
Cardiolipin-Containing Lipid Membranes Attract the Bacterial Cell Division Protein DivIVA

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
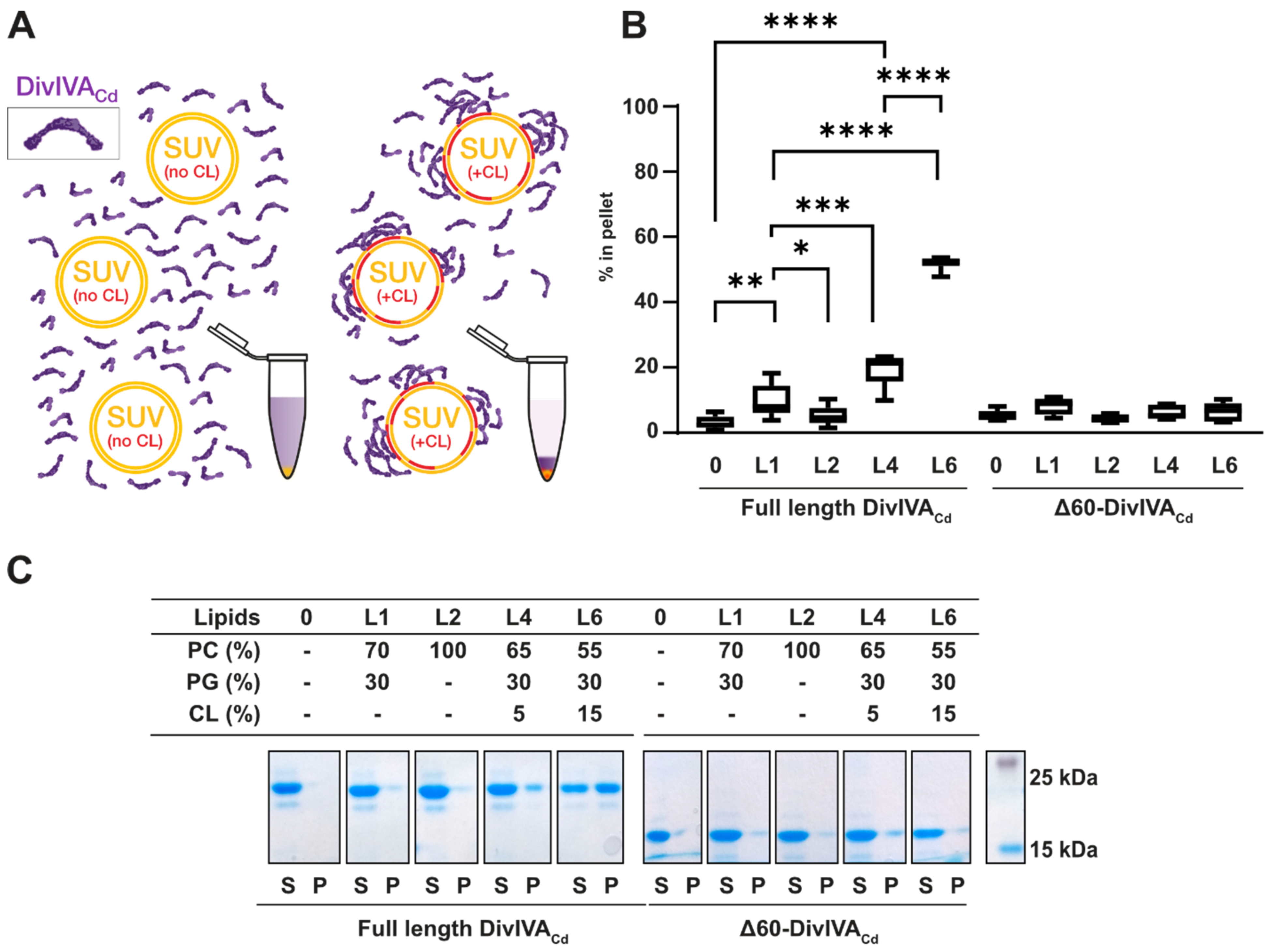
2.1. DivIVACd Binds More Efficiently to Cardiolipin-Containing Membranes In Vitro
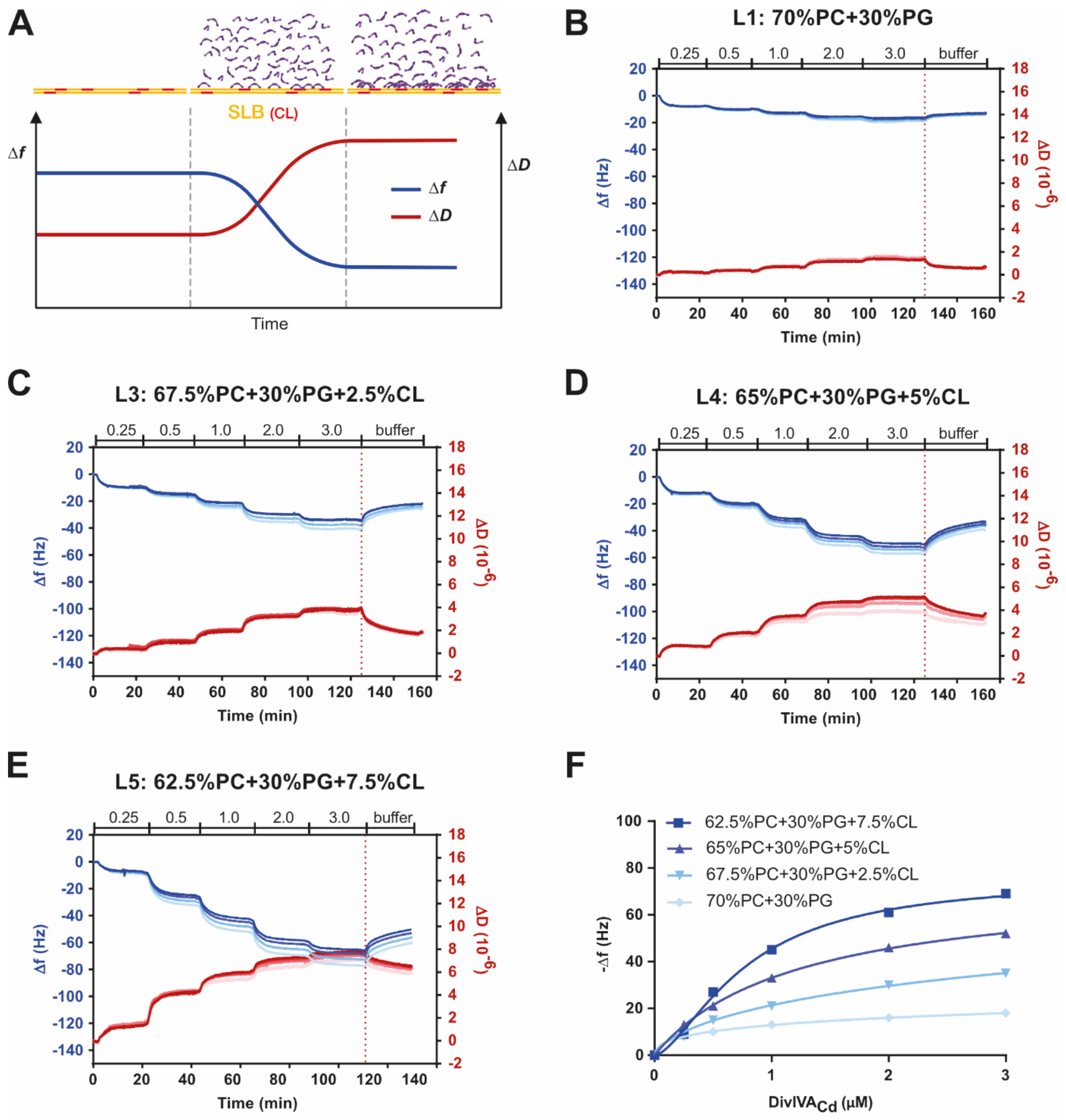
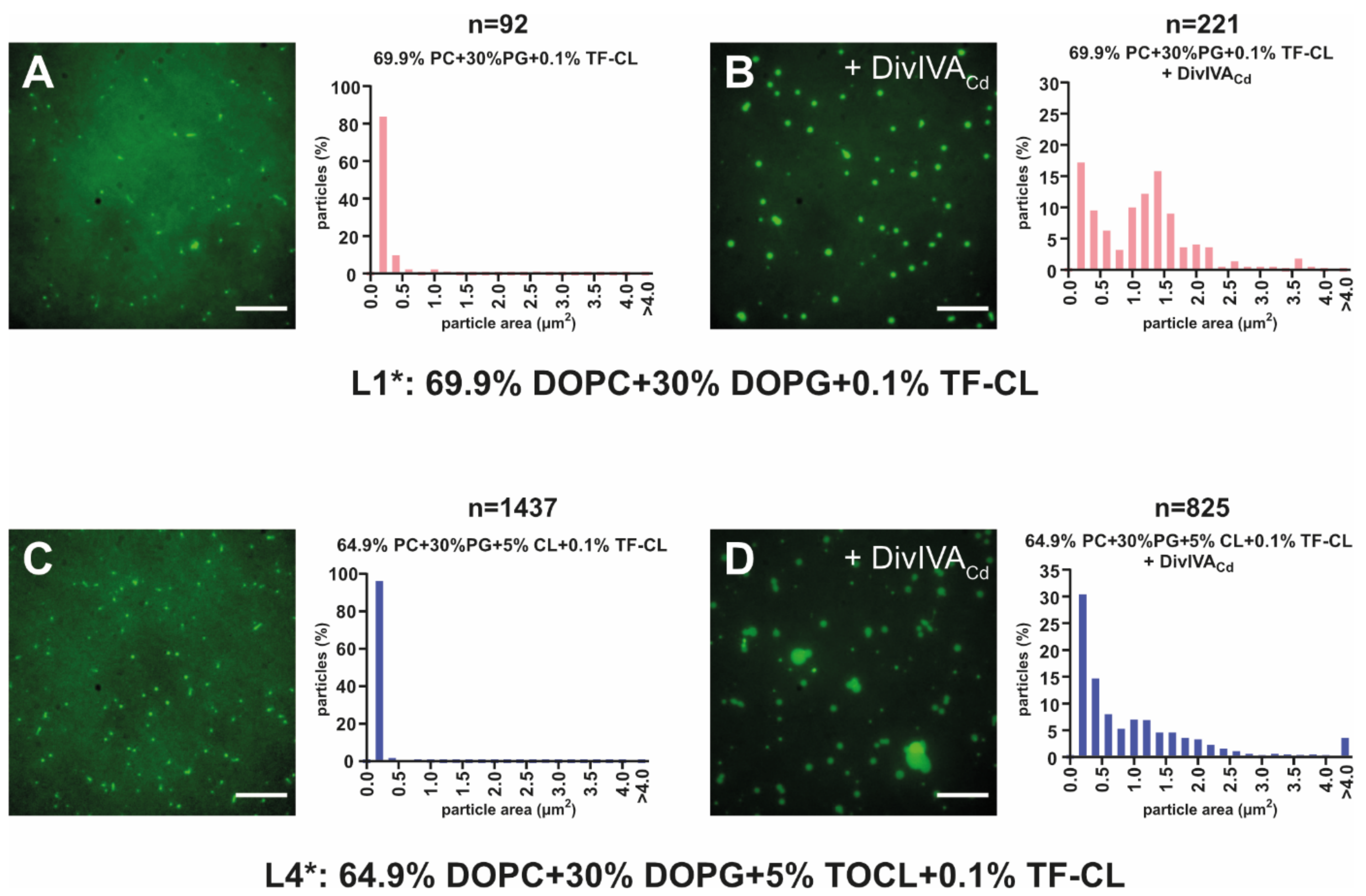
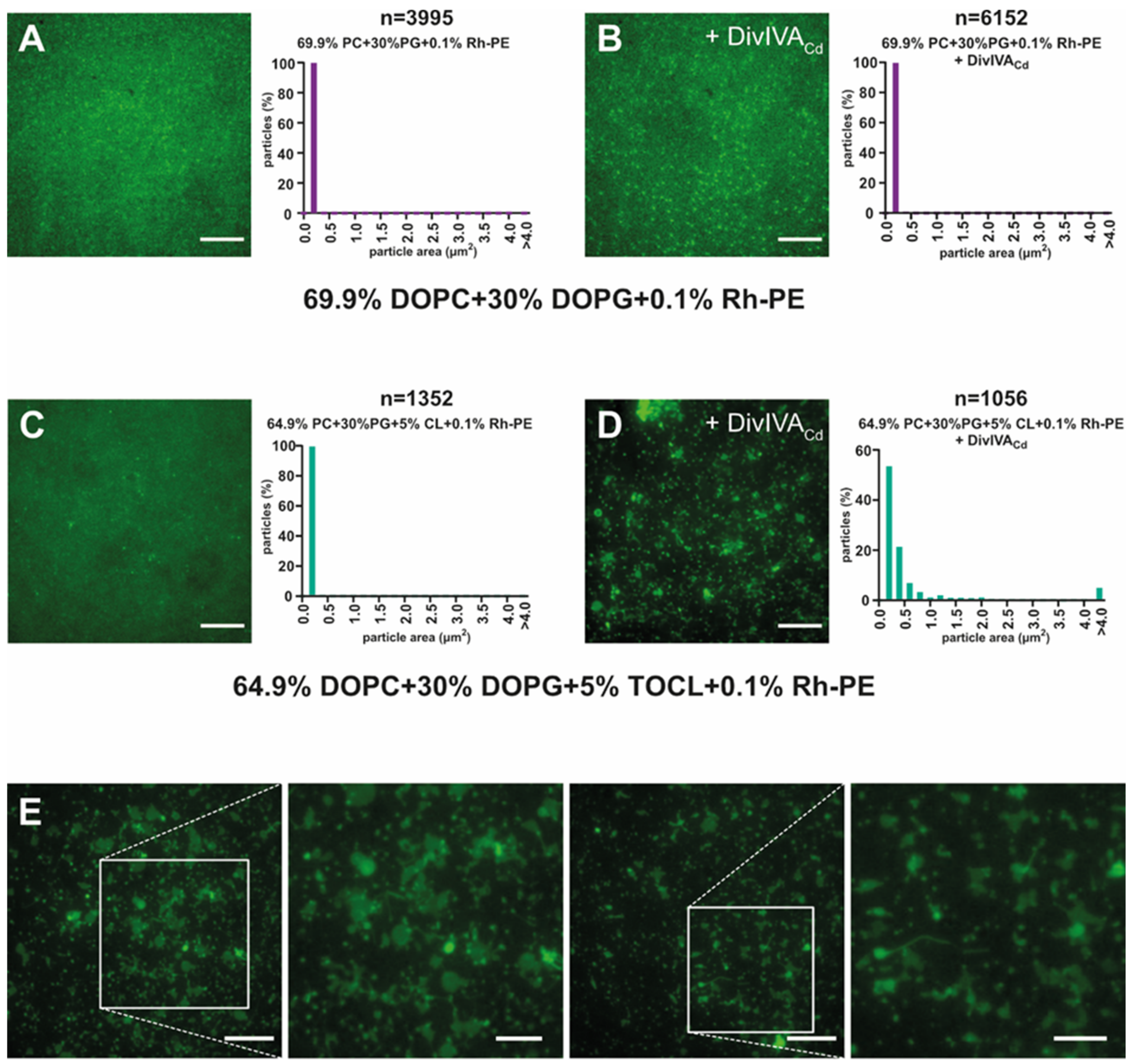
2.2. DivIVACd Binds to a Supported Lipid Bilayer
2.3. DivIVACd Deforms the Supported Lipid Bilayer
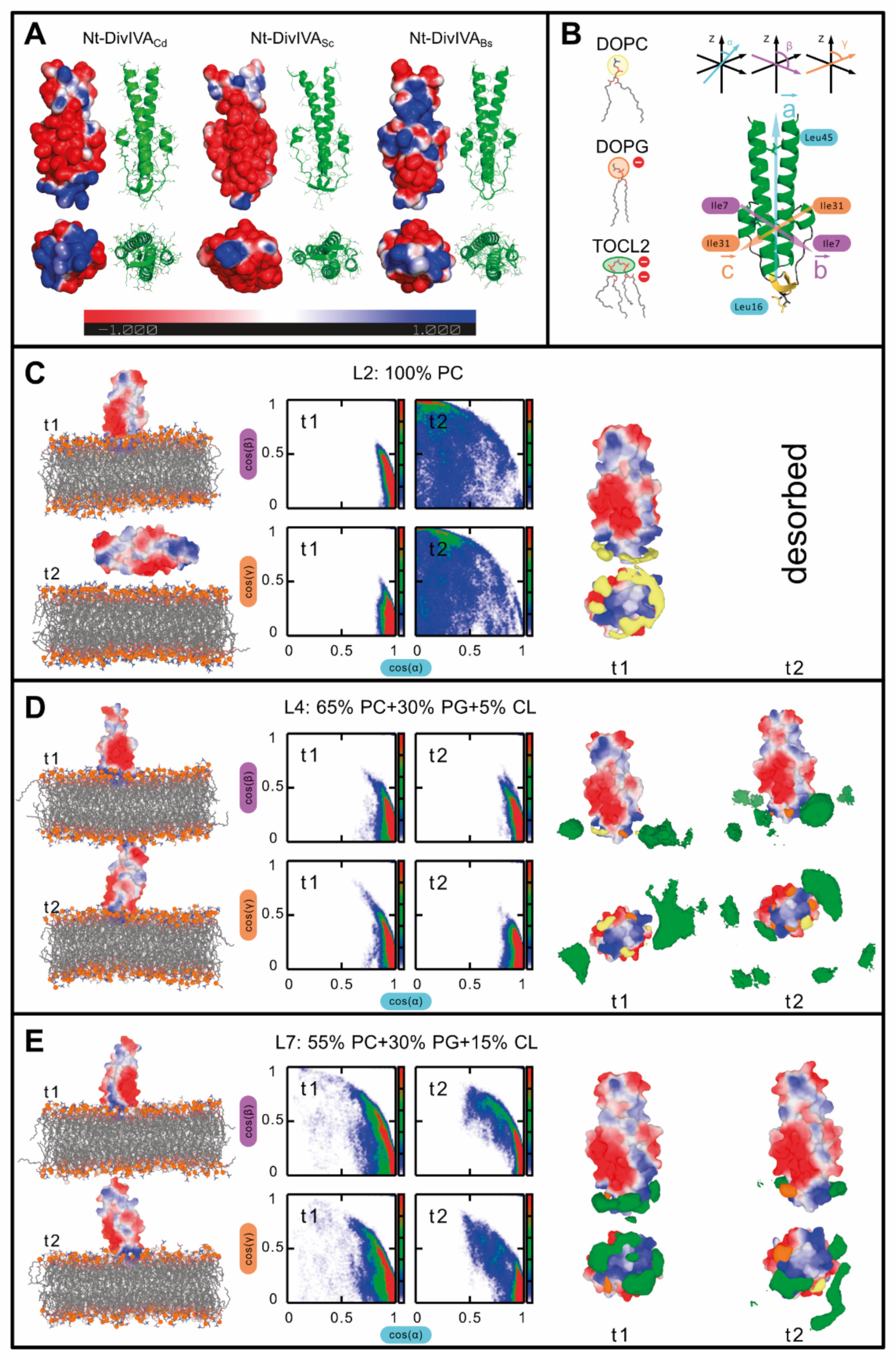
2.4. DivIVA Also Shows a Higher Affinity for Cardiolipin-Containing Membranes in Molecular Dynamics Simulations
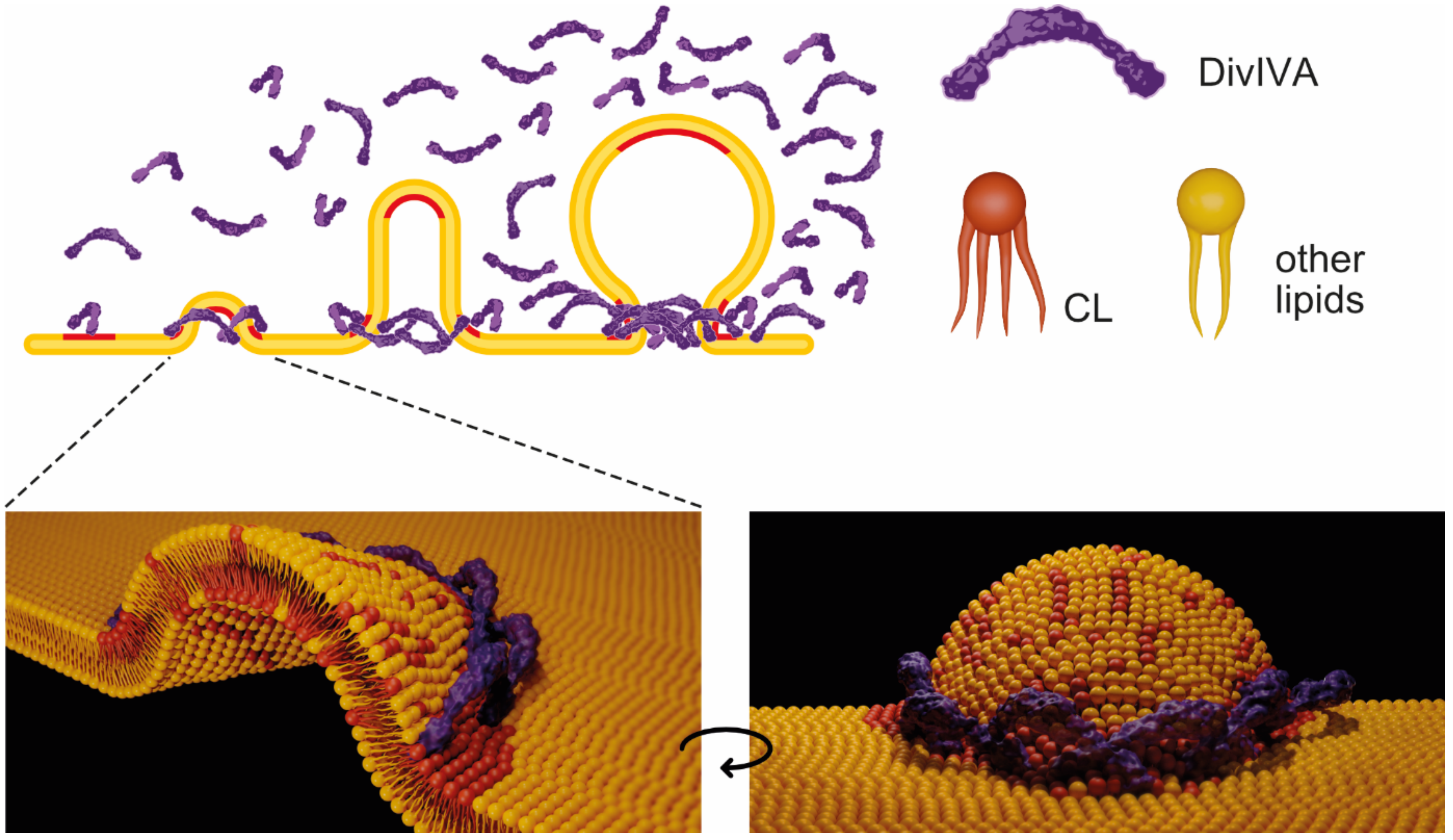
3. Discussion
4. Materials and Methods
4.1. General Methods
4.2. DivIVACd and Δ60-DivIVACd Protein Expression and Purification
4.3. Preparation of Small Unilamellar Vesicles (SUVs)
4.4. Sedimentation Assay
4.5. QCM-D Measurements
4.6. TIRF Microscopy and Image Analysis
4.7. Homology Modelling
4.8. Molecular Dynamics (MD) Simulations
4.8.1. System Preparation for MD Simulations
4.8.2. The Domain Orientation
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhu, D.; Sorg, J.A.; Sun, X. Clostridioides difficile biology: Sporulation, germination, and corresponding therapies for C. difficile infection. Front. Cell. Infect. Microbiol. 2018, 8, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guh, A.Y.; Mu, Y.; Winston, L.G.; Johnston, H.; Olson, D.; Farley, M.M.; Wilson, L.E.; Holzbauer, S.M.; Phipps, E.C.; Dumyati, G.K.; et al. Trends in U.S. burden of Clostridioides difficile infection and outcomes. N. Engl. J. Med. 2020, 382, 1320–1330. [Google Scholar] [CrossRef]
- Marra, A.R.; Perencevich, E.N.; Nelson, R.E.; Samore, M.; Khader, K.; Chiang, H.Y.; Chorazy, M.L.; Herwaldt, L.A.; Diekema, D.J.; Kuxhausen, M.F.; et al. Incidence and outcomes associated with Clostridium difficile infections: A systematic review and meta-analysis. JAMA Netw. Open 2020, 3, e1917597. [Google Scholar] [CrossRef] [Green Version]
- Rhea, S.; Jones, K.; Endres-Dighe, S.; Munoz, B.; Weber, D.J.; Hilscher, R.; MacFarquhar, J.; Sickbert-Bennett, E.; DiBiase, L.; Marx, A.; et al. Modeling inpatient and outpatient antibiotic stewardship interventions to reduce the burden of Clostridioides difficile infection in a regional healthcare network. PLoS ONE 2020, 15, e0234031. [Google Scholar] [CrossRef]
