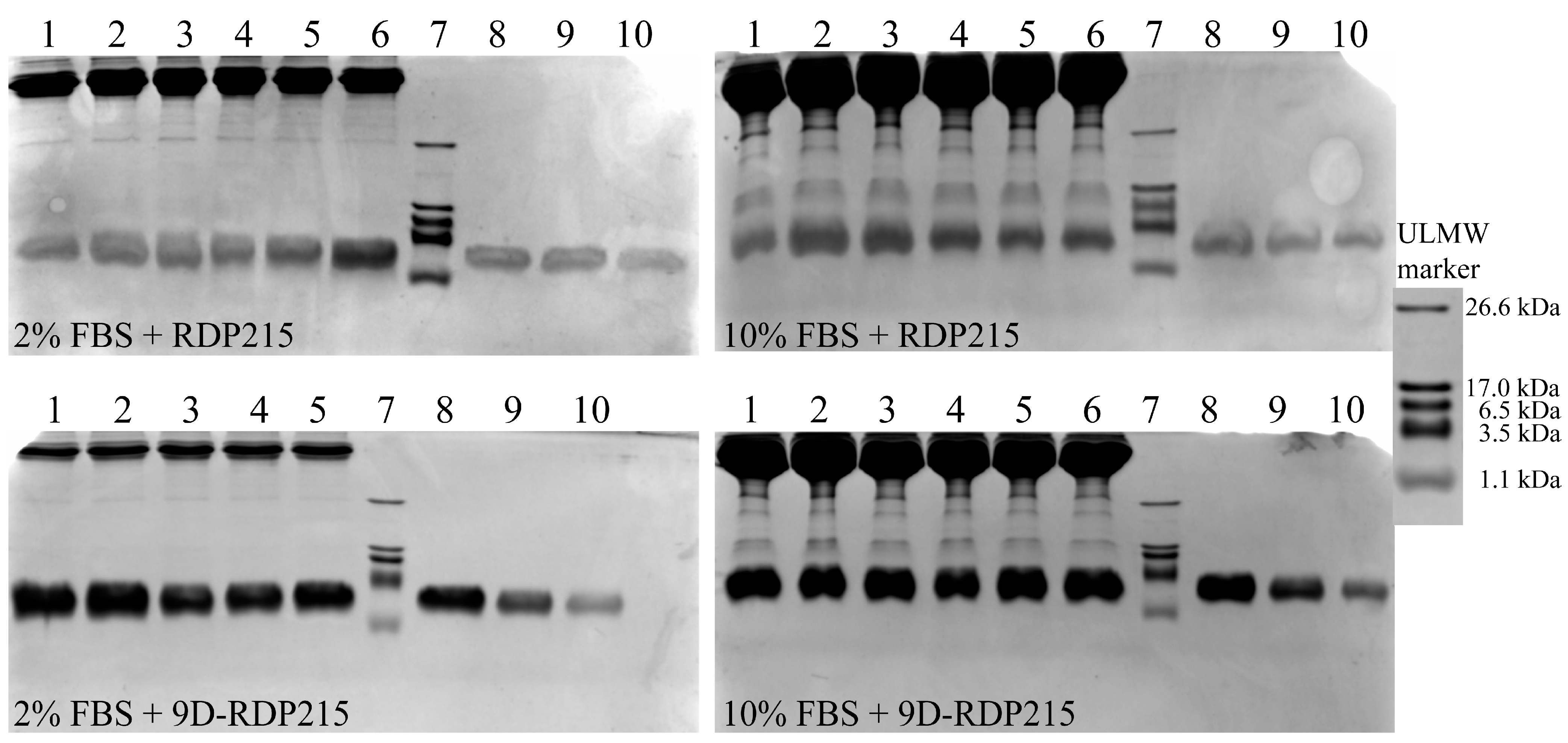
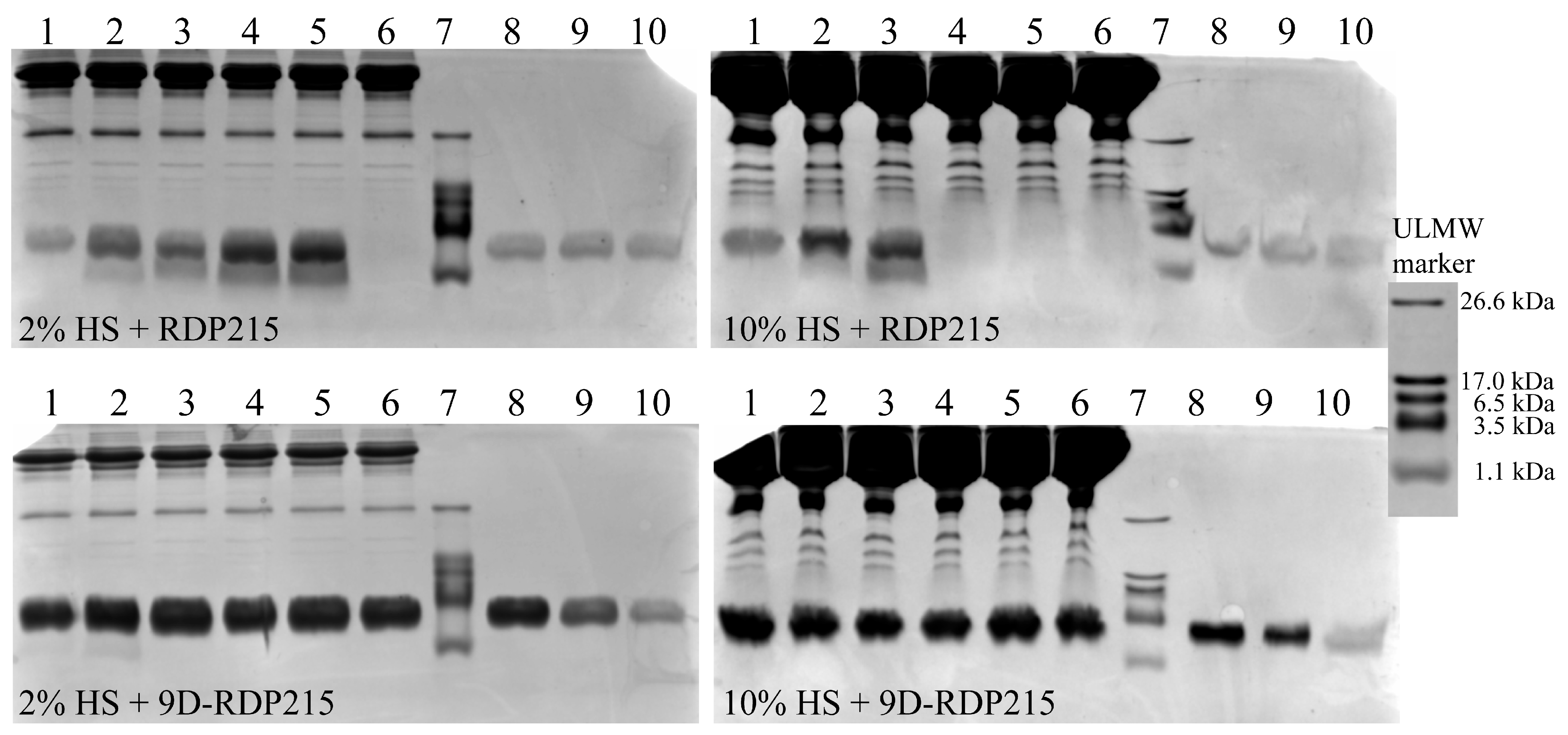
2.3. Effect of D-Amino Acids on Peptide Stability-Ex Vitro Studies
2.4. Effect of D-Amino Acids on Peptide Stability-In Vitro Studies in Melanoma
2.4.1. Peptide Toxicity and Specificity for Melanoma A375 and Normal Human Dermal Fibroblasts NHDF under Standard Serum Conditions
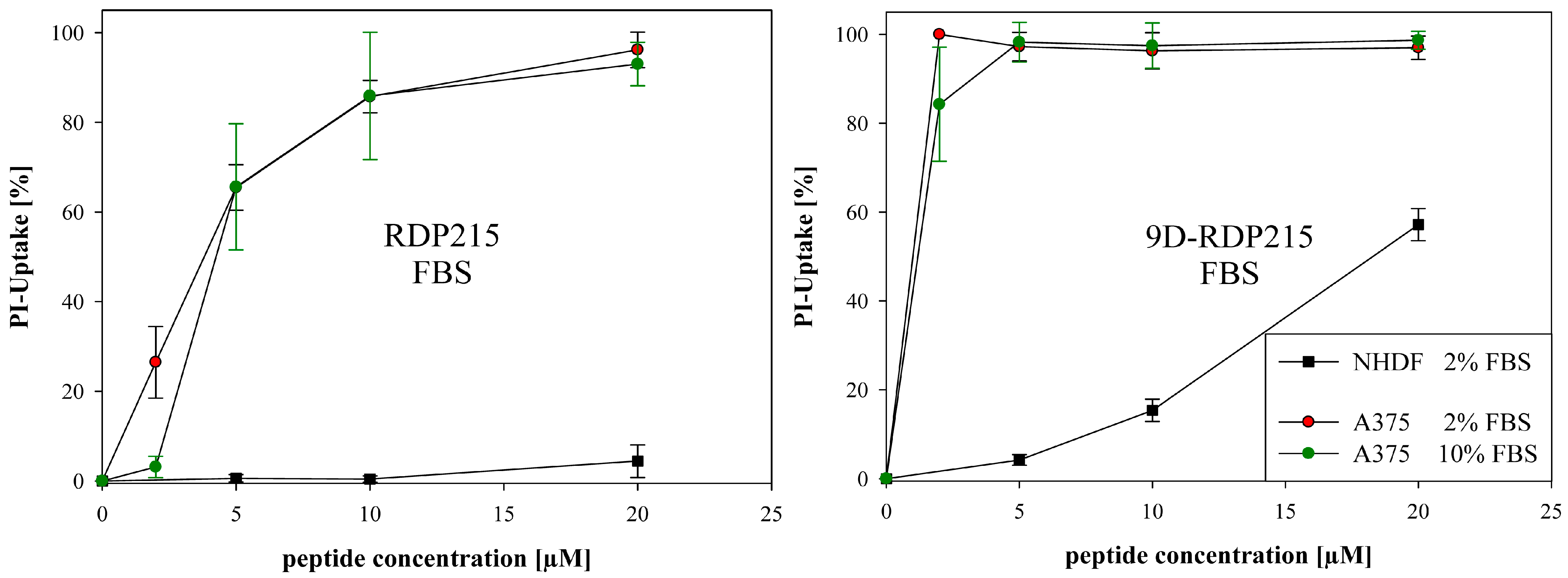
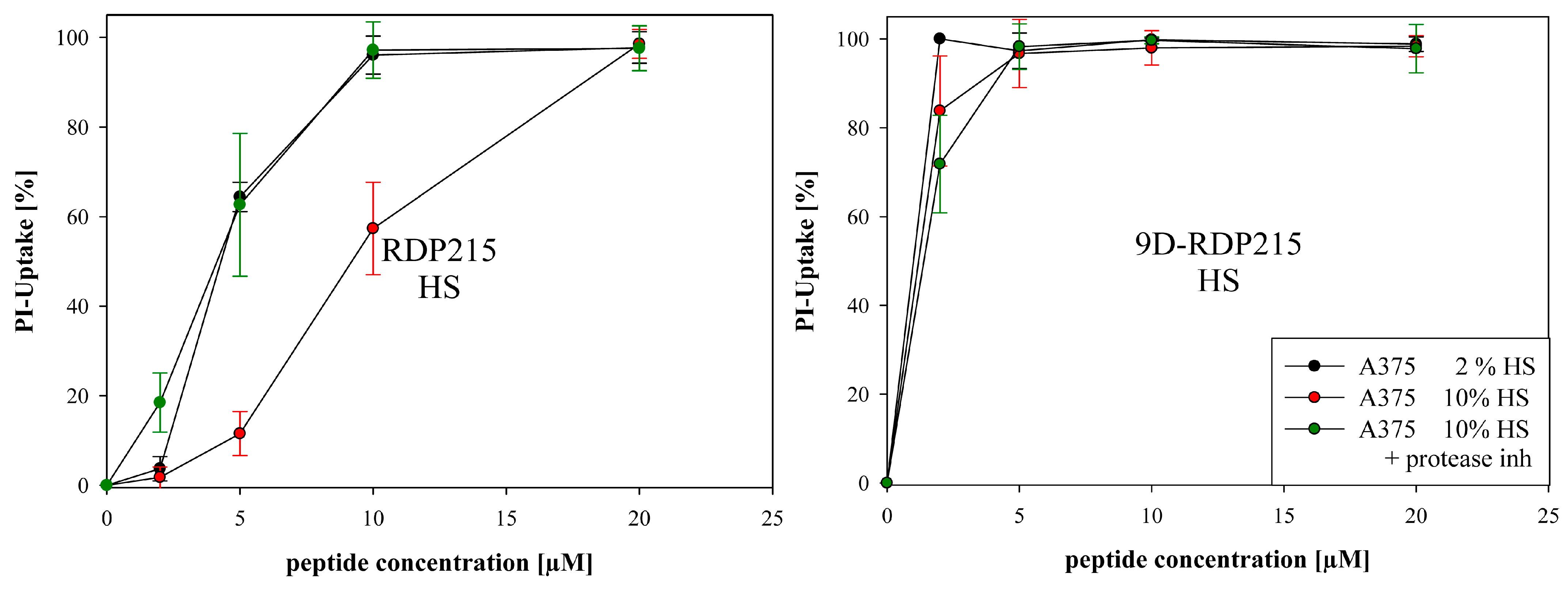
2.4.2. Peptide Toxicity and Stability in Presence of Different Concentrations of Fetal Bovine Serum FBS and Human Serum HS
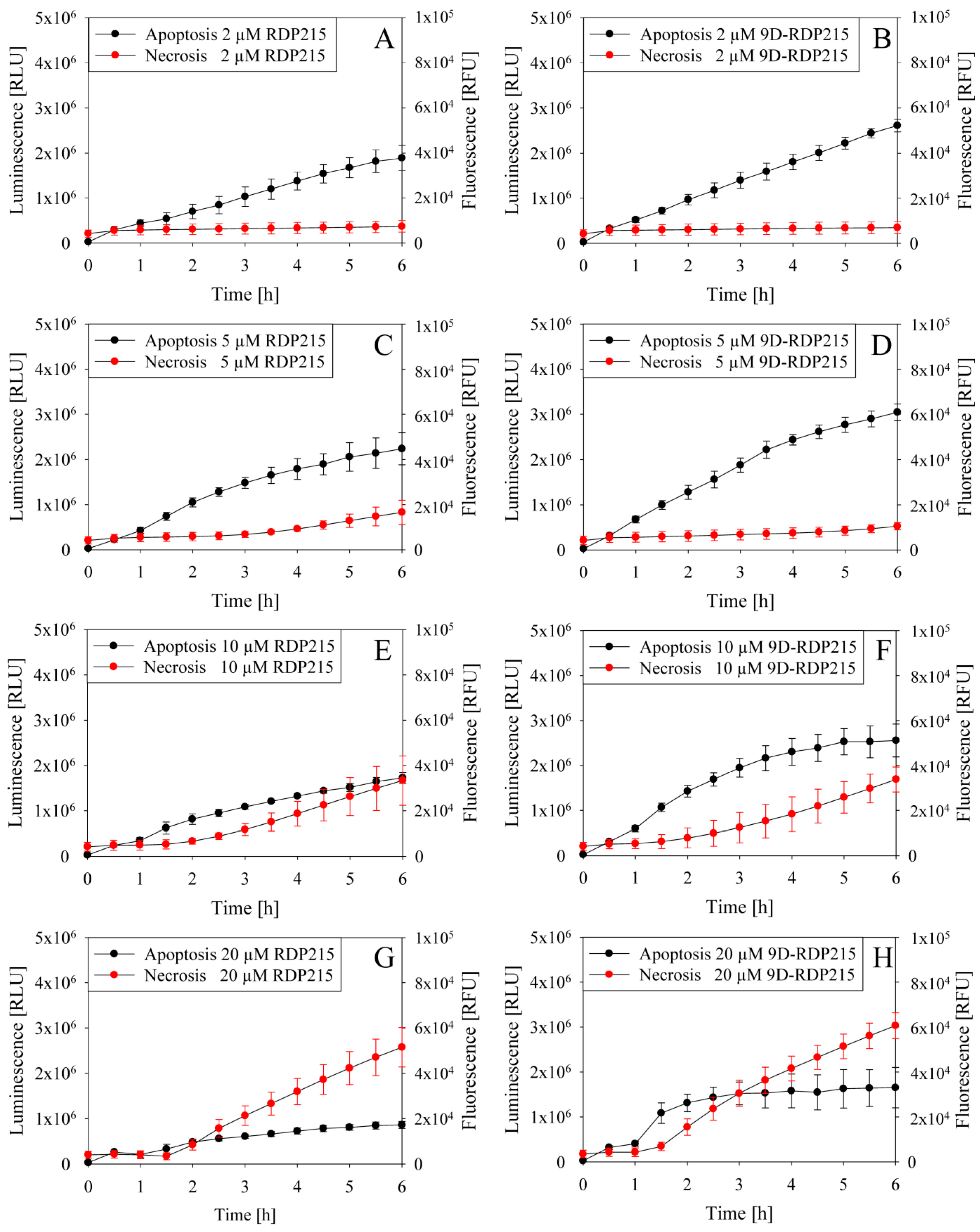
2.4.3. In Vitro Studies on Peptide Induced Killing Mechanism
2.5. Effect of D-Amino Acids on Peptide Toxicity—2D In Vitro Studies in Glioblastoma
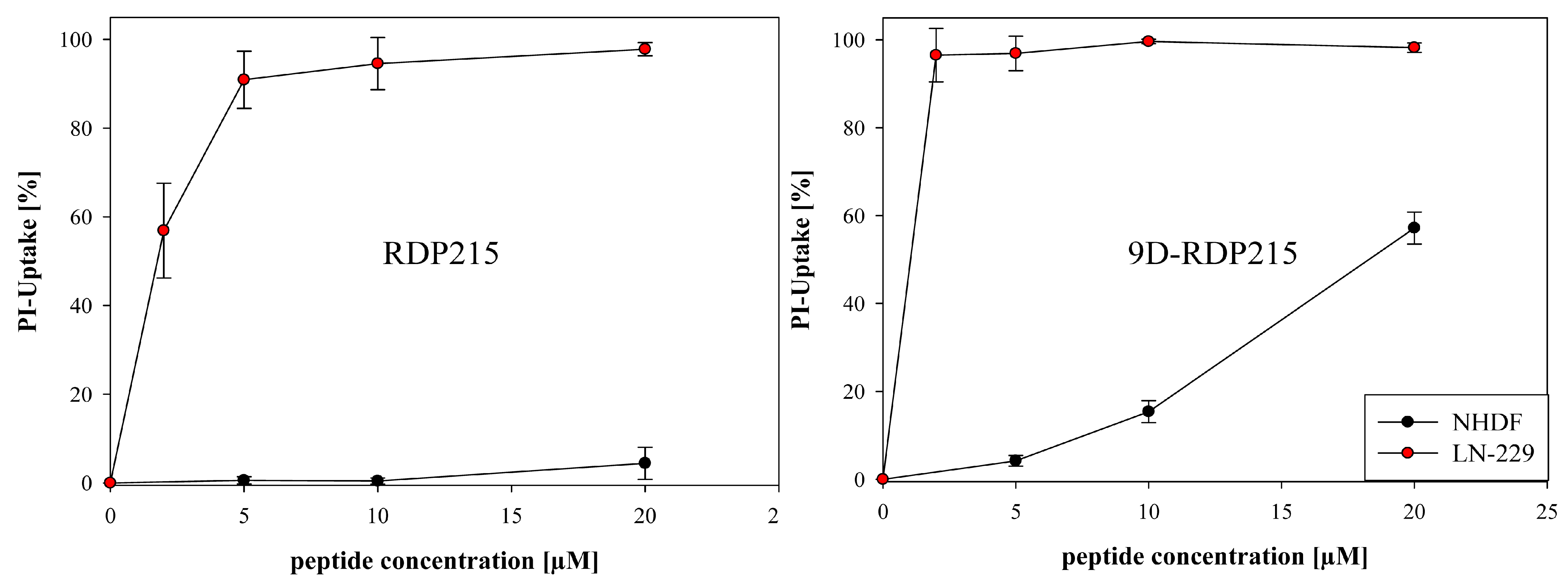
2.5.1. Peptide Toxicity and Specificity for Glioblastoma LN-229 and Normal Human Dermal Fibroblasts NHDF
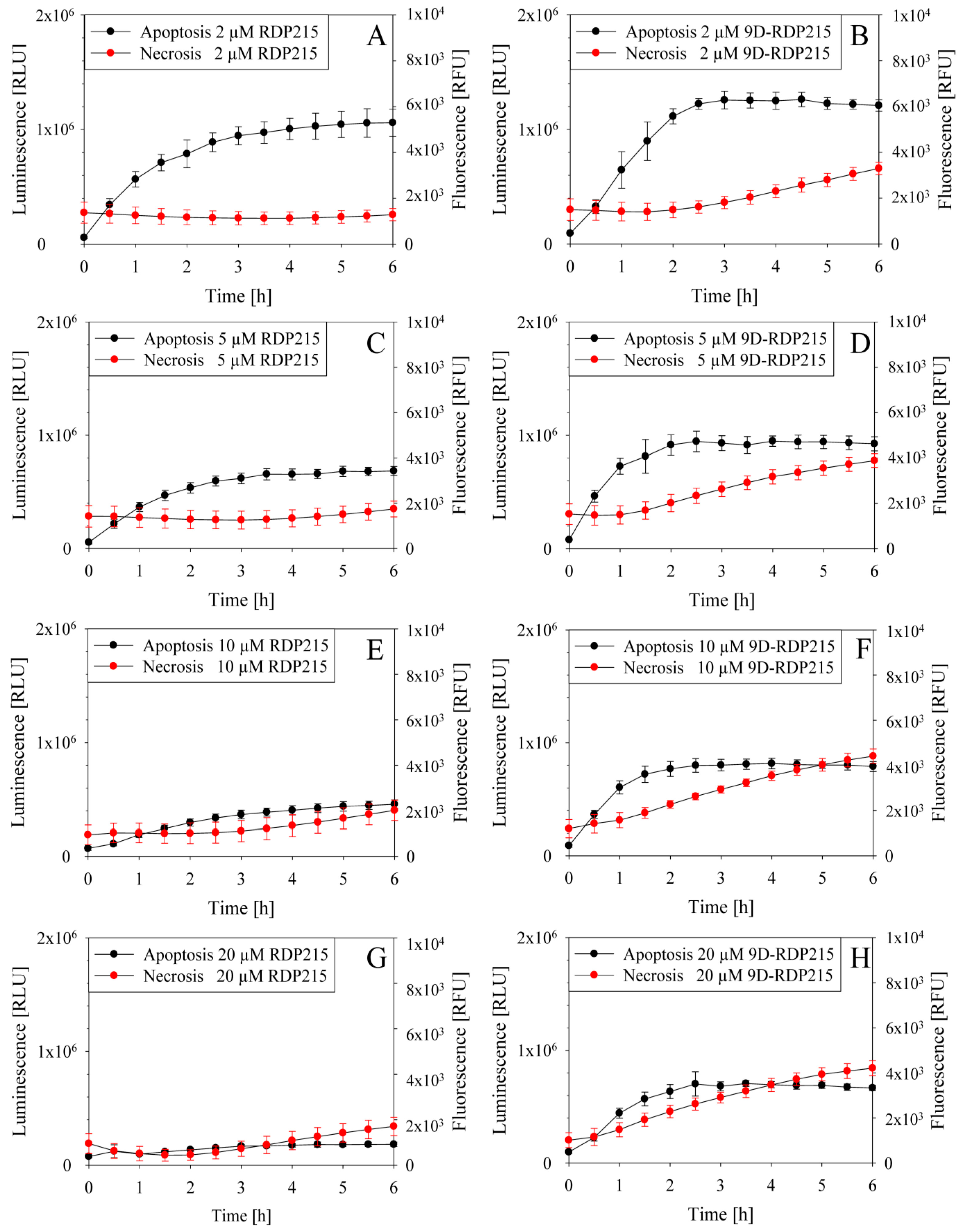
2.5.2. Studies on Peptide Induced Killing Mechanism
2.6. Effect of D-Amino Acids on Peptide Toxicity—3D In Vitro Studies on Glioblastoma Spheroids
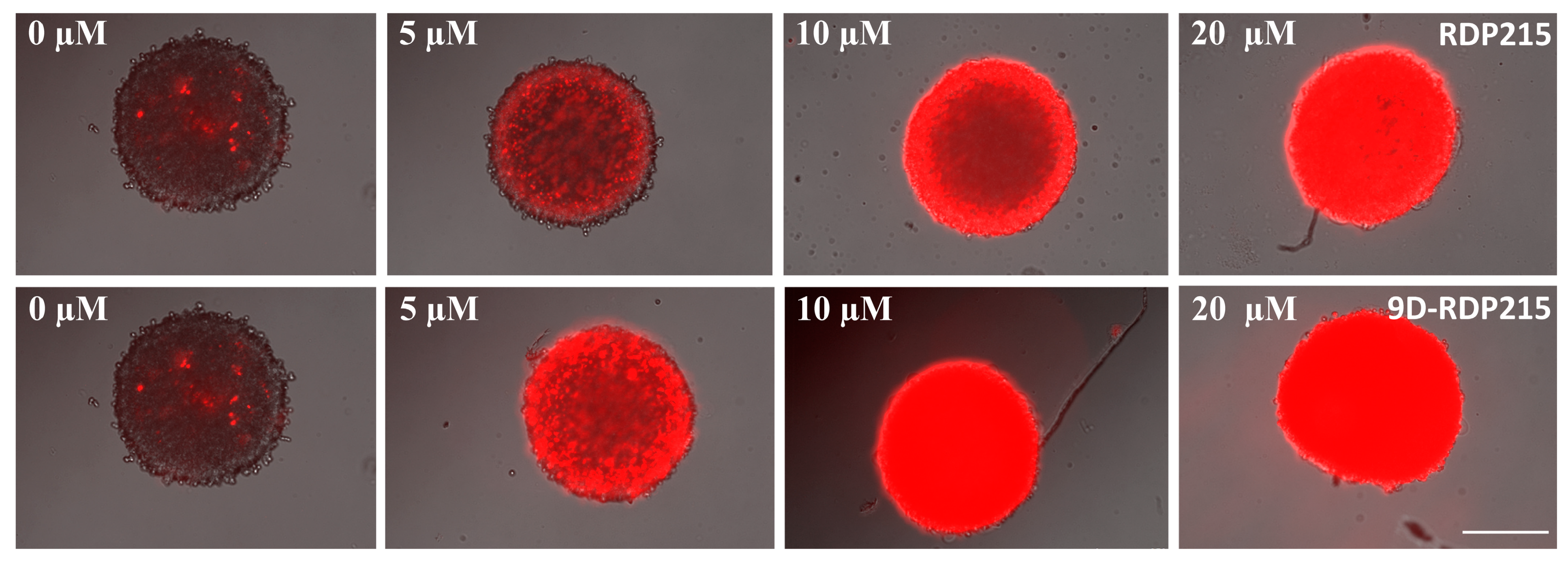
2.6.1. Direct Toxicity-PI-Uptake of MCTS and Concentration Dependence of Killing by RDP215 and 9D-RDP215
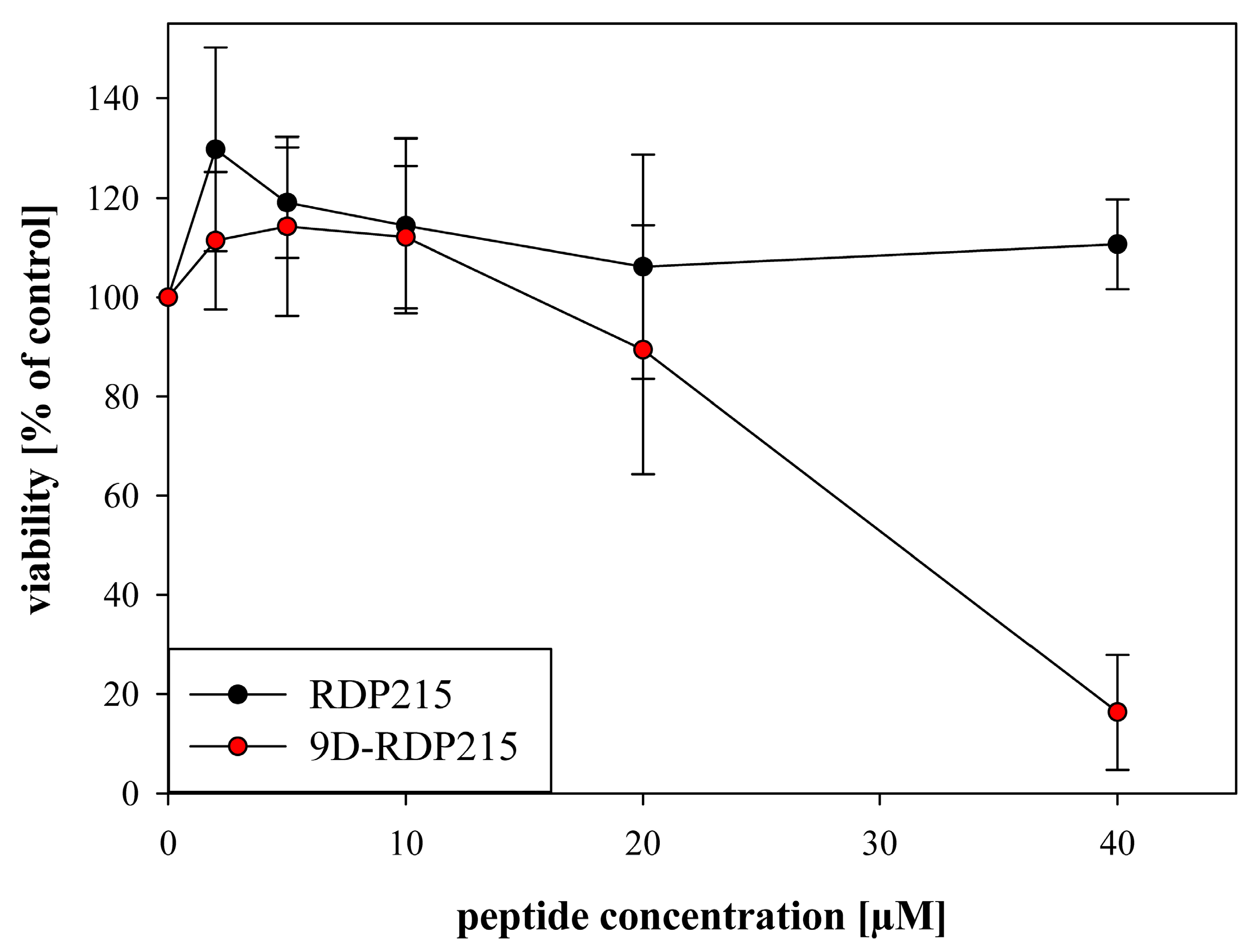
2.6.2. Effect of D-Amino Acids on Impairment of Viability of MCTS upon Passage through Blood Brain Barrier
3. Discussion
4. Materials and Methods
4.1. Peptides
4.2. Lipids
4.3. Cell Lines and Cell Culture
4.4. Liposomal Model Studies
4.4.1. Liposome Preparation
4.4.2. Circular Dichroism (CD)
4.4.3. ANTS/DPX Leakage Experiments
4.4.4. Differential Scanning Calorimetry
4.4.5. Tryptophan/Acrylamide Quenching
4.5. Ex Vitro Studies
Stability Study
4.6. In Vitro Experiments
4.6.1. Toxicity Assay—Propidium Iodide-Uptake Assay
4.6.2. Influence of Serum on Peptide Activity
4.6.3. Apoptosis-/Necrosis Assay
4.6.4. Caspase-3/7 Assay
4.6.5. 3D Cell Culture—Generation of Multicellular Tumor Spheroids (MCTS)
4.6.6. Fluorescence Microscopy: PI-Uptake of MCTS
4.6.7. 3D-Viability Assay
4.6.8. Stimulation of the Blood-Brain Barrier (BBB)
4.7. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021. [Google Scholar] [CrossRef]
- Shewach, D.S.; Kuchta, R.D. Introduction to cancer chemotherapeutics. Chem. Rev. 2009, 109, 2859–2861. [Google Scholar] [CrossRef] [Green Version]
- Mohammadpour, A.; Derakhshan, M.; Darabi, H.; Hedayat, P.; Momeni, M. Melanoma: Where we are and where we go. J. Cell. Physiol. 2019, 234, 3307–3320. [Google Scholar] [CrossRef]
- Omuro, A.; DeAngelis, L.M. Glioblastoma and other malignant gliomas: A clinical review. JAMA 2013, 310, 1842–1850. [Google Scholar] [CrossRef] [PubMed]
- Schadendorf, D.; Fisher, D.E.; Garbe, C.; Gershenwald, J.E.; Grob, J.-J.; Halpern, A.; Herlyn, M.; Marchetti, M.A.; McArthur, G.; Ribas, A.; et al. Melanoma. Nat. Rev. Dis. Primers 2015, 1, 15003. [Google Scholar] [CrossRef]
- Rastrelli, M.; Tropea, S.; Rossi, C.R.; Alaibac, M. Melanoma: Epidemiology, risk factors, pathogenesis, diagnosis and classifi-cation. In Vivo 2014, 28, 1005–1011. [Google Scholar] [PubMed]
