
Transcriptome Analysis of Flower Development and Mining of Genes Related to Flowering Time in Tomato (Solanum lycopersicum)

 ,
,
Abstract
:1. Introduction
2. Results
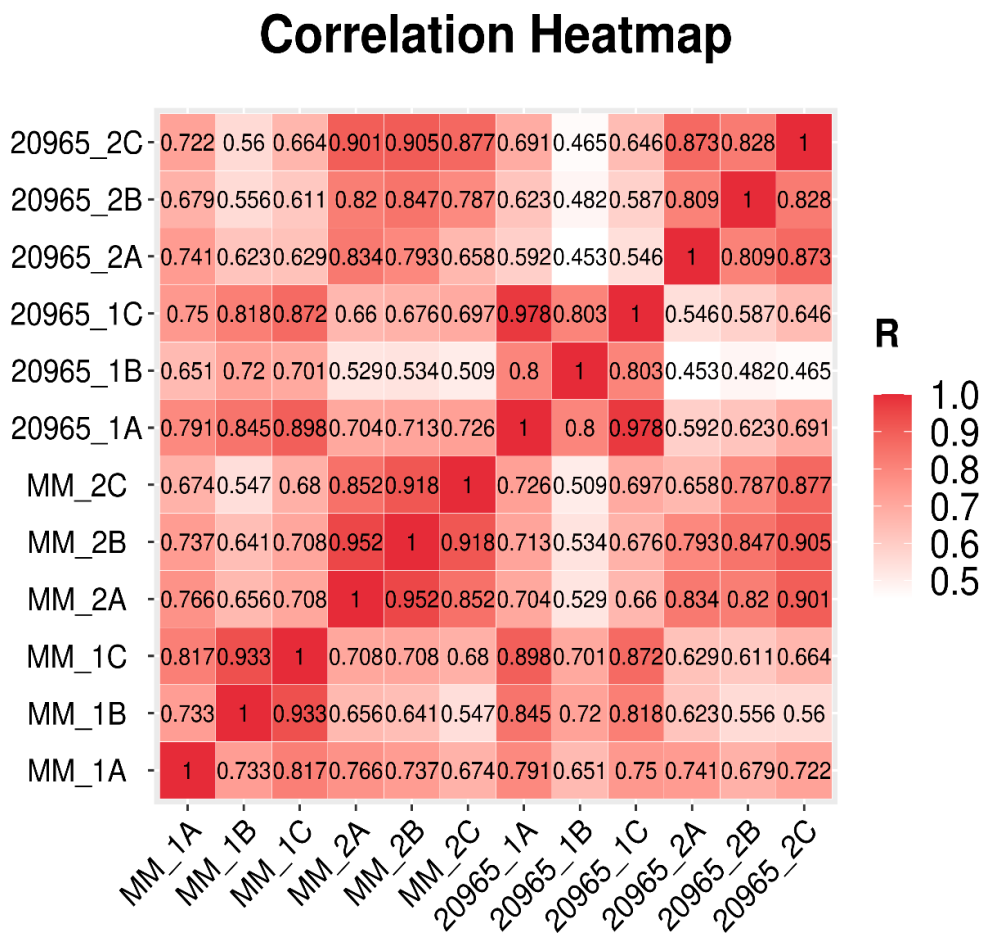
2.1. Statistics and Quality