
Purinergic Signalling in Allogeneic Haematopoietic Stem Cell Transplantation and Graft-versus-Host Disease

 ,
,  , ,
, ,
Abstract
:1. Introduction
2. Allogeneic Haematopoietic Stem Cell Transplantation and the Graft-Versus-Tumour Effect
3. Acute Graft-versus-Host Disease
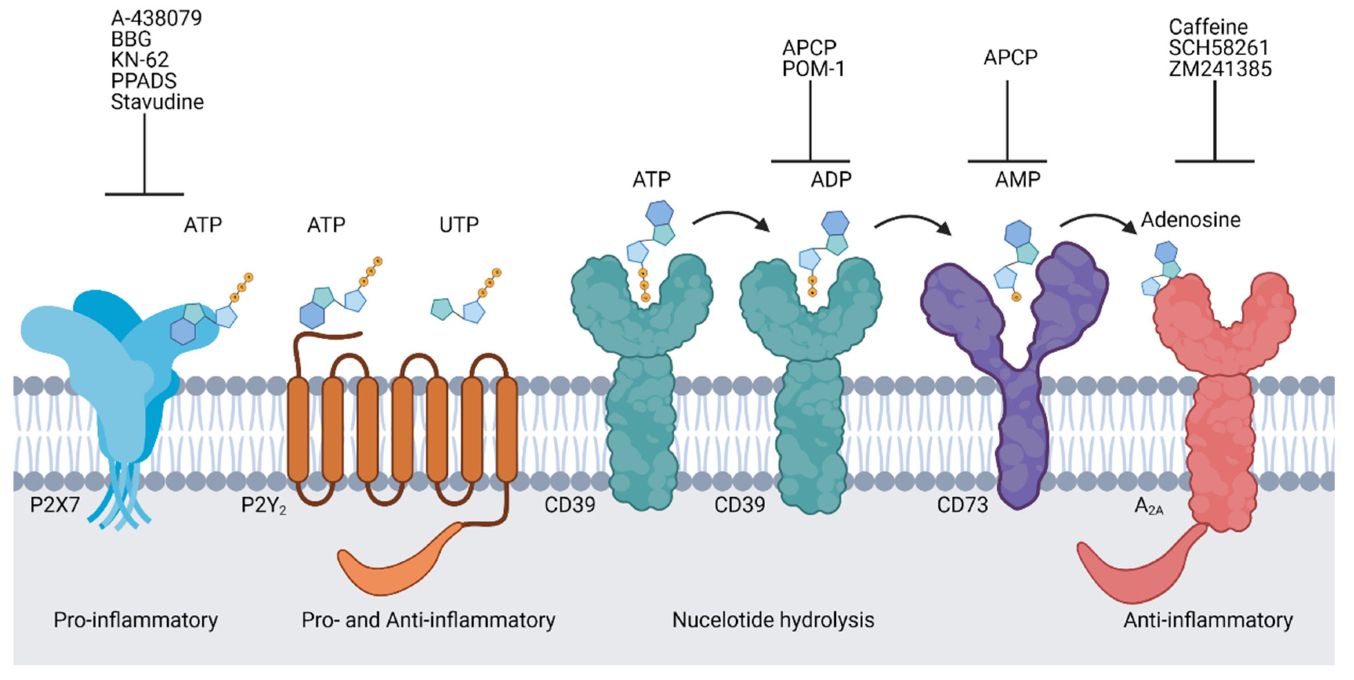
4. Purinergic Signalling
5. Purinergic Signalling in Allogeneic Haematopoietic Stem Cell Transplantation
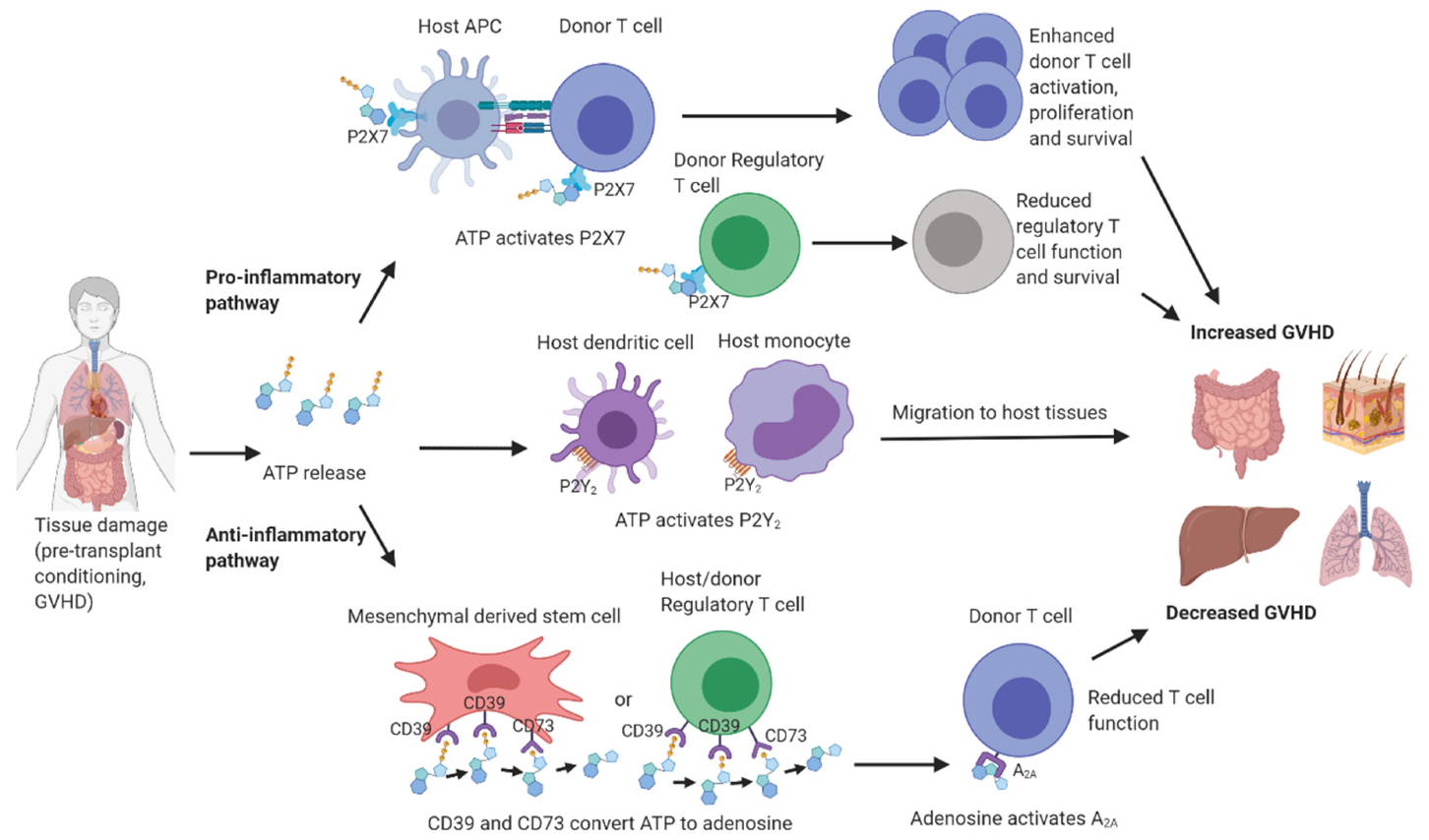
6. Purinergic Signalling in GVHD
7. P2X7 Receptor Signalling in GVHD
8. P2Y2 and P2Y12 Receptor Signalling in GVHD
9. Adenosine Receptor Signalling in GVHD
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviation
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics. CA Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [Green Version]
- Appelbaum, F.R. Hematopoietic-cell transplantation. N. Engl. J. Med. 2007, 357, 1472–1475. [Google Scholar] [CrossRef] [Green Version]
- Zeiser, R.; Blazar, B.R. Acute graft-versus-host disease—Biologic process, prevention, and therapy. N. Engl. J. Med. 2017, 377, 2167–2179. [Google Scholar] [CrossRef]
- Truitt, R.L.; Atasoylu, A.A. Contribution of CD4+ and CD8+ T cells to graft-versus-host disease and graft-versus-leukemia re-activity after transplantation of MHC-compatible bone marrow. Bone Marrow Transpl. 1991, 8, 51–58. [Google Scholar]
- Sprent, J.; Schaefer, M.; Gao, E.K.; Korngold, R. Role of T cell subsets in lethal graft-versus-host disease (GVHD) directed to class I versus class II H-2 differences. I. L3T4+ cells can either augment or retard GVHD elicited by Lyt-2+ cells in class I different hosts. J. Exp. Med. 1988, 167, 556–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Namdaroglu, S.; Kaya, A.H.; Batgi, H.; Kayikci, O.; Dal, M.S.; Iskender, D.; Cakar, M.K.; Tekgunduz, E.; Altuntas, F. Impacts of post-transplantation cyclophosphamide treatment after allogeneic hematopoietic stem cell transplantation in acute myeloid leukemia. Sci. Rep. 2019, 9, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burnstock, G. Purine and purinergic receptors. Brain Neurosci. Adv. 2018, 2. [Google Scholar] [CrossRef] [Green Version]
- Bach, F.H.; Albertini, R.J.; Joo, P.; Anderson, J.L.; Bortin, M.M. Bone-marrow transplantation in a patient with the Wiskott-Aldrich syndrome. Lancet 1968, 2, 1364–1366. [Google Scholar] [CrossRef]
- Thomas, E.D.; Storb, R.; Fefer, A.; Slichter, S.; Bryant, J.; Buckner, C.D.; Neiman, P.; Clift, R.; Funk, D.; Lerner, K. A plastic anaemia treated by marrow transplantation. Lancet 1972, 299, 284–289. [Google Scholar] [CrossRef]
- Lapidot, T.; Dar, A.; Kollet, O. How do stem cells find their way home? Blood 2005, 106, 1901–1910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bacigalupo, A.; Ballen, K.; Rizzo, D.; Giralt, S.; Lazarus, H.; Ho, V.; Apperley, J.; Slavin, S.; Pasquini, M.; Sandmaier, B.M.; et al. Defining the intensity of conditioning regimens: Working definitions. Biol. Blood Marrow Transplant. 2009, 15, 1628–1633. [Google Scholar] [CrossRef] [Green Version]
- Gyurkocza, B.; Sandmaier, B.M. Conditioning regimens for hematopoietic cell transplantation: One size does not fit all. Blood 2014, 124, 344–353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Copelan, E.A. Hematopoietic stem-cell transplantation. N. Engl. J. Med. 2006, 354, 1813–1826. [Google Scholar] [CrossRef]
- Bishop, M.R.; Tarantolo, S.R.; Geller, R.B.; Lynch, J.C.; Bierman, P.J.; Pavletic, Z.S.; Vose, J.M.; Kruse, S.; Dix, S.P.; Morris, M.E.; et al. A randomized, double-blind trial of filgrastim (granulocyte colony-stimulating factor) versus placebo following allogeneic blood stem cell transplantation. Blood 2000, 96, 80–85. [Google Scholar] [CrossRef]
- Barnes, D.W.H.; Loutit, J.F. Treatment of murine leukaemia with X-rays and homologous bone marrow. Br. J. Haematol. 1957, 3, 241–252. [Google Scholar] [CrossRef]
- Liang, Y.; Liu, C.; Djeu, J.Y.; Zhong, B.; Peters, T.; Scharffetter-Kochanek, K.; Anasetti, C.; Yu, X.-Z. β2 integrins separate graft-versus-host disease and graft-versus-leukemia effects. Blood 2008, 111, 954–962. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, X.; Song, Q.; Cassady, K.; Deng, R.; Jin, H.; Zhang, M.; Dong, H.; Forman, S.; Martin, P.J.; Chen, Y.-Z.; et al. PD-L1 interacts with CD80 to regulate graft-versus-leukemia activity of donor CD8+ T cells. J. Clin. Investig. 2017, 127, 1960–1977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ritacco, C.; Ehx, G.; Canti, L.; Dubois, S.; Vandenhove, B.; Servais, S.; Beguin, Y.; Humblet-Baron, S.; Baron, F. Ptcy prevents xenogeneic gvhd without abrogating gvl effects. Biol. Blood Marrow Transplant. 2020, 26, S168. [Google Scholar] [CrossRef]
- Delens, L.; Ehx, G.; Somja, J.; Vrancken, L.; Belle, L.; Seidel, L.; Grégoire, C.; Fransolet, G.; Ritacco, C.; Hannon, M.; et al. In vitro Th17-polarized human CD4+ T cells exacerbate xenogeneic graft-versus-host disease. Biol. Blood Marrow Transplant. 2019, 25, 204–215. [Google Scholar] [CrossRef] [Green Version]
- Ehx, G.; Fransolet, G.; De Leval, L.; D’Hondt, S.; Lucas, S.; Hannon, M.; Delens, L.; Dubois, S.; Drion, P.; Beguin, Y.; et al. Azacytidine prevents experimental xenogeneic graft-versus-host disease without abrogating graft-versus-leukemia effects. OncoImmunology 2017, 6, e1314425. [Google Scholar] [CrossRef] [Green Version]