- Malissen, N.; Grob, J.-J. Metastatic melanoma: Recent therapeutic progress and future perspectives. Drugs 2018, 78, 1197–1209. [Google Scholar] [CrossRef]
- Winder, M.; Virós, A. Mechanisms of drug resistance in melanoma. In Mechanisms of Drug Resistance in Cancer Therapy; Mandalà, M., Romano, E., Eds.; Handbook of Experimental Pharmacology; Springer Nature: Basel, Switzerland, 2018; Volume 249, pp. 91–108. [Google Scholar] [CrossRef]
- Arvold, N.D.; Reardon, D.A. Treatment options and outcomes for glioblastoma in the elderly patient. Clin. Interv. Aging 2014, 9, 357–367. [Google Scholar] [CrossRef] [Green Version]
- Ohgaki, H.; Kleihues, P. The definition of primary and secondary glioblastoma. Clin. Cancer Res. 2013, 19, 764–772. [Google Scholar] [CrossRef] [Green Version]
- Holland, E.C. Glioblastoma multiforme: The terminator. Proc. Natl. Acad. Sci. USA 2000, 97, 6242–6244. [Google Scholar] [CrossRef] [Green Version]
- Stupp, R.; Mason, W.P.; van den Bent, M.J.; Weller, M.; Fisher, B.; Taphoorn, M.J.B.; Belanger, K.; Brandes, A.A.; Marosi, C.; Bogdahn, U.; et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N. Engl. J. Med. 2005, 352, 987–996. [Google Scholar] [CrossRef]
- Jain, K.K. A critical overview of targeted therapies for glioblastoma. Front. Oncol. 2018, 8, 419. [Google Scholar] [CrossRef]
- Ostrom, Q.T.; Bauchet, L.; Davis, F.G.; Deltour, I.; Fisher, J.L.; Langer, C.E.; Pekmezci, M.; Schwartzbaum, J.A.; Turner, M.C.; Walsh, K.M.; et al. The epidemiology of glioma in adults: A “state of the science” review. Neuro Oncol. 2014, 16, 896–913. [Google Scholar] [CrossRef] [Green Version]
- Davis, M.E. Glioblastoma: Overview of disease and treatment. Clin. J. Oncol. Nurs. 2016, 20, S2–S8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanif, F.; Muzaffar, K.; Perveen, K.; Malhi, S.M.; Simjee, S.U. Glioblastoma multiforme: A review of its epidemiology and pathogenesis through clinical presentation and treatment. Asian Pac. J. Cancer Prev. 2017, 18, 3–9. [Google Scholar] [CrossRef]
- Iacob, G.; Dinca, E.B. Current data and strategy in glioblastoma multiforme. J. Med. Life 2009, 2, 386–393. [Google Scholar] [PubMed]
- Li, W.; Graeber, M.B. The molecular profile of microglia under the influence of glioma. Neuro Oncol. 2012, 14, 958–978. [Google Scholar] [CrossRef] [Green Version]
- Patel, M.M.; Patel, B.M. Crossing the blood-brain barrier: Recent advances in drug delivery to the brain. CNS Drugs 2017, 31, 109–133. [Google Scholar] [CrossRef]
- Kumar, P.; Wu, H.; McBride, J.L.; Jung, K.-E.; Kim, M.H.; Davidson, B.L.; Lee, S.K.; Shankar, P.; Manjunath, N. Transvascular delivery of small interfering RNA to the central nervous system. Nature 2007, 448, 39–43. [Google Scholar] [CrossRef]
- Oller-Salvia, B.; Teixidó, M.; Giralt, E. From venoms to BBB shuttles: Synthesis and blood-brain barrier transport assessment of apamin and a nontoxic analog. Biopolymers 2013, 100, 675–686. [Google Scholar] [CrossRef]