Verification of Transcriptome Sequencing Data
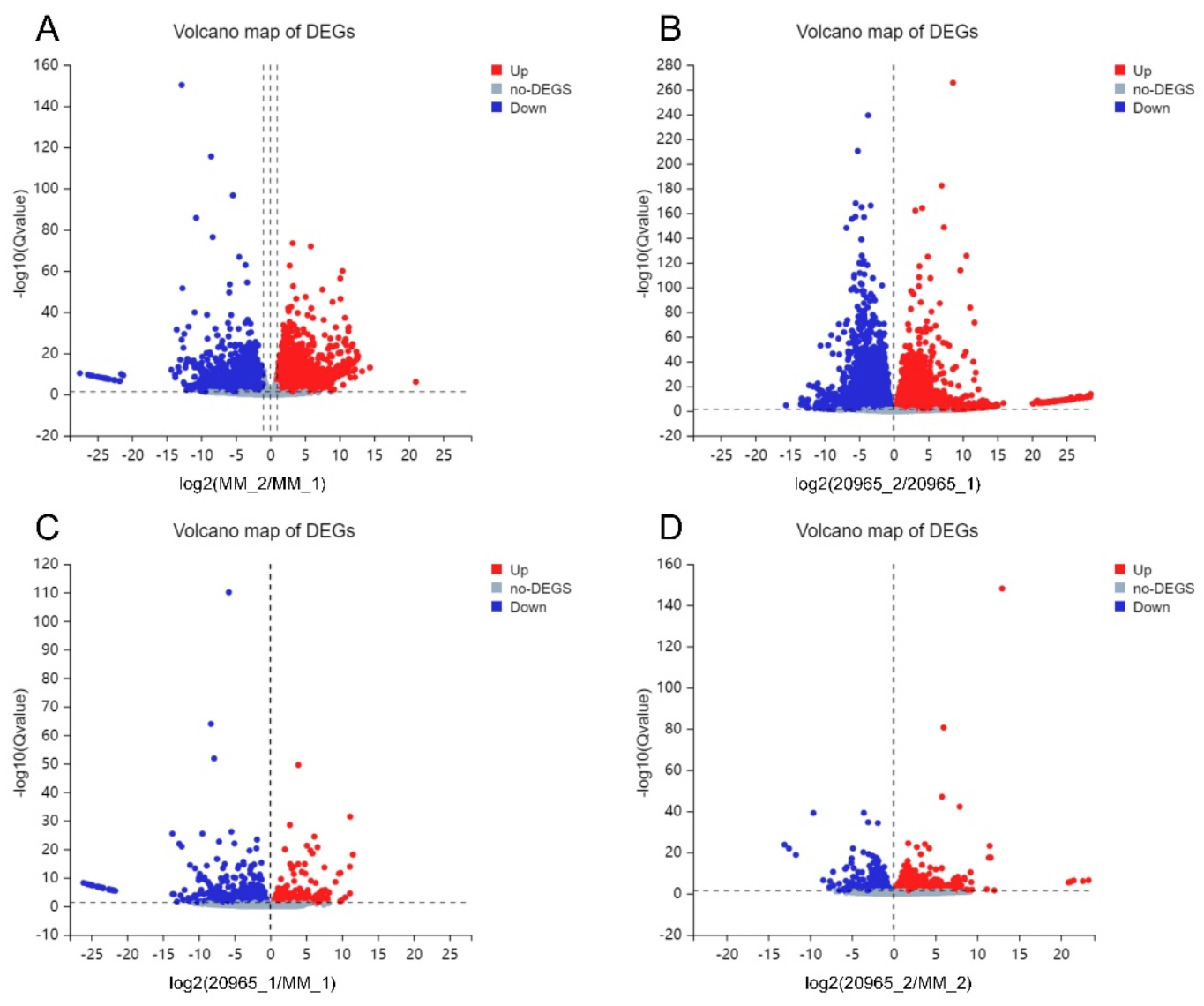
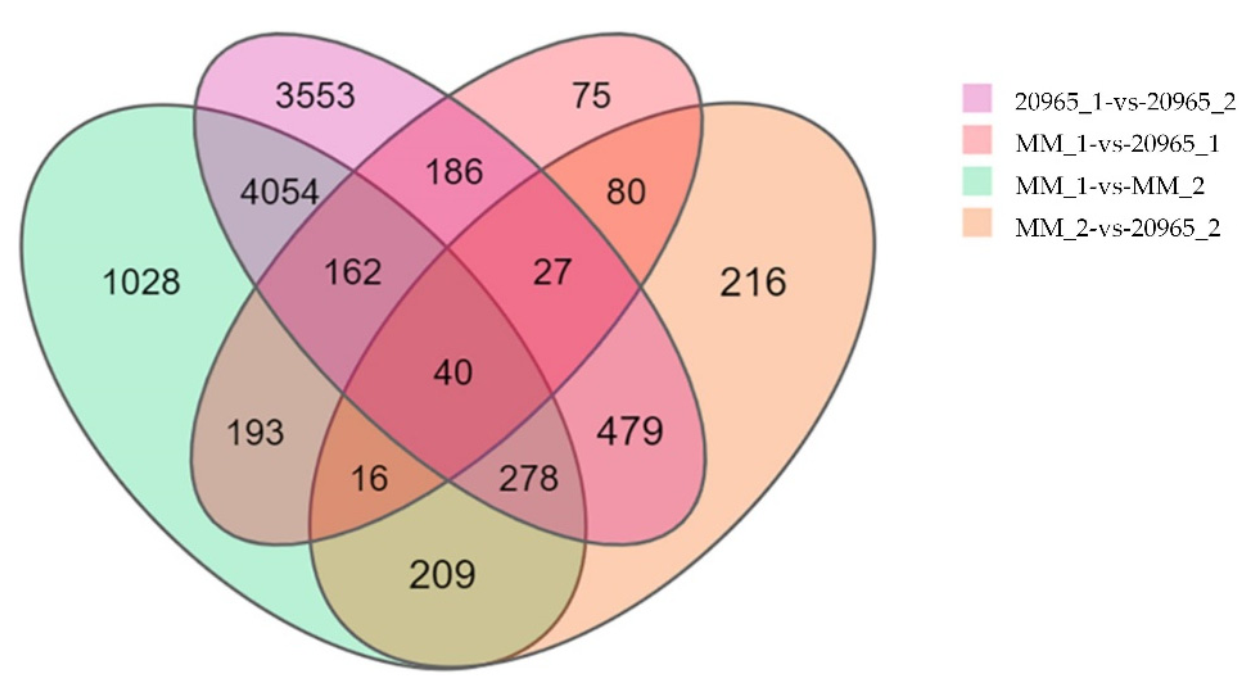