- Zeng, K.; Ma, H.; Popat, U.; Nieto, Y.; Ciurea, S.O.; Olson, A.L.; Lyu, M.-A.; Huang, M.; Nishimoto, M.; Qazilbash, M.H.; et al. Allogeneic cord blood regulatory t cells can prevent graft vs. host disease and preserve graft vs leukemia effect: Update on phase I/II clinical trial. Blood 2019, 134, 4547. [Google Scholar] [CrossRef]
- Versluis, J.; Kalin, B.; Zeijlemaker, W.; Passweg, J.; Graux, C.; Manz, M.G.; Vekemans, M.-C.; Biemond, B.J.; Legdeur, M.-C.J.; Kooy, M.V.M.; et al. Graft-Versus-leukemia effect of allogeneic stem-cell transplantation and minimal residual disease in patients with acute myeloid leukemia in first complete remission. JCO Precis. Oncol. 2017, 1–13. [Google Scholar] [CrossRef]
- Ferrara, J.L.; Levine, J.; Reddy, P.; Holler, E. Graft-versus-host disease. Lancet 2009, 373, 1550–1561. [Google Scholar] [CrossRef]
- Saber, W.; Opie, S.; Rizzo, J.D.; Zhang, M.-J.; Horowitz, M.M.; Schriber, J. Outcomes after matched unrelated donor versus identical sibling hematopoietic cell transplantation in adults with acute myelogenous leukemia. Blood 2012, 119, 3908–3916. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.J.; Klein, J.P.; Barrett, A.J.; Ringden, O.; Antin, J.H.; Cahn, J.-Y.; Carabasi, M.H.; Gale, R.P.; Giralt, S.; Hale, G.A.; et al. Severity of chronic graft-versus-host disease: Association with treatment-related mortality and relapse. Blood 2002, 100, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Apostolova, P.; Zeiser, R. The Role of purine metabolites as DAMPs in acute graft-versus-host disease. Front. Immunol. 2016, 7, 439. [Google Scholar] [CrossRef] [Green Version]
- Mosmann, T.R.; Cherwinski, H.; Bond, M.W.; Giedlin, M.A.; Coffman, R.L. Two types of murine helper T cell clone. I. Definition according to profiles of lymphokine activities and secreted proteins. J. Immunol. 1986, 136, 2348–2357. [Google Scholar] [PubMed]
- Park, H.; Li, Z.; Yang, X.O.; Chang, S.H.; Nurieva, R.; Wang, Y.-H.; Wang, Y.; Hood, L.; Zhu, Z.; Tian, Q.; et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin. Nat. Immunol. 2005, 6, 1133–1141. [Google Scholar] [CrossRef]
- Schwab, L.; Goroncy, L.; Palaniyandi, S.; Gautam, S.; Triantafyllopoulou, A.; Mócsai, A.; Reichardt, W.; Karlsson, F.J.; Radhakrishnan, S.V.; Hanke, K.; et al. Neutrophil granulocytes recruited upon translocation of intestinal bacteria enhance graft-versus-host disease via tissue damage. Nat. Med. 2014, 20, 648–654. [Google Scholar] [CrossRef]
- Hülsdünker, J.; Ottmüller, K.J.; Neeff, H.P.; Koyama, M.; Gao, Z.; Thomas, O.S.; Follo, M.; Al-Ahmad, A.; Prinz, G.; Duquesne, S.; et al. Neutrophils provide cellular communication between ileum and mesenteric lymph nodes at graft-versus-host disease onset. Blood 2018, 131, 1858–1869. [Google Scholar] [CrossRef]
- Hong, Y.-Q.; Wan, B.; Li, X.-F. Macrophage regulation of graft-vs-host disease. World J. Clin. Cases 2020, 8, 1793–1805. [Google Scholar] [CrossRef] [PubMed]
- Di Ianni, M.; Falzetti, F.; Carotti, A.; Terenzi, A.; Castellino, F.; Bonifacio, E.; Del Papa, B.; Zei, T.; Ostini, R.I.; Cecchini, D.; et al. Tregs prevent GVHD and promote immune reconstitution in HLA-haploidentical transplantation. Blood 2011, 117, 3921–3928. [Google Scholar] [CrossRef]
- Ruggeri, L.; Di Ianni, M.; Urbani, E.; Mancusi, A.; Falzetti, F.; Carotti, A.; Terenzi, A.; Massei, M.S.; Amico, L.; Zei, T.; et al. Tregs suppress GvHD at the periphery and unleash the Gvl effect in the bone marrow. Blood 2014, 124, 842. [Google Scholar] [CrossRef]
- Tanaka, M.; Kobayashi, S.; Numata, A.; Tachibana, T.; Takasaki, H.; Maruta, A.; Ishigatsubo, Y.; Kanamori, H. The impact of the dose of natural killer cells in the graft on severe acute graft-versus-host disease after unrelated bone marrow transplantation. Leuk. Res. 2012, 36, 699–703. [Google Scholar] [CrossRef]
- Kim, S.Y.; Lee, H.; Han, M.-S.; Shim, H.; Eom, H.-S.; Park, B.; Kong, S.-Y. Post-transplantation natural killer cell count: A predictor of acute graft-versus-host disease and survival outcomes after allogeneic hematopoietic stem cell transplantation. Clin. Lymphoma Myeloma Leuk. 2016, 16, 527–535.e2. [Google Scholar] [CrossRef]
- Xun, C.; Brown, S.A.; Jennings, C.D.; Henslee-Downey, P.J.; Thompson, J.S. Acute graft-versus-host-like disease induced by transplantation of human activated natural killer cells into SCID mice. Transplantation 1993, 56, 409–416. [Google Scholar] [CrossRef]
- Cooley, S.; McCullar, V.; Wangen, R.; Bergemann, T.; Spellman, S.; Weisdorf, D.J.; Miller, J.S. KIR reconstitution is altered by T cells in the graft and correlates with clinical outcomes after unrelated donor transplantation. Blood 2005, 106, 4370–4376. [Google Scholar] [CrossRef] [Green Version]
- Olson, J.A.; Leveson-Gower, D.B.; Gill, S.; Baker, J.; Beilhack, A.; Negrin, R.S. NK cells mediate reduction of GVHD by inhibiting activated, alloreactive T cells while retaining GVT effects. Blood 2010, 115, 4293–4301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroeder, M.A.; DiPersio, J.F. Mouse models of graft-versus-host disease: Advances and limitations. Dis. Model. Mech. 2011, 4, 318–333. [Google Scholar] [CrossRef] [Green Version]