- Gifford, J.L.; Hunter, H.N.; Vogel, H.J. Lactoferricin: A lactoferrin-derived peptide with antimicrobial, antiviral, antitumor and immunological properties. Cell. Mol. Life Sci. 2005, 62, 2588–2598. [Google Scholar] [CrossRef] [PubMed]
- Riedl, S.; Rinner, B.; Schaider, H.; Lohner, K.; Zweytick, D. Killing of melanoma cells and their metastases by human lactofer-ricin derivatives requires interaction with the cancer marker phosphatidylserine. Biometals 2014, 27, 981–997. [Google Scholar] [CrossRef] [Green Version]
- Riedl, S.; Leber, R.; Rinner, B.; Schaider, H.; Lohner, K.; Zweytick, D. Human lactoferricin derived di-peptides deploying loop structures induce apoptosis specifically in cancer cells through targeting membranous phosphatidylserine. Biochim. Biophys. Acta 2015, 1848, 2918–2931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riedl, S.; Rinner, B.; Schaider, H.; Liegl-Atzwanger, B.; Meditz, K.; Preishuber-Pflügl, J.; Grissenberger, S.; Lohner, K.; Zweytick, D. In vitro and in vivo cytotoxic activity of human lactoferricin derived antitumor peptide R-DIM-P-LF11-334 on human ma-lignant melanoma. Oncotarget 2017, 8, 71817–71832. [Google Scholar] [CrossRef] [Green Version]
- Grissenberger, S.; Riedl, S.; Rinner, B.; Leber, R.; Zweytick, D. Design of human lactoferricin derived antitumor peptides-activity and specificity against malignant melanoma in 2D and 3D model studies. Biochim. Biophys. Acta Biomembr. 2020, 1862, 183264. [Google Scholar] [CrossRef]
- Riedl, S.; Rinner, B.; Asslaber, M.; Schaider, H.; Walzer, S.; Novak, A.; Lohner, K.; Zweytick, D. In search of a novel target—Phosphatidylserine exposed by non-apoptotic tumor cells and metastases of malignancies with poor treatment efficacy. Bio-chim. Biophys. Acta 2011, 1808, 2638–2645. [Google Scholar] [CrossRef] [Green Version]
- Riedl, S.; Zweytick, D.; Lohner, K. Membrane-active host defense peptides—Challenges and perspectives for the development of novel anticancer drugs. Chem. Phys. Lipids 2011, 164, 766–781. [Google Scholar] [CrossRef] [Green Version]
- Soengas, M.S.; Capodieci, P.; Polsky, D.; Mora, J.; Esteller, M.; Opitz-Araya, X.; McCombie, R.; Herman, J.G.; Gerald, W.L.; Lazebnik, Y.A.; et al. Inactivation of the apoptosis effector Apaf-1 in malignant melanoma. Nature 2001, 409, 207–211. [Google Scholar] [CrossRef]
- Di, L. Strategic approaches to optimizing peptide ADME properties. AAPS J. 2015, 17, 134–143. [Google Scholar] [CrossRef]
- Böttger, R.; Hoffmann, R.; Knappe, D. Differential stability of therapeutic peptides with different proteolytic cleavage sites in blood, plasma and serum. PLoS ONE 2017, 12, e0178943. [Google Scholar] [CrossRef]
- Li, H.; Anuwongcharoen, N.; Malik, A.A.; Prachayasittikul, V.; Wikberg, J.E.S.; Nantasenamat, C. Roles of d-amino acids on the bioactivity of host defense peptides. Int. J. Mol. Sci. 2016, 17, 23. [Google Scholar] [CrossRef] [Green Version]
- Werle, M.; Bernkop-Schnürch, A. Strategies to improve plasma half life time of peptide and protein drugs. Amino Acids 2006, 30, 351–367. [Google Scholar] [CrossRef]