- Lessa, F.C.; Gould, C.V.; Clifford McDonald, L. Current status of Clostridium difficile infection epidemiology. Clin. Infect. Dis. 2012, 55, 65–70. [Google Scholar] [CrossRef] [Green Version]
- Lessa, F.C.; Mu, Y.; Bamberg, W.M.; Beldavs, Z.G.; Dumyati, G.K.; Dunn, J.R.; Farley, M.M.; Holzbauer, S.M.; Meek, J.I.; Phipps, E.C.; et al. Burden of Clostridium difficile Infection in the United States. N. Engl. J. Med. 2015, 372, 825–834. [Google Scholar] [CrossRef] [Green Version]
- Gamba, P.; Veening, J.W.; Saunders, N.J.; Hamoen, L.W.; Daniel, R.A. Two-step assembly dynamics of the Bacillus subtilis divisome. J. Bacteriol. 2009, 191, 4186–4194. [Google Scholar] [CrossRef] [Green Version]
- Mukherjee, A.; Dai, K.; Lutkenhaus, J. Escherichia coli cell division protein FtsZ is a guanine nucleotide binding protein. Proc. Natl. Acad. Sci. USA 1993, 90, 1053–1057. [Google Scholar] [CrossRef] [Green Version]
- De Boer, P.; Crossley, R.; Rothfield, L. The essential bacterial cell-division protein FtsZ is a GTPase. Nature 1992, 359, 254–256. [Google Scholar] [CrossRef]
- Edwards, D.H.; Errington, J. The Bacillus subtilis DivIVA protein targets to the division septum and controls the site specificity of cell division. Mol. Microbiol. 1997, 24, 905–915. [Google Scholar] [CrossRef]
- Eswaramoorthy, P.; Erb, M.L.; Gregory, J.A.; Silverman, J.; Pogliano, K.; Pogliano, J.; Ramamurthi, K.S. Cellular architecture mediates DivIVA ultrastructure and regulates Min activity in Bacillus subtilis. MBio 2011, 2, e00257-11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marston, A.L.; Errington, J. Selection of the midcell division site in Bacillus subtilis through MinD-dependent polar localization and activation of MinC. Mol. Microbiol. 1999, 33, 84–96. [Google Scholar] [CrossRef] [PubMed]
- Bramkamp, M.; Emmins, R.; Weston, L.; Donovan, C.; Daniel, R.A.; Errington, J. A novel component of the division-site selection system of Bacillus subtilis and a new mode of action for the division inhibitor MinCD. Mol. Microbiol. 2008, 70, 1556–1569. [Google Scholar] [CrossRef]
- Bramkamp, M.; van Baarle, S. Division site selection in rod-shaped bacteria. Curr. Opin. Microbiol. 2009, 12, 683–688. [Google Scholar] [CrossRef]
- Patrick, J.E.; Kearns, D.B. MinJ (YvjD) is a topological determinant of cell division in Bacillus subtilis. Mol. Microbiol. 2008, 70, 1166–1179. [Google Scholar] [CrossRef] [PubMed]
- Hammond, L.R.; White, M.L.; Eswara, P.J. ¡vIVA la DivIVA! J. Bacteriol. 2019, 201, e00245-19. [Google Scholar] [CrossRef]