- Walsh, N.C.; Kenney, L.L.; Jangalwe, S.; Aryee, K.-E.; Greiner, D.L.; Brehm, M.A.; Shultz, L.D. Humanized mouse models of clinical disease. Annu. Rev. Pathol. Mech. Dis. 2017, 12, 187–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kawasaki, Y.; Sato, K.; Hayakawa, H.; Takayama, N.; Nakano, H.; Ito, R.; Mashima, K.; Oh, I.; Minakata, D.; Yamasaki, R.; et al. Comprehensive analysis of the activation and proliferation kinetics and effector functions of human lymphocytes, and antigen presentation capacity of antigen-presenting cells in xenogeneic graft-versus-host disease. Biol. Blood Marrow Transplant. 2018, 24, 1563–1574. [Google Scholar] [CrossRef] [Green Version]
- King, M.A.; Covassin, L.; Brehm, M.; Racki, W.; Pearson, T.; Leif, J.; Laning, J.; Fodor, W.; Foreman, O.; Burzenski, L.; et al. Human peripheral blood leucocyte non-obese diabetic-severe combined immunodeficiency interleukin-2 receptor gamma chain gene mouse model of xenogeneic graft-versus-host-like disease and the role of host major histocompatibility complex. Clin. Exp. Immunol. 2009, 157, 104–118. [Google Scholar] [CrossRef]
- Cooke, K.R.; Kobzik, L.; Martin, T.R.; Brewer, J.; Delmonte, J.; Crawford, J.M.; Ferrara, J.L. An experimental model of idiopathic pneumonia syndrome after bone marrow transplantation: I. The roles of minor H antigens and endotoxin. Blood 1996, 88, 3230–3239. [Google Scholar] [CrossRef] [Green Version]
- Ehx, G.; Somja, J.; Warnatz, H.-J.; Ritacco, C.; Hannon, M.; Delens, L.; Fransolet, G.; Delvenne, P.; Muller, J.; Beguin, Y.; et al. Xenogeneic graft-versus-host disease in humanized NSG and NSG-HLA-A2/HHD mice. Front. Immunol. 2018, 9, 1943. [Google Scholar] [CrossRef] [PubMed]
- Cuthbertson, P.; Adhikary, S.; Geraghty, N.J.; Guy, T.V.; Hadjiashrafi, A.; Fuller, S.J.; Ly, D.; Watson, D.; Sluyter, R. Increased P2X7 expression in the gastrointestinal tract and skin in a humanised mouse model of graft-versus-host disease. Clin. Sci. 2020, 134, 207–223. [Google Scholar] [CrossRef]
- Hannon, M.; Lechanteur, C.; Lucas, S.; Somja, J.; Seidel, L.; Belle, L.; Bruck, F.; Baudoux, E.; Giet, O.; Chantillon, A.-M.; et al. Infusion of clinical-grade enriched regulatory T cells delays experimental xenogeneic graft-versus-host disease. Transfusion 2013, 54, 353–363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Achita, P.; Dervovic, D.; Ly, D.; Lee, J.B.; Haug, T.; Joe, B.; Hirano, N.; Zhang, L. Infusion of ex-vivo expanded human TCR-αβ+ double-negative regulatory T cells delays onset of xenogeneic graft-versus -host disease. Clin. Exp. Immunol. 2018, 193, 386–399. [Google Scholar] [CrossRef] [Green Version]
- Kanakry, C.G.; Ganguly, S.; Zahurak, M.; Bolaños-Meade, J.; Thoburn, C.; Perkins, B.; Fuchs, E.J.; Jones, R.J.; Hess, A.D.; Luznik, L. Aldehyde dehydrogenase expression drives human regulatory T cell resistance to posttransplantation cyclophosphamide. Sci. Transl. Med. 2013, 5, 211ra157. [Google Scholar] [CrossRef] [Green Version]
- Adhikary, S.R.; Cuthbertson, P.; Nicholson, L.; Bird, K.M.; Sligar, C.; Hu, M.; O’Connell, P.J.; Sluyter, R.; Alexander, S.I.; Watson, D. Post-transplant cyclophosphamide limits reactive donor T cells and delays the development of graft-versus-host disease in a humanized mouse model. Immunology 2021. [Google Scholar] [CrossRef] [PubMed]
- Giuliani, A.L.; Sarti, A.C.; Di Virgilio, F. Extracellular nucleotides and nucleosides as signalling molecules. Immunol. Lett. 2019, 205, 16–24. [Google Scholar] [CrossRef] [PubMed]
- Idzko, M.; Ferrari, D.; Eltzschig, H.K. Nucleotide signalling during inflammation. Nature 2014, 509, 310–317. [Google Scholar] [CrossRef] [Green Version]
- Borea, P.A.; Gessi, S.; Merighi, S.; Vincenzi, F.; Varani, K. Pharmacology of adenosine receptors: The state of the art. Physiol. Rev. 2018, 98, 1591–1625. [Google Scholar] [CrossRef]
- Burnstock, G. P2X ion channel receptors and inflammation. Purinergic Signal. 2016, 12, 59–67. [Google Scholar] [CrossRef] [Green Version]
- Le Duc, D.; Schulz, A.; Lede, V.; Schulze, A.; Thor, D.; Brüser, A.; Schöneberg, T. P2Y Receptors in immune response and inflammation. Adv. Immunol. 2017, 136, 85–121. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, K.A.; Delicado, E.G.; Gachet, C.; Kennedy, C.; Von Kügelgen, I.; Li, B.; Miras-Portugal, M.T.; Novak, I.; Schöneberg, T.; Perez-Sen, R.; et al. Update of P2Y receptor pharmacology: IUPHAR Review. Br. J. Pharmacol. 2020, 177, 2413–2433. [Google Scholar] [CrossRef]
- Yegutkin, G. Enzymes involved in metabolism of extracellular nucleotides and nucleosides: Functional implications and measurement of activities. Crit. Rev. Biochem. Mol. Biol. 2014, 49, 473–497. [Google Scholar] [CrossRef] [PubMed]
- Zimmermann, H.; Zebisch, M.; Sträter, N. Cellular function and molecular structure of ecto-nucleotidases. Purinergic Signal. 2012, 8, 437–502. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonioli, L.; Pacher, P.; Vizi, E.S.; Haskó, G. CD39 and CD73 in immunity and inflammation. Trends Mol. Med. 2013, 19, 355–367. [Google Scholar] [CrossRef] [Green Version]
- Filippin, K.; De Souza, K.F.S.; Júnior, R.T.D.A.; Torquato, H.F.V.; Dias, D.A.; Parisotto, E.B.; Ferreira, A.T.; Paredes-Gamero, E.J. Involvement of P2 receptors in hematopoiesis and hematopoietic disorders, and as pharmacological targets. Purinergic Signal. 2019, 16, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, M.Z.; Adamiak, M.; Bujko, K.; Thapa, A.; Pensato, V.; Kucia, M.; Ratajczak, J.; Ulrich, H. Innate immunity orchestrates the mobilization and homing of hematopoietic stem/progenitor cells by engaging purinergic signaling—An update. Purinergic Signal. 2020, 16, 153–166. [Google Scholar] [CrossRef]