- Feng, Z.; Xu, B. Inspiration from the mirror: D-amino acid containing peptides in biomedical approaches. Biomol. Concepts 2016, 7, 179–187. [Google Scholar] [CrossRef]
- Papo, N.; Braunstein, A.; Eshhar, Z.; Shai, Y. Suppression of human prostate tumor growth in mice by a cytolytic D-, L-amino acid peptide: Membrane lysis, increased necrosis, and inhibition of prostate-specific antigen secretion. Cancer Res. 2004, 64, 5779–5786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avrahami, D.; Oren, Z.; Shai, Y. Effect of multiple aliphatic amino acids substitutions on the structure, function, and mode of action of diastereomeric membrane active peptides 6. Biochemistry 2001, 40, 12591–12603. [Google Scholar]
- Shen, Y.; Maupetit, J.; Derreumaux, P.; Tufféry, P. Improved PEP-FOLD approach for peptide and miniprotein structure prediction. J. Chem. Theory Comput. 2014, 10, 4745–4758. [Google Scholar] [CrossRef]
- Thévenet, P.; Shen, Y.; Maupetit, J.; Guyon, F.; Derreumaux, P.; Tufféry, P. PEP-FOLD: An updated de novo structure prediction server for both linear and disulfide bonded cyclic peptides. Nucleic Acids Res. 2012, 40, W288–W293. [Google Scholar] [CrossRef] [Green Version]
- Maupetit, J.; Derreumaux, P.; Tuffery, P. PEP-FOLD: An online resource for de novo peptide structure prediction. Nucleic Acids Res. 2009, 37, W498–W503. [Google Scholar] [CrossRef] [Green Version]
- Kaur, H.; Garg, A.; Raghava, G.P.S. PEPstr: A de novo method for tertiary structure prediction of small bioactive peptides. Protein Pept. Lett. 2007, 14, 626–631. [Google Scholar] [CrossRef]
- Singh, S.; Singh, H.; Tuknait, A.; Chaudhary, K.; Singh, B.; Kumaran, S.; Raghava, G.P.S. PEPstrMOD: Structure prediction of peptides containing natural, non-natural and modified residues. Biol. Direct 2015, 10, 73. [Google Scholar] [CrossRef] [Green Version]
- Bevers, E.M.; Comfurius, P.; Dekkers, D.W.; Harmsma, M.; Zwaal, R.F. Transmembrane phospholipid distribution in blood cells: Control mechanisms and pathophysiological significance. Biol. Chem. 2005, 379, 973–986. [Google Scholar]
- Utsugi, T.; Schroit, A.J.; Connor, J.; Bucana, C.D.; Fidler, I.J. Elevated expression of phosphatidylserine in the outer membrane leaflet of human tumor cells and recognition by activated human blood monocytes. Cancer Res. 1991, 51, 3062–3066. [Google Scholar]
- Papo, N.; Shai, Y. Host defense peptides as new weapons in cancer treatment. Cell. Mol. Life Sci. 2005, 62, 784–790. [Google Scholar] [CrossRef]
- Uratani, Y.; Kobayashi, M.; Yokoyama, Y.; Maeda, T.; Mitaku, S.; Hoshino, T. Phospholipids stabilize the secondary structure of the sodium-coupled branched-chain amino acid carrier of Pseudomonas aeruginosa. Biochim. Biophys. Acta 1999, 1435, 71–83. [Google Scholar] [CrossRef]
- Epand, R.M.; Bottega, R. Determination of the phase behaviour of phosphatidylethanolamine admixed with other lipids and the effects of calcium chloride: Implications for protein kinase C regulation. Biochim. Biophys Acta 2005, 944, 144–154. [Google Scholar] [CrossRef]