- Migocki, M.D.; Freeman, M.K.; Wake, R.G.; Harry, E.J. The Min system is not required for precise placement of the midcell Z ring in Bacillus subtilis. EMBO Rep. 2002, 3, 1163–1167. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, C.D.A.; Harry, E.J. The Min system and nucleoid occlusion are not required for identifying the division site in Bacillus subtilis but ensure its efficient utilization. PLoS Genet. 2012, 8, e1002561. [Google Scholar] [CrossRef]
- Thomaides, H.B.; Freeman, M.; El Karoui, M.; Errington, J. Division site selection protein DivIVA of Bacillus subtilis has a second distinct function in chromosome segregation during sporulation. Genes Dev. 2001, 15, 1662–1673. [Google Scholar] [CrossRef] [Green Version]
- Kaval, K.G.; Halbedel, S. Architecturally the same, but playing a different game: The diverse species-specific roles of DivIVA proteins. Virulence 2012, 3, 406–408. [Google Scholar] [CrossRef] [Green Version]
- Dos Santos, V.T.; Bisson-Filho, A.W.; Gueiros-Filho, F.J. DivIVA-mediated polar localization of ComN, a posttranscriptional regulator of Bacillus subtilis. J. Bacteriol. 2012, 194, 3661–3669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Yehuda, S.; Rudner, D.Z.; Losick, R. RacA, a bacterial protein that anchors chromosomes to the cell poles. Science 2003, 299, 532–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Baarle, S.; Celik, I.N.; Kaval, K.G.; Bramkamp, M.; Hamoen, L.W.; Halbedel, S. Protein-protein interaction domains of Bacillus subtilis DivIVA. J. Bacteriol. 2013, 195, 1012–1021. [Google Scholar] [CrossRef] [Green Version]
- Briley, K.; Prepiak, P.; Dias, M.J.; Hahn, J.; Dubnau, D. Maf acts downstream of ComGA to arrest cell division in competent cells of B. subtilis. Mol. Microbiol. 2011, 81, 23–39. [Google Scholar] [CrossRef] [Green Version]
- Cha, J.H.; Stewart, G.C. The divIVA minicell locus of Bacillus subtilis. J. Bacteriol. 1997, 179, 1671–1683. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eswaramoorthy, P.; Winter, P.W.; Wawrzusin, P.; York, A.G.; Shroff, H.; Ramamurthi, K.S. Asymmetric division and differential gene expression during a bacterial developmental program requires DivIVA. PLoS Genet. 2014, 10, e1004526. [Google Scholar] [CrossRef] [Green Version]
- Kang, C.M.; Nyayapathy, S.; Lee, J.Y.; Suh, J.W.; Husson, R.N. Wag31, a homologue of the cell division protein DivIVA, regulates growth, morphology and polar cell wall synthesis in mycobacteria. Microbiology 2008, 154, 725–735. [Google Scholar] [CrossRef] [Green Version]