2.2. Differential Gene Expression Analysis
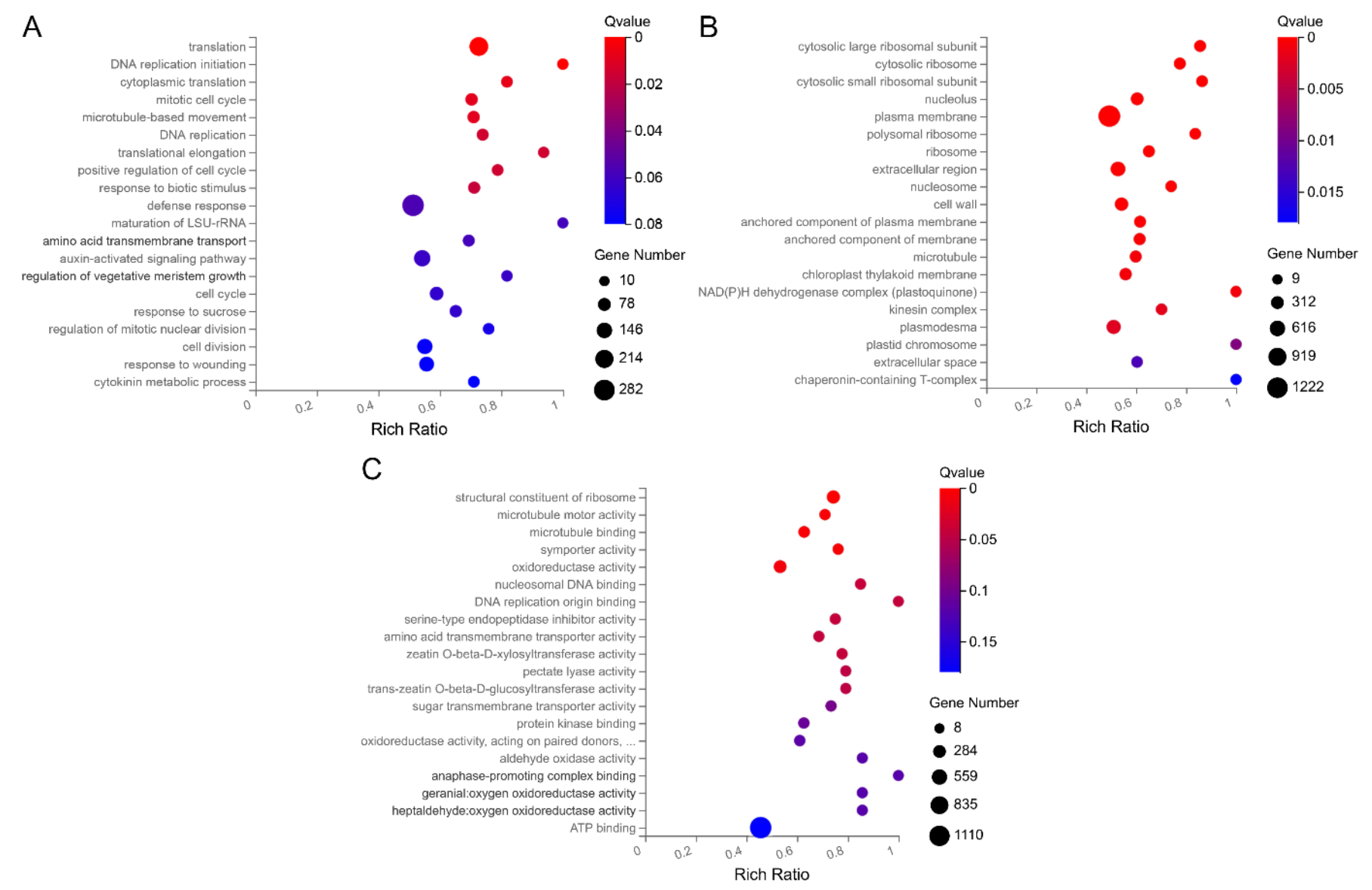
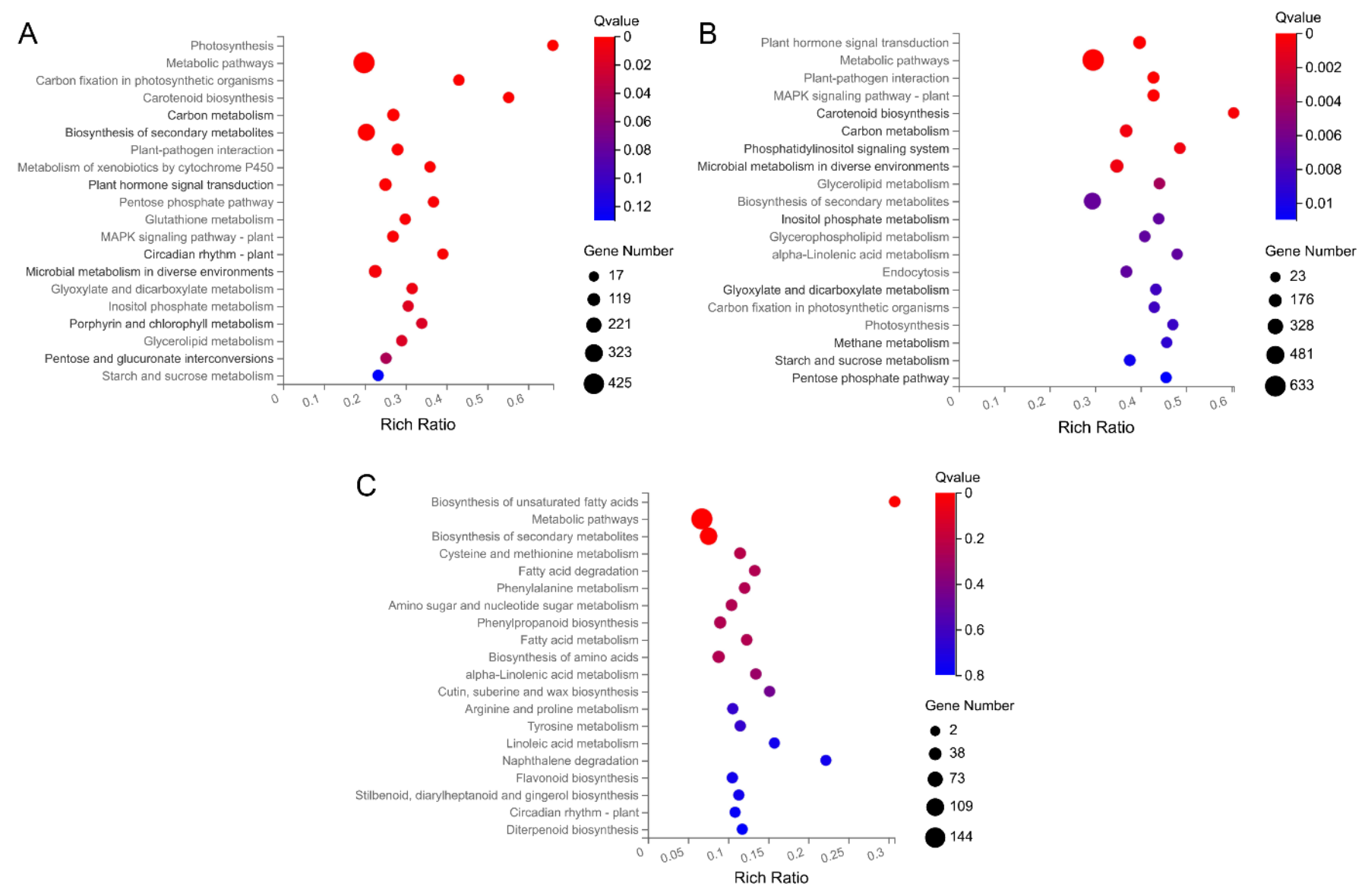