- Zweytick, D.; Pabst, G.; Abuja, P.M.; Jilek, A.; Blondelle, S.E.; Andrä, J.; Jerala, R.; Monreal, D.; Martinez de Tejada, G.; Lohner, K. Influence of N-acylation of a peptide derived from human lactoferricin on membrane selectivity. Biochim. Biophys. Acta 2006, 1758, 1426–1435. [Google Scholar] [CrossRef] [Green Version]
- Papo, N.; Shahar, M.; Eisenbach, L.; Shai, Y. A novel lytic peptide composed of DL-amino acids selectively kills cancer cells in culture and in mice. J. Biol. Chem. 2003, 278, 21018–21023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, S.; Zhu, R.; Zhao, Y.; An, X.; Jia, F.; Peng, J.; Ma, Z.; Zhu, Y.; Wang, J.; Su, J.; et al. Antimicrobial activity and stability of protonectin with D-amino acid substitutions. J. Pept. Sci. 2017, 23, 392–402. [Google Scholar] [CrossRef] [PubMed]
- Grieco, P.; Carotenuto, A.; Auriemma, L.; Saviello, M.R.; Campiglia, P.; Gomez-Monterrey, I.M.; Marcellini, L.; Luca, V.; Barra, D.; Novellino, E.; et al. The effect of d-amino acid substitution on the selectivity of temporin L towards target cells: Identification of a potent anti-Candida peptide. Antimicrob. Pept. Cell Membr. Microb. Surf. Interact. 2013, 1828, 652–660. [Google Scholar] [CrossRef] [PubMed]
- Altenbach, C.; Froncisz, W.; Hyde, J.S.; Hubbell, W.L. Conformation of spin-labeled melittin at membrane surfaces investigated by pulse saturation recovery and continuous wave power saturation electron paramagnetic resonance. Biophys. J. 2005, 56, 1183–1191. [Google Scholar] [CrossRef] [Green Version]
- Elmore, S. Apoptosis: A review of programmed cell death. Toxicol. Pathol. 2007, 35, 495–516. [Google Scholar] [CrossRef] [PubMed]
- Kupcho, K.; Shultz, J.; Hurst, R.; Hartnett, J.; Zhou, W.; Machleidt, T.; Grailer, J.; Worzella, T.; Riss, T.; Lazar, D.; et al. A real-time, bioluminescent annexin V assay for the assessment of apoptosis. Apoptosis 2019, 24, 184–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cesarz, Z.; Tamama, K. Spheroid culture of mesenchymal stem cells. Stem Cells Int. 2016, 2016, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Patel, N.R.; Aryasomayajula, B.; Abouzeid, A.H.; Torchilin, V.P. Cancer cell spheroids for screening of chemotherapeutics and drug-delivery systems. Ther. Deliv. 2015, 6, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Zhao, Z.; Montagne, A.; Nelson, A.R.; Zlokovic, B.V. Blood-brain barrier: From physiology to disease and back. Physiol. Rev. 2019, 99, 21–78. [Google Scholar] [CrossRef]
- Abbott, N.J.; Patabendige, A.A.K.; Dolman, D.E.M.; Yusof, S.R.; Begley, D.J. Structure and function of the blood-brain barrier. Neurobiol. Dis. 2010, 37, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Sherman, H.; Rossi, A.E. A novel three-dimensional glioma blood-brain barrier model for high-throughput testing of tumoricidal capability. Front. Oncol. 2019, 9, 351. [Google Scholar] [CrossRef] [Green Version]