- Ramos, A.; Honrubia, M.P.; Valbuena, N.; Vaquera, J.; Mateos, L.M.; Gil, J.A. Involvement of DivIVA in the morphology of the rod-shaped actinomycete Brevibacterium lactofermentum. Microbiology 2003, 149, 3531–3542. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.; Scherr, N.; Gatfield, J.; Walburger, A.; Pieters, J.; Thompson, C.J. Antigen 84, an effector of pleiomorphism in Mycobactenum smegmatis. J. Bacteriol. 2007, 189, 7896–7910. [Google Scholar] [CrossRef] [Green Version]
- Flärdh, K. Essential role of DivIVA in polar growth and morphogenesis in Streptomyces coelicolor A3(2). Mol. Microbiol. 2003, 49, 1523–1536. [Google Scholar] [CrossRef]
- Makroczyová, J.; Jamroškovič, J.; Krascsenitsová, E.; Labajová, N.; Barák, I. Oscillating behavior of Clostridium difficile Min proteins in Bacillus subtilis. Microbiol. Open 2016, 5, 387–401. [Google Scholar] [CrossRef] [Green Version]
- Rowlett, V.W.; Margolin, W. The bacterial Min system. Curr. Biol. 2013, 23, R553–R556. [Google Scholar] [CrossRef] [Green Version]
- Jamroškovič, J.; Pavlendová, N.; Muchová, K.; Wilkinson, A.J.; Barák, I. An oscillating Min system in Bacillus subtilis influences asymmetrical septation during sporulation. Microbiology 2012, 158, 1972–1981. [Google Scholar] [CrossRef] [PubMed]
- Valenčíková, R.; Krascsenitsová, E.; Labajová, N.; Makroczyová, J.; Barák, I. Clostridial DivIVA and MinD interact in the absence of MinJ. Anaerobe 2018, 50, 22–31. [Google Scholar] [CrossRef]
- Lenarcic, R.; Halbedel, S.; Visser, L.; Shaw, M.; Wu, L.J.; Errington, J.; Marenduzzo, D.; Hamoen, L.W. Localisation of DivIVA by targeting to negatively curved membranes. EMBO J. 2009, 28, 2272–2282. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramamurthi, K.S.; Losick, R. Negative membrane curvature as a cue for subcellular localization of a bacterial protein. Proc. Natl. Acad. Sci. USA 2009, 106, 13541–13545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jurásek, M.; Flärdh, K.; Vácha, R. Effect of membrane composition on DivIVA-membrane interaction. Biochim. Biophys. Acta BBA Biomembr. 2019, 1862, 183144. [Google Scholar] [CrossRef] [PubMed]
- Oliva, M.; Halbedel, S.; Freund, S.M.; Dutow, P.; Leonard, T.A.; Veprintsev, D.B.; Hamoen, L.W.; Löwe, J. Features critical for membrane binding revealed by DivIVA crystal structure. EMBO J. 2010, 29, 1988–2001. [Google Scholar] [CrossRef]