- Rossi, L.; Salvestrini, V.; Ferrari, D.; Di Virgilio, F.; Lemoli, R.M. The sixth sense: Hematopoietic stem cells detect danger through purinergic signaling. Blood 2012, 120, 2365–2375. [Google Scholar] [CrossRef] [Green Version]
- Antonioli, L.; Fornai, M.; Blandizzi, C.; Pacher, P.; Haskó, G. Adenosine signaling and the immune system: When a lot could be too much. Immunol. Lett. 2019, 205, 9–15. [Google Scholar] [CrossRef]
- Lenkiewicz, A.M.; Adamiak, M.; Thapa, A.; Bujko, K.; Pedziwiatr, D.; Abdel-Latif, A.K.; Kucia, M.; Ratajczak, J.; Ratajczak, M.Z. The Nlrp3 inflammasome orchestrates mobilization of bone marrow-residing stem cells into peripheral blood. Stem Cell Rev. Rep. 2019, 15, 391–403. [Google Scholar] [CrossRef] [Green Version]
- Koldej, R.; Collins, J.; Ritchie, D. P2X7 polymorphisms and stem cell mobilisation. Leukemia 2018, 32, 2724–2726. [Google Scholar] [CrossRef]
- Adamiak, M.; Bujko, K.; Cymer, M.; Plonka, M.; Glaser, T.; Kucia, M.; Ratajczak, J.; Ulrich, H.; Abdel-Latif, A.; Ratajczak, M.Z. Novel evidence that extracellular nucleotides and purinergic signaling induce innate immunity-mediated mobilization of hematopoietic stem/progenitor cells. Leukemia 2018, 32, 1920–1931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ratajczak, M.Z.; Adamiak, M.; Plonka, M.; Abdel-Latif, A.; Ratajczak, J. Mobilization of hematopoietic stem cells as a result of innate immunity-mediated sterile inflammation in the bone marrow microenvironment—The involvement of extracellular nucleotides and purinergic signaling. Leukemia 2018, 32, 1116–1123. [Google Scholar] [CrossRef] [Green Version]
- Rossi, L.; Manfredini, R.; Bertolini, F.; Ferrari, D.; Fogli, M.; Zini, R.; Salati, S.; Salvestrini, V.; Gulinelli, S.; Adinolfi, E.; et al. The extracellular nucleotide UTP is a potent inducer of hematopoietic stem cell migration. Blood 2006, 109, 533–542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, J.; Yusuf, R.; Kook, S.; Attar, E.; Lee, N.; Park, B.; Cheng, T.; Scadden, D.T.; Lee, B.C. Purinergic P2Y14 receptor modulates stress-induced hematopoietic stem/progenitor cell senescence. J. Clin. Investig. 2014, 124, 3159–3171. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbosa, C.M.V.; Leon, C.M.M.P.; Nogueira-Pedro, A.; Wasinsk, F.; Araújo, R.C.; Miranda, A.; Ferreira, A.T.; Paredes-Gamero, E.J. Differentiation of hematopoietic stem cell and myeloid populations by ATP is modulated by cytokines. Cell Death Dis. 2011, 2, e165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirata, Y.; Furuhashi, K.; Ishii, H.; Li, H.W.; Pinho, S.; Ding, L.; Robson, S.C.; Frenette, P.S.; Fujisaki, J. CD150 high bone marrow tregs maintain hematopoietic stem cell quiescence and immune privilege via adenosine. Cell Stem Cell 2018, 22, 445–453.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deotare, U.; Al-Dawsari, G.; Couban, S.; Lipton, J.H. G-CSF-primed bone marrow as a source of stem cells for allografting: Revisiting the concept. Bone Marrow Transplant. 2015, 50, 1150–1156. [Google Scholar] [CrossRef] [Green Version]
- Wilhelm, K.; Ganesan, J.; Müller, T.; Dürr, C.; Grimm, M.; Beilhack, A.; Krempl, C.D.; Sorichter, S.; Gerlach, U.V.; Jüttner, E.; et al. Graft-versus-host disease is enhanced by extracellular ATP activating P2X7R. Nat. Med. 2010, 16, 1434–1438. [Google Scholar] [CrossRef]
- Dürr, C.; Follo, M.; Idzko, M.; Reichardt, W.; Zeiser, R. Graft-versus-host disease reduces regulatory T-cell migration into the tumour tissue. Immunology 2012, 137, 80–88. [Google Scholar] [CrossRef] [Green Version]
- Cymer, M.; Brzeźniakiewicz-Janus, K.; Bujko, K.; Thapa, A.; Ratajczak, J.; Anusz, K.; Tracz, M.; Jackowska-Tracz, A.; Ratajczak, M.Z.; Adamiak, M. Pannexin-1 channel “fuels” by releasing ATP from bone marrow cells a state of sterile inflammation required for optimal mobilization and homing of hematopoietic stem cells. Purinergic Signal. 2020, 16, 313–325. [Google Scholar] [CrossRef]
- Granell, M.; Urbano-Ispizua, Á.; Pons, A.; Aróstegui, J.I.; Gel, B.; Navarro, A.; Jansa, S.; Artells, R.; Gaya, A.; Talarn, C.; et al. Common variants in NLRP2 and NLRP3 genes are strong prognostic factors for the outcome of HLA-identical sibling allogeneic stem cell transplantation. Blood 2008, 112, 4337–4342. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.-H.; Park, S.S.; Kim, I.; Kim, J.H.; Ra, E.K.; Yoon, S.-S.; Hong, Y.-C.; Kim, B.K. P2X7 receptor polymorphism and clinical outcomes in HLA-matched sibling allogeneic hematopoietic stem cell transplantation. Haematologica 2007, 92, 651–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karaesmen, E.; Rizvi, A.A.; Preus, L.M.; McCarthy, P.L.; Pasquini, M.C.; Onel, K.; Zhu, X.; Spellman, S.; Haiman, C.A.; Stram, D.O.; et al. Replication and validation of genetic polymorphisms associated with survival after allogeneic blood or marrow transplant. Blood 2017, 130, 1585–1596. [Google Scholar] [CrossRef] [Green Version]
- Koldej, R.M.; Perera, T.; Collins, J.; Ritchie, D.S. Association between P2X7 polymorphisms and post-transplant outcomes in allogeneic haematopoietic stem cell transplantation. Int. J. Mol. Sci. 2020, 21, 3772. [Google Scholar] [CrossRef] [PubMed]