- Ran, S.; Downes, A.; Thorpe, P.E. Increased exposure of anionic phospholipids on the surface of tumor blood vessels. Cancer Res. 2002, 62, 6132–6140. [Google Scholar]
- Zwaal, R.F.; Schroit, A.J. Pathophysiologic implications of membrane phospholipid asymmetry in blood cells. Blood 1997, 89, 1121–1132. [Google Scholar] [CrossRef]
- Vlieghe, P.; Lisowski, V.; Martinez, J.; Khrestchatisky, M. Synthetic therapeutic peptides: Science and market. Drug Discov. Today 2010, 15, 40–56. [Google Scholar] [CrossRef]
- Pokorny, A.; Birkbeck, T.H.; Almeida, P.F.F. Mechanism and kinetics of delta-lysin interaction with phospholipid vesicles. Biochemistry 2002, 41, 11044–11056. [Google Scholar] [CrossRef]
- Heitzer, E.; Groenewoud, A.; Meditz, K.; Lohberger, B.; Liegl-Atzwanger, B.; Prokesch, A.; Kashofer, K.; Behrens, D.; Haybaeck, J.; Kolb-Lenz, D.; et al. Human melanoma brain metastases cell line MUG-Mel1, isolated clones and their detailed characterization. Sci. Rep. 2019, 9, 4096. [Google Scholar] [CrossRef] [Green Version]
- Wodlej, C.; Riedl, S.; Rinner, B.; Leber, R.; Drechsler, C.; Voelker, D.R.; Choi, J.-Y.; Lohner, K.; Zweytick, D. Interaction of two antitumor peptides with membrane lipids—Influence of phosphatidylserine and cholesterol on specificity for melanoma cells. PLoS ONE 2019, 14, e0211187. [Google Scholar] [CrossRef] [PubMed]
- Papo, N.; Shai, Y. Effect of drastic sequence alteration and D-amino acid incorporation on the membrane binding behavior of lytic peptides. Biochemistry 2004, 43, 6393–6403. [Google Scholar] [CrossRef]
- Oren, Z.; Shai, Y. Selective lysis of bacteria but not mammalian cells by diastereomers of melittin: Structure-function study. Biochemistry 1997, 36, 1826–1835. [Google Scholar] [CrossRef] [PubMed]
- Diao, L.; Meibohm, B. Pharmacokinetics and pharmacokinetic-pharmacodynamic correlations of therapeutic peptides. Clin. Pharmacokinet. 2013, 52, 855–868. [Google Scholar] [CrossRef]
- Pietrowska, M.; Wlosowicz, A.; Gawin, M.; Widlak, P. MS-based proteomic analysis of serum and plasma: Problem of high abundant components and lights and shadows of albumin removal. Adv. Exp. Med. Biol. 2019, 1073, 57–76. [Google Scholar] [CrossRef] [PubMed]
- Heger, J.I.; Froehlich, K.; Pastuschek, J.; Schmidt, A.; Baer, C.; Mrowka, R.; Backsch, C.; Schleußner, E.; Markert, U.R.; Schmidt, A. Human serum alters cell culture behavior and improves spheroid formation in comparison to fetal bovine serum. Exp. Cell Res. 2018, 365, 57–65. [Google Scholar] [CrossRef]
- Sumann, I. Studies on Stability and Ability of Antitumor Peptides to Treat Cancer of Malignant Melanoma. Master’s Thesis, Karl-Franzens-University, Graz, Austria, 2017. [Google Scholar]
- Broussard, L.; Howland, A.; Ryu, S.; Song, K.; Norris, D.; Armstrong, C.A.; Song, P.I. Melanoma cell death mechanisms. Chonnam Med. J. 2018, 54, 135–142. [Google Scholar] [CrossRef] [Green Version]
- Escamilla-Ramírez, A.; Castillo-Rodríguez, R.A.; Zavala-Vega, S.; Jimenez-Farfan, D.; Anaya-Rubio, I.; Briseño, E.; Palencia, G.; Guevara, P.; Cruz-Salgado, A.; Sotelo, J.; et al. Autophagy as a potential therapy for malignant glioma. Pharmaceuticals 2020, 13, 156. [Google Scholar] [CrossRef]