- Vecchiarelli, A.G.; Li, M.; Mizuuchi, M.; Mizuuchi, K. Differential affinities of MinD and MinE to anionic phospholipid influence Min patterning dynamics in vitro. Mol. Microbiol. 2014, 93, 453–463. [Google Scholar] [CrossRef] [Green Version]
- Ramm, B.; Glock, P.; Schwille, P. In vitro reconstitution of self-organizing protein patterns on supported lipid bilayers. J. Vis. Exp. 2018, 2018, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Guan, Z.; Katzianerb, D.; Zhub, J.; Goldfine, H. Clostridium difficile contains plasmalogen species of phospholipids and glycolipids. Biochim. Biophys. Acta 2014, 1842, 1353–1359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reviakine, I.; Johannsmann, D.; Richter, R.P. Hearing what you cannot see and visualizing what you hear: Interpreting quartz crystal microbalance data from solvated interfaces. Anal. Chem. 2011, 83, 8838–8848. [Google Scholar] [CrossRef] [PubMed]
- Dixon, M.C. Quartz crystal microbalance with dissipation monitoring: Enabling real-time characterization of biological materials and their interactions. J. Biomol. Tech. 2008, 19, 151–158. [Google Scholar] [PubMed]
- De Kerchove, A.J.; Elimelech, M. Structural growth and viscoelastic properties of adsorbed alginate layers in monovalent and divalent salts. Macromolecules 2006, 39, 6558–6564. [Google Scholar] [CrossRef]
- Renner, L.D.; Weibel, D.B. MinD and MinE interact with anionic phospholipids and regulate division plane formation in Escherichia coli. J. Biol. Chem. 2012, 287, 38835–38844. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Bai, Q.; Hu, J.; Hou, D. A practical model of quartz crystal microbalance in actual applications. Sensors 2017, 17, 1785. [Google Scholar] [CrossRef] [Green Version]
- Lozeau, L.D.; Rolle, M.W.; Camesano, T.A. A QCM-D study of the concentration- and time-dependent interactions of human LL37 with model mammalian lipid bilayers. Colloids Surf. B Biointerfaces 2018, 167, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Kachel, K.; Asuncion-Punzalan, E.; London, E. The location of fluorescence probes with charged groups in model membranes. Biochim. Biophys. Acta BBA Biomembr. 1998, 1374, 63–76. [Google Scholar] [CrossRef] [Green Version]
- McMahon, H.T.; Boucrot, E. Membrane curvature at a glance. J. Cell Sci. 2015, 128, 1065–1070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, K.C.; Ramamurthi, K.S. Macromolecules that prefer their membranes curvy: MicroReview. Mol. Microbiol. 2010, 76, 822–832. [Google Scholar] [CrossRef] [Green Version]