- Vaisitti, T.; Arruga, F.; Guerra, G.; Deaglio, S. Ectonucleotidases in blood malignancies: A tale of surface markers and therapeutic targets. Front. Immunol. 2019, 10, 2301. [Google Scholar] [CrossRef]
- De Marchi, E.; Pegoraro, A.; Adinolfi, E. P2X7 receptor in hematological malignancies. Front. Cell Dev. Biol. 2021, 9, 645605. [Google Scholar] [CrossRef]
- Linden, J.; Koch-Nolte, F.; Dahl, G. Purine release, metabolism, and signaling in the inflammatory response. Annu. Rev. Immunol. 2019, 37, 325–347. [Google Scholar] [CrossRef]
- Apostolova, P.; Zeiser, R. The role of danger signals and ectonucleotidases in acute graft-versus-host disease. Hum. Immunol. 2016, 77, 1037–1047. [Google Scholar] [CrossRef]
- Cekic, C.; Linden, J. Purinergic regulation of the immune system. Nat. Rev. Immunol. 2016, 16, 177–192. [Google Scholar] [CrossRef] [PubMed]
- Sáez, P.J.; Vargas, P.; Shoji, K.F.; Harcha, P.A.; Lennon-Duménil, A.-M.; Sáez, J.C. ATP promotes the fast migration of dendritic cells through the activity of pannexin 1 channels and P2X7 receptors. Sci. Signal. 2017, 10, eaah7107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Englezou, P.C.; Rothwell, S.W.; Ainscough, J.S.; Brough, D.; Landsiedel, R.; Verkhratsky, A.; Kimber, I.; Dearman, R.J. P2X7R activation drives distinct IL-1 responses in dendritic cells compared to macrophages. Cytokine 2015, 74, 293–304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yip, L.; Woehrle, T.; Corriden, R.; Hirsh, M.; Chen, Y.; Inoue, Y.; Ferrari, V.; Insel, P.A.; Junger, W.G. Autocrine regulation of T-cell activation by ATP release and P2X 7 receptors. FASEB J. 2009, 23, 1685–1693. [Google Scholar] [CrossRef] [Green Version]
- Müller, T.; Robaye, B.; Vieira, R.P.; Ferrari, D.; Grimm, M.; Jakob, T.; Martin, S.F.; Di Virgilio, F.; Boeynaems, J.-M.; Virchow, J.C.; et al. The purinergic receptor P2Y2 receptor mediates chemotaxis of dendritic cells and eosinophils in allergic lung inflammation. Allergy 2010, 65, 1545–1553. [Google Scholar] [CrossRef]
- Stachon, P.; Geis, S.; Peikert, A.; Heidenreich, A.; Michel, N.A.; Ünal, F.; Hoppe, N.; Dufner, B.; Schulte, L.; Marchini, T.; et al. Extracellular ATP induces vascular inflammation and atherosclerosis via purinergic receptor Y2 in mice. Arter. Thromb. Vasc. Biol. 2016, 36, 1577–1586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higgins, K.R.; Kovacevic, W.; Stokes, L. Nucleotides regulate secretion of the inflammatory chemokine CCL2 from human macrophages and monocytes. Mediat. Inflamm. 2014, 2014, 1–13. [Google Scholar] [CrossRef] [Green Version]
- De La Rosa, G.; Gómez, A.I.; Baños, M.C.; Pelegrín, P. Signaling through purinergic receptor P2Y2 enhances macrophage IL-1β production. Int. J. Mol. Sci. 2020, 21, 4686. [Google Scholar] [CrossRef]
- Lappas, C.M.; Rieger, J.M.; Linden, J. A2A adenosine receptor induction inhibits IFN-γ production in murine CD4+T cells. J. Immunol. 2005, 174, 1073–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sorrentino, C.; Hossain, F.; Rodriguez, P.C.; Sierra, R.A.; Pannuti, A.; Hatfield, S.; Osborne, B.A.; Minter, L.M.; Miele, L.; Morello, S. Adenosine A2A receptor stimulation inhibits TCR-induced Notch1 activation in CD8+T-cells. Front. Immunol. 2019, 10, 162. [Google Scholar] [CrossRef]
- Alnouri, M.W.; Jepards, S.; Casari, A.; Schiedel, A.C.; Hinz, S.; Müller, C.E. Selectivity is species-dependent: Characterization of standard agonists and antagonists at human, rat, and mouse adenosine receptors. Purinergic Signal. 2015, 11, 389–407. [Google Scholar] [CrossRef] [Green Version]
- Donnelly-Roberts, D.L.; Namovic, M.T.; Han, P.; Jarvis, M.F. Mammalian P2X7 receptor pharmacology: Comparison of recombinant mouse, rat and human P2X7 receptors. Br. J. Pharmacol. 2009, 157, 1203–1214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sluyter, R. The P2X7 receptor. Adv. Exp. Med. Biol. 2017, 1051, 17–53. [Google Scholar] [CrossRef]
- Qiu, F.; Dahl, G. A permeant regulating its permeation pore: Inhibition of pannexin 1 channels by ATP. Am. J. Physiol. Physiol. 2009, 296, C250–C255. [Google Scholar] [CrossRef]
- Tokumitsu, H.; Chijiwa, T.; Hagiwara, M.; Mizutani, A.; Terasawa, M.; Hidaka, H. KN-62, 1-[N,O-bis(5-isoquinolinesulfonyl)-N-methyl-L-tyrosyl]-4-phenylpiperazi ne, a specific inhibitor of Ca2+/calmodulin-dependent protein kinase II. J. Biol. Chem. 1990, 265, 4315–4320. [Google Scholar] [CrossRef]
- Gargett, C.E.; Wiley, J. The isoquinoline derivative KN-62 a potent antagonist of the P2Z-receptor of human lymphocytes. Br. J. Pharmacol. 1997, 120, 1483–1490. [Google Scholar] [CrossRef] [Green Version]
- Spaulding, A.; Rutherford, G.W.; Siegfried, N. Stavudine or zidovudine in three-drug combination therapy for initial treatment of HIV infection in antiretroviral-naïve individuals. Cochrane Database Syst. Rev. 2010, CD008651. [Google Scholar] [CrossRef]
- Fowler, B.J.; Gelfand, B.D.; Kim, Y.; Kerur, N.; Tarallo, V.; Hirano, Y.; Amarnath, S.; Fowler, D.H.; Radwan, M.; Young, M.T.; et al. Nucleoside reverse transcriptase inhibitors possess intrinsic anti-inflammatory activity. Science 2014, 346, 1000–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Khalidi, R.; Panicucci, C.; Cox, P.; Chira, N.; Róg, J.; Young, C.N.J.; McGeehan, R.E.; Ambati, K.; Ambati, J.; Zablocki, K.; et al. Zidovudine ameliorates pathology in the mouse model of Duchenne muscular dystrophy via P2RX7 purinoceptor antagonism. Acta Neuropathol. Commun. 2018, 6, 1–17. [Google Scholar] [CrossRef]