- McArthur, G.A.; Chapman, P.B.; Robert, C.; Larkin, J.; Haanen, J.B.; Dummer, R.; Ribas, A.; Hogg, D.; Hamid, O.; Ascierto, P.A.; et al. Safety and efficacy of vemurafenib in BRAFV600E and BRAFV600K mutation-positive melanoma (BRIM-3): Extended follow-up of a phase 3, randomised, open-label study. Lancet Oncol. 2014, 15, 323–332. [Google Scholar] [CrossRef] [Green Version]
- Halaban, R.; Zhang, W.; Bacchiocchi, A.; Cheng, E.; Parisi, F.; Ariyan, S.; Krauthammer, M.; McCusker, J.P.; Kluger, Y.; Sznol, M. PLX4032, a selective BRAFV600E kinase inhibitor, activates the ERK pathway and enhances cell migration and proliferation of BRAFWT melanoma cells. Pigment Cell Melanoma Res. 2010, 23, 190–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, P.H.; Marais, R. Cancer: Melanoma troops massed. Nature 2009, 459, 336–337. [Google Scholar] [CrossRef]
- Bartlett, G.R. Colorimetric assay methods for free and phosphorylated glyceric acids. J. Biol. Chem. 1959, 234, 469–471. [Google Scholar] [CrossRef]
- Broekhuyse, R.M. Phospholipids in tissues of the eye. I. Isolation, characterization and quantitative analysis by two-dimensional thin-layer chromatography of diacyl and vinyl-ether phospholipids. Biochim. Biophys. Acta 2005, 152, 307–315. [Google Scholar] [CrossRef]
- Yang, J.T.; Wu, C.S.; Martinez, H.M. Calculation of protein conformation from circular dichroism. Methods Enzymol. 1986, 130, 208–269. [Google Scholar]
- McElhaney, R.N. The use of differential scanning calorimetry and differential thermal analysis in studies of model and biological membranes. Chem. Phys. Lipids 1982, 30, 229–259. [Google Scholar] [CrossRef]
- Tao, T.; Cho, J. Fluorescence lifetime quenching studies on the accessibilities of actin sulfhydryl sites. Biochemistry 1979, 18, 2759–2765. [Google Scholar] [CrossRef] [PubMed]
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide. | Sequence | Amino Acids | Molecular Weight (g/mol) | Net Charge |
|---|---|---|---|---|
| R-DIM-P-LF11-215 (RDP215) | H-FWRIRIRR-P-RRIRIRWF-NH2 | 17 | 2483.03 | +9 |
| 9D-R-DIM-P-LF11-215 (9D-RDP215) | H-fWrIrIrr-P-rrIrIrWf-NH2 | 17 | 2483.03 | +9 |
| ANTS/DPX Leakage (%) | ||
|---|---|---|
| peptide Concentration (µM) | RDP215 | 9D-RDP215 |
| 0.5 | 7.9 ± 0.9 | 2.6 ± 0.3 |
| 1.0 | 14.2 ± 1.9 | 3.2 ± 0.7 |
| 2.0 | 31.5 ± 4.8 | 22.3 ± 2.5 |
| 4.0 | 83.3 ± 7.1 | 81.2 ± 0.5 |
| 8.0 | 99.2 ± 0.7 | ± 0.6 |
| ΔHpre or * [kcal/mol] | Tpre or * (°C) | ΔHm 1/2/3 (Total) (kcal/mol) | Tm 1/2/3 (°C) | T1/2 1/2/3 (°C) | |
|---|---|---|---|---|---|
| DPPS | - | - | 10.8 | 52.9 | 0.34 |
| +RDP215 100:1 | 0.1 (*) | 39.1 (*) | 2.2/10.3 (12.5) | 47.5/52.9 | 1.60/0.36 |
| +RDP215 50:1 | 0.4 (*) | 39.5 (*) | 4.8/1.2/4.2 (10.2) | 48.5/49.0/52.8 | 1.49/0.66/0.39 |
| +RDP215 25:1 | 0.9 (*) | 39.5 (*) | 5.9/1.2/2.4 (9.5) | 48.6/49.2/52.9 | 1.62/1.13/0.34 |
| +9D-RDP215 100:1 | 0.9 (*) | 39.4 (*) | 0.6/9.5 (10.1) | 51.3/52.9 | 1.25/1.34/0.35 |