- Beltrán-Heredia, E.; Tsai, F.C.; Salinas-Almaguer, S.; Cao, F.J.; Bassereau, P.; Monroy, F. Membrane curvature induces cardiolipin sorting. Commun. Biol. 2019, 2, 1–7. [Google Scholar] [CrossRef]
- Kusaka, J.; Shuto, S.; Imai, Y.; Ishikawa, K.; Saito, T.; Natori, K.; Matsuoka, S.; Hara, H.; Matsumoto, K. Septal localization by membrane targeting sequences and a conserved sequence essential for activity at the COOH-terminus of Bacillus subtilis cardiolipin synthase. Res. Microbiol. 2016, 167, 202–214. [Google Scholar] [CrossRef]
- Kawai, F.; Shoda, M.; Harashima, R.; Sadaie, Y.; Hara, H.; Matsumoto, K. Cardiolipin domains in Bacillus subtilis Marburg membranes. J. Bacteriol. 2004, 186, 1475–1483. [Google Scholar] [CrossRef] [Green Version]
- Mileykovskaya, E.; Dowhan, W. Visualization of phospholipid domains in Escherichia coli by using the cardiolipin-specific fluorescent dye 10-N-nonyl acridine orange. J. Bacteriol. 2000, 182, 1172–1175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pogmore, A.; Seistrup, K.H.; Strahl, H. The Gram-positive model organism Bacillus subtilis does not form microscopically detectable cardiolipin-specific lipid domains Growth conditions. Microbiology 2018, 164, 475–482. [Google Scholar] [CrossRef] [PubMed]
- Koppelman, C.M.; Den Blaauwen, T.; Duursma, M.C.; Heeren, R.M.A.; Nanninga, N. Escherichia coli minicell membranes are enriched in cardiolipin. J. Bacteriol. 2001, 183, 6144–6147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawai, F.; Hara, H.; Takamatsu, H.; Watabe, K.; Matsumoto, K. Cardiolipin enrichment in spore membranes and its involvement in germination of Bacillus subtilis Marburg. Genes Genet. Syst. 2006, 81, 69–76. [Google Scholar] [CrossRef] [Green Version]
- Sambrook, J.; Green, M.R. Molecular Cloning: A Laboratory Manual, III; Cold Spring Harbor Press: Cold Spring Harbor, NY, USA, 2001; Volume 1. [Google Scholar]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Press: Cold Spring Harbor, NY, USA, 1989. [Google Scholar]
- Ausubel, F.M.; Brent, R.; Kingston, R.E.; Moore, D.D.; Seidman, J.G.; Smith, J.A.; Struhl, K. Current Protocols in Molecular Biology; John Wiley & Sons Inc.: Hoboken, NJ, USA, 1988; Volume 1–2. [Google Scholar]