- Bhattarai, S.; Freundlieb, M.; Pippel, J.; Meyer, A.; Abdelrahman, A.; Fiene, A.; Lee, S.-Y.; Zimmermann, H.; Yegutkin, G.; Sträter, N.; et al. α,β-methylene-ADP (AOPCP) derivatives and analogues: Development of potent and selective ecto-5′-nucleotidase (CD73) inhibitors. J. Med. Chem. 2015, 58, 6248–6263. [Google Scholar] [CrossRef] [PubMed]
- Covarrubias, R.; Chepurko, E.; Reynolds, A.; Huttinger, Z.M.; Huttinger, R.; Stanfill, K.; Wheeler, D.G.; Novitskaya, T.; Robson, S.C.; Dwyer, K.M.; et al. Role of the CD39/CD73 purinergic pathway in modulating arterial thrombosis in mice. Arter. Thromb. Vasc. Biol. 2016, 36, 1809–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, P.; Dale, N. Modulation of K+ currents in Xenopus spinal neurons by p2y receptors: A role for ATP and ADP in motor pattern generation. J. Physiol. 2002, 540, 843–850. [Google Scholar] [CrossRef]
- Müller, C.E.; Iqbal, J.; Baqi, Y.; Zimmermann, H.; Röllich, A.; Stephan, H. Polyoxometalates—A new class of potent ecto-nucleoside triphosphate diphosphohydrolase (NTPDase) inhibitors. Bioorganic Med. Chem. Lett. 2006, 16, 5943–5947. [Google Scholar] [CrossRef]
- Pimenta-Dos-Reis, G.; Torres, E.J.L.; Quintana, P.G.; Vidal, L.O.; Dos Santos, B.A.F.; Lin, C.-S.; Heise, N.; Persechini, P.M.; Schachter, J. POM-1 inhibits P2 receptors and exhibits anti-inflammatory effects in macrophages. Purinergic Signal. 2017, 13, 611–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pizzirani, C.; Falzoni, S.; Govoni, M.; La Corte, R.; Donadei, S.; Di Virgilio, F.; Trotta, F.; Monaco, A.L. Dysfunctional inflammasome in Schnitzler’s syndrome. Rheumatology 2009, 48, 1304–1308. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Chen, Z.; Liu, C.; Lu, X.; Yang, C.; Qiu, S. Distributive differences of P2Xs between the forelimb and hind limb of adjuvant arthritis rats and intervention by Notopterygh rhizoma et radix. Pharm. Biol. 2019, 57, 81–88. [Google Scholar] [CrossRef] [Green Version]
- Denlinger, L.C.; Manthei, D.M.; Seibold, M.A.; Ahn, K.; Bleecker, E.; Boushey, H.A.; Calhoun, W.J.; Castro, M.; Chinchili, V.M.; Fahy, J.V.; et al. P2X7-regulated protection from exacerbations and loss of control is independent of asthma maintenance therapy. Am. J. Respir. Crit. Care Med. 2013, 187, 28–33. [Google Scholar] [CrossRef] [Green Version]
- Koehn, B.H.; Saha, A.; McDonald-Hyman, C.; Loschi, M.; Thangavelu, G.; Ma, L.; Zaiken, M.C.; Dysthe, J.; Krepps, W.; Panthera, J.; et al. Danger-associated extracellular ATP counters MDSC therapeutic efficacy in acute GVHD. Blood 2019, 134, 1670–1682. [Google Scholar] [CrossRef]
- Tsukamoto, H.; Chernogorova, P.; Ayata, K.; Gerlach, U.V.; Rughani, A.; Ritchey, J.W.; Ganesan, J.; Follo, M.; Zeiser, R.; Thompson, L.F.; et al. Deficiency of CD73/ecto-5′-nucleotidase in mice enhances acute graft-versus-host disease. Blood 2012, 119, 4554–4564. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Fan, J.; Chen, S.; Zhang, Y.; Curiel, T.J.; Zhang, B. Graft-versus-host disease is enhanced by selective CD73 blockade in mice. PLoS ONE 2013, 8, e58397. [Google Scholar] [CrossRef]
- Geraghty, N.J.; Watson, D.; Sluyter, R. Pharmacological blockade of the CD39/CD73 pathway but not adenosine receptors augments disease in a humanized mouse model of graft- versus -host disease. Immunol. Cell Biol. 2019, 97, 597–610. [Google Scholar] [CrossRef]
- Zhong, X.; Zhu, F.; Qiao, J.; Zhao, K.; Zhu, S.; Zeng, L.; Chen, X.; Xu, K. The impact of P2X7 receptor antagonist, brilliant blue G on graft-versus-host disease in mice after allogeneic hematopoietic stem cell transplantation. Cell. Immunol. 2016, 310, 71–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Smith, B.; Iype, J.; Prestipino, A.; Pfeifer, D.; Grundmann, S.; Schmitt-Graeff, A.; Idzko, M.; Beck, Y.; Prinz, G.; et al. MicroRNA-155-deficient dendritic cells cause less severe GVHD through reduced migration and defective inflammasome activation. Blood 2015, 126, 103–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, F.; Chen, M.; Chen, W.; Gu, J.; Yuan, J.; Xue, Y.; Dang, J.; Su, W.; Wang, J.; Zadeh, H.; et al. Human gingiva-derived mesenchymal stem cells inhibit xeno-graft-versus-host disease via CD39-CD73-Adenosine and IDO Signals. Front. Immunol. 2017, 8, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cuthbertson, P.; Geraghty, N.J.; Adhikary, S.R.; Casolin, S.; Watson, D.; Sluyter, R. P2X7 receptor antagonism increases regulatory T cells and reduces clinical and histological graft-versus-host disease in a humanised mouse model. Clin. Sci. 2021, 135, 495–513. [Google Scholar] [CrossRef]
- Geraghty, N.J.; Belfiore, L.; Ly, D.; Adhikary, S.; Fuller, S.J.; Varikatt, W.; Sanderson-Smith, M.; Sluyter, V.; Alexander, S.I.; Watson, D. The P2X7 receptor antagonist Brilliant Blue G reduces serum human interferon-γ in a humanized mouse model of graft-versus-host disease. Clin. Exp. Immunol. 2017, 190, 79–95. [Google Scholar] [CrossRef] [Green Version]