2.3. GO and KEGG Enrichment Analyses of DEGs
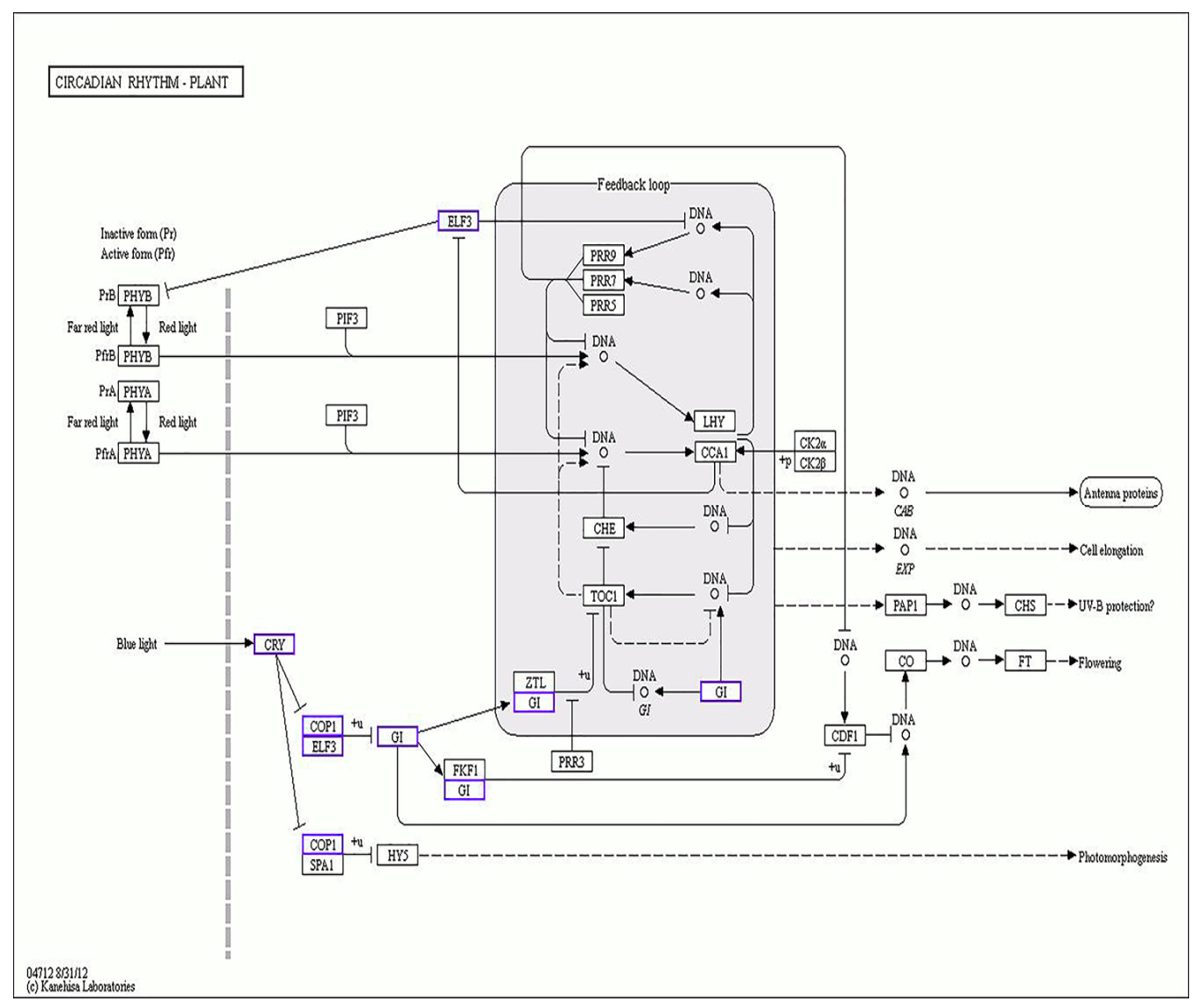
2.4. Analysis of