- Bendezú, F.O.; De Boer, P.A.J. Conditional lethality, division defects, membrane involution, and endocytosis in mre and mrd shape mutants of Escherichia coli. J. Bacteriol. 2008, 190, 1792–1811. [Google Scholar] [CrossRef] [Green Version]
- Stahlberg, H.; Kutejová, E.; Muchová, K.; Gregorini, M.; Lustig, A.; Müller, S.A.; Olivieri, V.; Engel, A.; Wilkinson, A.J.; Barák, I. Oligomeric structure of the Bacillus subtilis cell division protien DivIVA determined by transmission microscopy. Mol. Microbiol. 2004, 52, 1281–1290. [Google Scholar] [CrossRef]
- Rodahl, M.; Höök, F.; Krozer, A.; Brzezinski, P.; Kasemo, B. Quartz crystal microbalance setup for frequency and Q-factor measurements in gaseous and liquid environments. Rev. Sci. Instrum. 1995, 66, 3924–3930. [Google Scholar] [CrossRef] [Green Version]
- Keller, C.A.; Kasemo, B. Surface specific kinetics of lipid vesicle adsorption measured with a quartz crystal microbalance. Biophys. J. 1998, 75, 1397–1402. [Google Scholar] [CrossRef] [Green Version]
- Richter, R.; Mukhopadhyay, A.; Brisson, A. Pathways of Lipid Vesicle Deposition on Solid Surfaces: A Combined QCM-D and AFM Study. Biophys. J. 2003, 85, 3035–3047. [Google Scholar] [CrossRef] [Green Version]
- Loose, M.; Fischer-Friedrich, E.; Ries, J.; Kruse, K.; Schwille, P. Spatial regulators for bacterial cell division self-organize into surface waves in vitro. Science 2008, 320, 789–792. [Google Scholar] [CrossRef] [PubMed]
- Schindelin, J.; Arganda-Carreras, I.; Frise, E.; Kaynig, V.; Longair, M.; Pietzsch, T.; Preibisch, S.; Rueden, C.; Saalfeld, S.; Schmid, B.; et al. Fiji: An open-source platform for biological-image analysis. Nat. Methods 2012, 9, 676–682. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biasini, M.; Bienert, S.; Waterhouse, A.; Arnold, K.; Studer, G.; Schmidt, T.; Kiefer, F.; Cassarino, T.G.; Bertoni, M.; Bordoli, L.; et al. SWISS-MODEL: Modelling protein tertiary and quaternary structure using evolutionary information. Nucleic Acids Res. 2014, 42, 252–258. [Google Scholar] [CrossRef]
- Abraham, M.J.; Murtola, T.; Schulz, R.; Páll, S.; Smith, J.C.; Hess, B.; Lindah, E. Gromacs: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 2015, 1–2, 19–25. [Google Scholar] [CrossRef] [Green Version]
- Best, R.B.; Zhu, X.; Shim, J.; Lopes, P.E.M.; Mittal, J.; Feig, M.; MacKerell, A.D. Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ1 and χ2 Dihedral Angles. J. Chem. Theory Comput. 2012, 8, 3257–3273. [Google Scholar] [CrossRef] [Green Version]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926–935. [Google Scholar] [CrossRef]