- Geraghty, N.; Watson, D.; Sluyter, R. Long-term treatment with the P2X7 receptor antagonist Brilliant Blue G reduces liver inflammation in a humanized mouse model of graft-versus-host disease. Cell. Immunol. 2019, 336, 12–19. [Google Scholar] [CrossRef] [Green Version]
- Adhikary, S.; Geraghty, N.; Cuthbertson, P.; Sluyter, R.; Watson, D. Altered donor P2X7 activity in human leukocytes correlates with P2RX7 genotype but does not affect the development of graft-versus-host disease in humanised mice. Purinergic Signal. 2019, 15, 177–192. [Google Scholar] [CrossRef]
- Da Silva, H.B.; Beura, L.K.; Wang, H.; Hanse, E.A.; Gore, R.; Scott, M.C.; Walsh, D.A.; Block, K.E.; Fonseca, R.; Yan, Y.; et al. The purinergic receptor P2RX7 directs metabolic fitness of long-lived memory CD8+ T cells. Nature 2018, 559, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, H.B.; Peng, C.; Wang, H.; Wanhainen, K.M.; Ma, C.; Lopez, S.; Khoruts, A.; Zhang, N.; Jameson, S.C. Sensing of ATP via the purinergic receptor P2RX7 promotes CD8+ Trm cell generation by enhancing their sensitivity to the cytokine TGF-β. Immunity 2020, 53, 158–171.e6. [Google Scholar] [CrossRef]
- Schenk, U.; Frascoli, M.; Proietti, M.; Geffers, R.; Traggiai, E.; Buer, J.; Ricordi, C.; Westendorf, A.M.; Grassi, F. ATP inhibits the generation and function of regulatory T cells through the activation of purinergic P2X receptors. Sci. Signal. 2011, 4, ra12. [Google Scholar] [CrossRef] [PubMed]
- Koehn, B.H.; Apostolova, P.; Haverkamp, J.M.; Miller, J.S.; McCullar, V.; Tolar, J.; Munn, D.; Murphy, W.J.; Brickey, W.J.; Serody, J.S.; et al. GVHD-associated, inflammasome-mediated loss of function in adoptively transferred myeloid-derived suppressor cells. Blood 2015, 126, 1621–1628. [Google Scholar] [CrossRef] [Green Version]
- Klämbt, V.; Wohlfeil, S.; Schwab, L.; Hülsdünker, J.; Ayata, K.; Apostolova, P.; Schmitt-Graeff, A.; Dierbach, H.; Prinz, G.; Follo, M.; et al. A Novel function for P2Y2 in myeloid recipient-derived cells during graft-versus-host disease. J. Immunol. 2015, 195, 5795–5804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munir, S.Z.; Aylward, J. A review of ocular graft-versus-host disease. Optom. Vis. Sci. 2017, 94, 545–555. [Google Scholar] [CrossRef]
- Pupovac, A.; Sluyter, R. Roles of extracellular nucleotides and P2 receptors in ectodomain shedding. Cell. Mol. Life Sci. 2016, 73, 4159–4173. [Google Scholar] [CrossRef] [Green Version]
- Sakimoto, T.; Ohnishi, T.; Ishimori, A. Significance of ectodomain shedding of TNF receptor 1 in ocular surface. Investig. Opthalmology Vis. Sci. 2014, 55, 2419–2423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohashi, Y.; Munesue, M.; Shimazaki, J.; Takamura, E.; Yokoi, N.; Watanabe, H.; Nomura, A.; Shimada, F. Long-term safety and effectiveness of diquafosol for the treatment of dry eye in a real-world setting: A prospective observational study. Adv. Ther. 2020, 37, 707–717. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Sheng, X.; Xiang, D.; Wei, X.; Chen, T.; Yang, Z.; Zhang, Y. CD150 high treg cells may attenuate graft versus host disease and intestinal cell apoptosis after hematopoietic stem cell transplantation. Am. J. Transl. Res. 2019, 11, 1299–1310. [Google Scholar]
- Rissiek, A.; Baumann, I.; Cuapio, A.; Mautner, A.; Kolster, M.; Arck, P.C.; Dodge-Khatami, A.; Mittrücker, H.-W.; Koch-Nolte, F.; Haag, F.; et al. The expression of CD39 on regulatory T cells is genetically driven and further upregulated at sites of inflammation. J. Autoimmun. 2015, 58, 12–20. [Google Scholar] [CrossRef]
- Adhikary, S.R.; Cuthbertson, P.; Turner, R.J.; Sluyter, R.; Watson, D. A single-nucleotide polymorphism in the human ENTPD1 gene encoding CD39 is associated with worsened graft-versus-host disease in a humanized mouse model. Immunol. Cell Biol. 2020, 98, 397–410. [Google Scholar] [CrossRef] [PubMed]
- Lappas, C.M.; Liu, P.-C.; Linden, J.; Kang, E.M.; Malech, H. Adenosine A2A receptor activation limits graft-versus-host disease after allogenic hematopoietic stem cell transplantation. J. Leukoc. Biol. 2009, 87, 345–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, K.L.; Thomas, S.V.M.; Koontz, S.M.; Changpriroa, C.M.; Ha, S.-K.; Malech, H.L.; Kang, E.M. Adenosine A2A receptor agonist-mediated increase in donor-derived regulatory t cells suppresses development of graft-versus-host disease. J. Immunol. 2012, 190, 458–468. [Google Scholar] [CrossRef] [Green Version]
- Amarnath, S.; Foley, J.E.; Farthing, D.E.; Gress, R.E.; Laurence, A.; Eckhaus, M.A.; Métais, J.-Y.; Rose, J.J.; Hakim, F.T.; Felizardo, T.C.; et al. Bone marrow-derived mesenchymal stromal cells harness purinergenic signaling to tolerize human Th1 cells in vivo. Stem Cells 2015, 33, 1200–1212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geraghty, N.; Adhikary, S.; Watson, D.; Sluyter, R. The A2A receptor agonist CGS 21680 has beneficial and adverse effects on disease development in a humanised mouse model of graft-versus-host disease. Int. Immunopharmacol. 2019, 72, 479–486. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Model | Graft | Host | Target | Drug Regime | Outcomes | Ref. |
|---|---|---|---|---|---|---|
| Allogeneic | 5 × 106 TCD a BM b cells + 1.6 × 106 (FVB/N) or 1 × 106 CD4+ /CD8+ (C57BL/6) (i.v. c) | BALB/c | eATP d | 4U Apyrase (hydrolysis catalyst) i.p. e days 0–2, 6–8 | ↑f Survival ↓g Apoptosis and inflammation in the gastrointestinal tract ↓ Serum IFN h-γ | [72] |