| +9D-RDP215 50:1 | 1.8 (*) | 39.7 (*) | 0.6/6.9 (7.5) | 50.5/52.9 | 0.70/0.93/0.57 |
| +9D-RDP215 25:1 | 3.9 (*) | 40.0 (*) | 0.7/2.8/0.2 (3.7) | 50.5/52.4/52.8 | 0.32/2.52 |
| POPS | - | - | 5.3 | 11.8 | 1.76 |
| +RDP215 100:1 | - | - | 0.7/3.4 (4.1) | 8.1/11.7 | 1.95/1.84 |
| +RDP215 50:1 | - | - | 0.9/2.2 (3.1) | 8.3/11.8 | 2.31/1.83 |
| +RDP215 25:1 | - | - | 0.1/0.7 (0.8) | 8.2/11.8 | 1.35/1.72 |
| +9D-RDP215 100:1 | - | - | 3.9 | 11.6 | 1.73 |
| +9D-RDP215 50:1 | - | - | 2.5 | 11.8 | 1.78 |
| +9D-RDP215 25:1 | - | - | 0.5 | 11.7 | 1.91 |
| DPPC | 1.9 | 35.9 | 10.3 | 41.6 | 0.14 |
| +RDP215 100:1 | 1.6 | 35.8 | 9.8 | 41.6 | 0.14 |
| +RDP215 50:1 | 1.6 | 35.6 | 9.9 | 41.6 | 0.15 |
| +RDP215 25:1 | 1.3 | 35.6 | 8.9 | 41.6 | 0.14 |
| +9D-RDP215 100:1 | 1.7 | 35.9 | 10.9 | 41.6 | 0.16 |
| +9D-RDP215 50:1 | 2.0 | 35.8 | 12.0 | 41.6 | 0.15 |
| +9D-RDP215 25:1 | 2.0 | 35.8 | 11.1 | 41.6 | 0.14 |
| RDP215 | 9D-RDP215 | |||
|---|---|---|---|---|
| λ em,max [nm] | KSV (M−1) | λ em,max [nm] | KSV (M−1) | |
| PBS | 356 ± 0 | 24.7 ± 5.4 | 354 ± 0.5 | 27.0 ± 2.8 |
| Δλem,max [nm] | KSV (M−1) | Δλem,max [nm] | KSV (M−1) | |
| DPPS | −20 ± 0 | 4.0 ± 0.3 | −14 ± 1 | 3.9 ± 0.5 |
| POPS | −21 ± 1 | 4.8 ± 1.2 | −16 ± 1 | 4.1 ± 0.7 |
| DPPC | −1 ± 0 | 19.5 ± 1.0 | −1 ± 0 | 21.7 ± 0.4 |
| A375 | LC50 (µM) after 4 h | Specificity NHDF/A375 | LC50 (µM) after 8 h | Specificity NHDF/A375 |
| RDP215 | 17.1 ± 1.5 | 5.8 | 3.8 ± 0.3 | 18.0 |
| 9D-RDP215 | 7.0 ± 1.0 | 8.4 | 1.4 ± 0.1 | 13.5 |
| SBcl-2 | LC50 (µM) after 8 h | Specificity NHDF/SBcl-2 | ||
| RDP215 | 4.7 ± 0.4 | 14.5 | ||
| 9D-RDP215 | 1.8 ± 0.1 | 10.5 | ||
| NHDF | LC50 (µM) after 4 h | LC50 (µM) after 8 h | ||
| RDP215 | 99.1 ± 0.1 | 68.2 ± 1.8 | ||
| 9D-RDP215 | 58.8 ± 4.2 | 18.9 ± 1.0 |
| LN-229 | LC50 (µM) after 8 h | Specificity NHDF/LN-229 |
| RDP215 | 1.7 ± 0.1 | 40.1 |
| 9D-RDP215 | 1.4 ± 5.1 | 13.5 |
| U-87 mg | LC50 (µM) after 8 h | Specificity NHDF/U-87 mg |
| RDP215 | 3.0 ± 0.1 | 22.7 |
| 9D-RDP215 | 1.6 ± 0.1 | 11.8 |
| NHDF | LC50 (µM) after 8 h | |
| RDP215 | 68.2 ± 1.8 | |
| 9D-RDP215 | 18.9 ± 1.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Maxian, T.; Gerlitz, L.; Riedl, S.; Rinner, B.; Zweytick, D. Effect of L- to D-Amino Acid Substitution on Stability and Activity of Antitumor Peptide RDP215 against Human Melanoma and Glioblastoma. Int. J. Mol. Sci. 2021, 22, 8469. https://doi.org/10.3390/ijms22168469
Maxian T, Gerlitz L, Riedl S, Rinner B, Zweytick D. Effect of L- to D-Amino Acid Substitution on Stability and Activity of Antitumor Peptide RDP215 against Human Melanoma and Glioblastoma. International Journal of Molecular Sciences. 2021; 22(16):8469. https://doi.org/10.3390/ijms22168469
Chicago/Turabian StyleMaxian, Theresa, Lisa Gerlitz, Sabrina Riedl, Beate Rinner, and Dagmar Zweytick. 2021. "Effect of L- to D-Amino Acid Substitution on Stability and Activity of Antitumor Peptide RDP215 against Human Melanoma and Glioblastoma" International Journal of Molecular Sciences 22, no. 16: 8469. https://doi.org/10.3390/ijms22168469