- Nosé, S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 1983, 52, 255–268. [Google Scholar] [CrossRef]
- Hoover, W.G. Canonical dynamics: Equilibrium phase-space distributions. Phys. Rev. A 1985, 31, 1695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N·log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Hess, B.; Bekker, H.; Berendsen, H.J.C.; Fraaije, J.G.E.M. LINCS: A Linear Constraint Solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Jo, S.; Lim, J.B.; Klauda, J.B.; Im, W. CHARMM-GUI membrane builder for mixed bilayers and its application to yeast membranes. Biophys. J. 2009, 97, 50–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, E.L.; Cheng, X.; Jo, S.; Rui, H.; Song, K.C.; Dávila-Contreras, E.M.; Qi, Y.; Lee, J.; Monje-Galvan, V.; Venable, R.M.; et al. CHARMM-GUI membrane builder toward realistic biological membrane simulations. J. Comput. Chem. 2014, 35, 1997–2004. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Cheng, X.; Swails, J.M.; Yeom, M.S.; Eastman, P.K.; Lemkul, J.A.; Wei, S.; Buckner, J.; Jeong, J.C.; Qi, Y.; et al. CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J. Chem. Theory Comput. 2016, 12, 405–413. [Google Scholar] [CrossRef]
- Jo, S.; Kim, T.; Iyer, V.; Im, W. CHARMM-GUI: A web-based graphical user interface for CHARMM. J. Comput. Chem. 2008, 29, 1859–1865. [Google Scholar] [CrossRef]
- Grossfield, A.; Feller, S.E.; Pitman, M.C. Convergence of molecular dynamics simulations of membrane proteins. Proteins Struct. Funct. Genet. 2007, 67, 31–40. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Labajová, N.; Baranova, N.; Jurásek, M.; Vácha, R.; Loose, M.; Barák, I. Cardiolipin-Containing Lipid Membranes Attract the Bacterial Cell Division Protein DivIVA. Int. J. Mol. Sci. 2021, 22, 8350. https://doi.org/10.3390/ijms22158350
Labajová N, Baranova N, Jurásek M, Vácha R, Loose M, Barák I. Cardiolipin-Containing Lipid Membranes Attract the Bacterial Cell Division Protein DivIVA. International Journal of Molecular Sciences. 2021; 22(15):8350. https://doi.org/10.3390/ijms22158350
Chicago/Turabian StyleLabajová, Naďa, Natalia Baranova, Miroslav Jurásek, Robert Vácha, Martin Loose, and Imrich Barák. 2021. "Cardiolipin-Containing Lipid Membranes Attract the Bacterial Cell Division Protein DivIVA" International Journal of Molecular Sciences 22, no. 15: 8350. https://doi.org/10.3390/ijms22158350