| Allogeneic | 10 × 106 CD25 depleted T cells (C57BL/6) (injection route not disclosed) | BALB/c | eATP | 4U Apyrase (hydrolysis catalyst) i.p. days 0–4 | ↑ Survival | [110] |
| Allogeneic | 5 × 106 BM cells and 2 × 105 CD4+/CD8+ splenic T cells (i.v.) | C57BL/6 | CD73 | 50 mg/kg APCP i (antagonist) i.p. days 0–6 | ↑ Mortality | [111] |
| Allogeneic | 5 × 106 BALB/c splenocytes ± BM cells or splenocytes (i.v.) | C57BL/6 | CD73 | 20 mg/kg APCP (antagonist) i.v. twice weekly | ↑ Mortality ↑ Splenic CD4+ and CD8+ T cells ↑ Serum IFN-γ and ILj-6. | [112] |
| Humanised | 10 × 106 hPBMCs k (i.p.) | NSG l | CD39 and CD73 | 50 mg/kg APCP (antagonist) i.p. days 0–6 | ↑ Weight loss ↑ Histological damage ↑ Serum human IL-2 | [113] |
| Humanised | 20 × 106 CD25-depleted hPBMC + PBS, GMSCs m, human fibroblasts or nTregs n (i.v.) | NOD.Cg-Prkdcscid | CD39 | 100 μM POM-1 o (antagonist) was used to pretreat GMSCs | ↑ Mortality ↑ Histological damage ↑ Serum IL-4, IL-17, IFN-γ, IL-2 and TNFp-α. | [116] |
| Model | Graft | Host | Target | Drug Regime (All Antagonists) | Outcomes | Ref. |
|---|---|---|---|---|---|---|
| Allogeneic | 5 × 106 TCD a BM b cells + 1.6 × 106 (FVB/N) or 1 × 106 CD4+ /CD8+ (C57BL/6) (i.v. c) | BALB/c | P2X7 | 10 µmol PPADS d i.p. e days 0–10 | ↑f Survival ↑ Tregsg, ↓ T cell expansion ↓h Serum IFNi-γ ↓ GVHD severity | [72] |
| Allogeneic | 5 × 106 TCD BM cells + 1 × 106 CD4+ /CD8+ (C57BL/6) (i.v.) | BALB/c | P2X7 | 1 µmol KN62 i.p. days 0–10 | ↑ Survival | [72] |
| Allogeneic | 10 × 106 CD25 depleted T cells (C57BL/6) (injection route not disclosed) | BALB/c | P2X7 | 80 mg/kg A-438079 i.p. day 0–4 | ↑ Survival | [110] |
| Allogeneic | 10 × 106 TCD BM cells + 2.5 × 106 CD4+ T cells (C57BL/6) (injection route not disclosed) | BALB/c | P2X7 | 25 mg/kg Stavudine (d4T) i.p. twice daily from day −1 or 0 | ↑ Survival (when started from day −1) ↓ Serum IFN-γ, TNFj-α and ILk-6 ↓ Liver inflammation | [100] |
| Allogeneic | 5 × 106 BM cells + 5 × 106 splenic cells (C57BL/6) (i.v.) | BALB/c | P2X7 | 50 mg/kg or 75mg/kg BBGl i.p. twice weekly for 4 weeks | ↓ Weight loss ↓ Liver inflammation ↓ CXCL8 and CCL2, Il1B and Il18 | [114] |
| Humanised | 10 × 106 hPBMCsm (i.p.) | NSGn | P2X7 | 50 mg/kg BBG i.p. days 0, 2, 4, 6, 8, 10 | ↓ Serum IFN-γ ↓ Liver, skin and small intestine inflammation | [118] |
| Humanised | 10 × 106 hPBMCs (i.p.) | NSG | P2X7 | 50 mg/kg BBG i.p. thrice weekly until endpoint | ↓ Liver inflammation | [119] |
| Humanised | 10 × 106 hPBMCs (i.p.) | NSG | P2X7 | 50 mg/kg BBG i.p. days 0–10 | ↓ Clinical disease ↓ Liver and skin inflammation ↓ Serum IFN-γ ↑ Tregs and B cells | [117] |
| Humanised | 10 × 106 hPBMCs (i.p.) | NSG | P2X7 | 300 mg/kg PPADS i.p. days 0–10 | ↑ Tregs | [117] |
| Model | Graft | Host | Target | Drug Regime | Outcomes | Ref. |
|---|---|---|---|---|---|---|
| Allogeneic | 5 × 106 BMa cells and 2 × 105 CD4+/CD8+ splenic T cells (i.v.b) | C57BL/6 | Adenosine receptors | 10 mg/kg Caffeine (antagonist) i.p.c days 0–6 then 3–5 times a week | ↑d Mortality | [111] |
| Allogeneic | 5 × 106 BALB/c splenocytes ± BM cells or splenocytes (i.v.) | C57BL/6 | A2A | 2 mg/kg SCH58261 (antagonist) i.p. days −2–12 | ↑ Mortality ↑ Splenic CD4+ and CD8+ T cells ↑ Serum IFNe-γ, TNFf-α and ILg-6 | [112] |
| Allogeneic | 5 × 106 BALB/c splenocytes ± BM cells or splenocytes (i.v.) | C57BL/6 | A2B | 2 mg/kg MRS1754 (antagonist) i.p. days −2–12 | No differences | [112] |
| Allogeneic | 10 × 106 C57BL/6 splenocytes ± 10 × 106 T cells (i.v.) | B6D2F1/J | A2A | 10 ng/kg ATL-146e (agonist) s.c. days 0–14 | ↑ Survival ↑ Serum IL-10 ↓h Splenic CD4+ and CD8+ T cells ↓ T cell infiltration into target organs ↓ Serum IFN-γ and IL-6 | [133] |
| Humanised | 5 × 106 human Th1i cells and 3 × 106 human monocytes (i.p.) | NSGj | A2A | 1.5 mg/kg ZM241385 (antagonist) i.p. daily from disease onset until endpoint | ↑ Mortality ↑ Histological damage ↑ IFN-γ and TNF-α producing cells | [135] |
| Humanised | 10 × 106 hPBMCsk (i.p.) | NSG | Adenosine receptors | 10 mg/kg Caffeine (antagonist) i.p. days 0–14 | No differences | [113] |
| Humanised | 10 × 106 hPBMCs (i.p.) | NSG | A2A | 0.1 mg/kg CGS21680 (agonist) i.p. day −2–11 | ↑ Weight loss ↑ Serum human IL-6 ↓ Histological damage ↓ Serum human TNF-α | [136] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cuthbertson, P.; Geraghty, N.J.; Adhikary, S.R.; Bird, K.M.; Fuller, S.J.; Watson, D.; Sluyter, R. Purinergic Signalling in Allogeneic Haematopoietic Stem Cell Transplantation and Graft-versus-Host Disease. Int. J. Mol. Sci. 2021, 22, 8343. https://doi.org/10.3390/ijms22158343
Cuthbertson P, Geraghty NJ, Adhikary SR, Bird KM, Fuller SJ, Watson D, Sluyter R. Purinergic Signalling in Allogeneic Haematopoietic Stem Cell Transplantation and Graft-versus-Host Disease. International Journal of Molecular Sciences. 2021; 22(15):8343. https://doi.org/10.3390/ijms22158343
Chicago/Turabian StyleCuthbertson, Peter, Nicholas J. Geraghty, Sam R. Adhikary, Katrina M. Bird, Stephen J. Fuller, Debbie Watson, and Ronald Sluyter. 2021. "Purinergic Signalling in Allogeneic Haematopoietic Stem Cell Transplantation and Graft-versus-Host Disease" International Journal of Molecular Sciences 22, no. 15: 8343. https://doi.org/10.3390/ijms22158343