Genes Related to Tomato Flowering Time and Flower Development
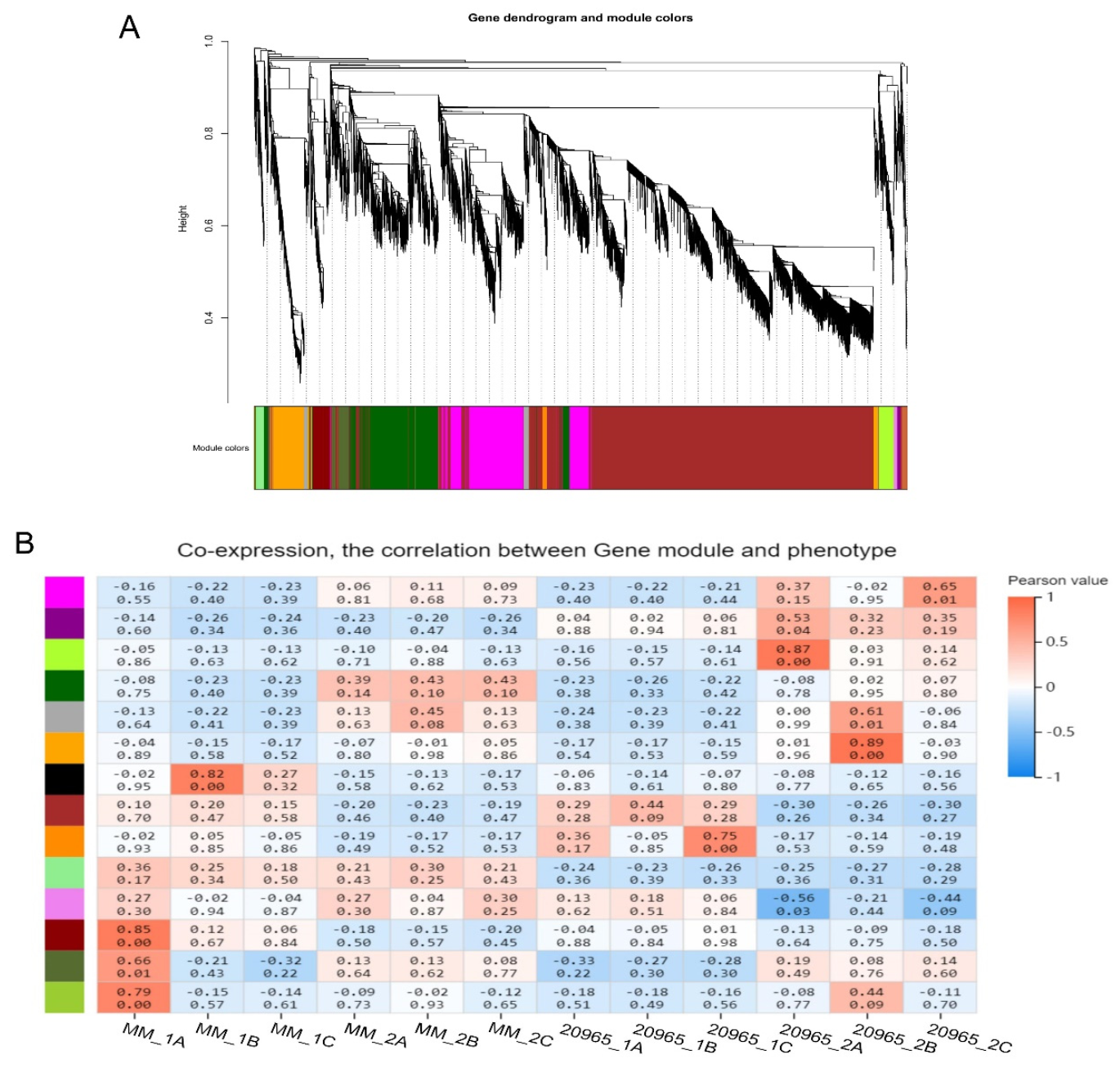
2.5. WGCNA of DEGs
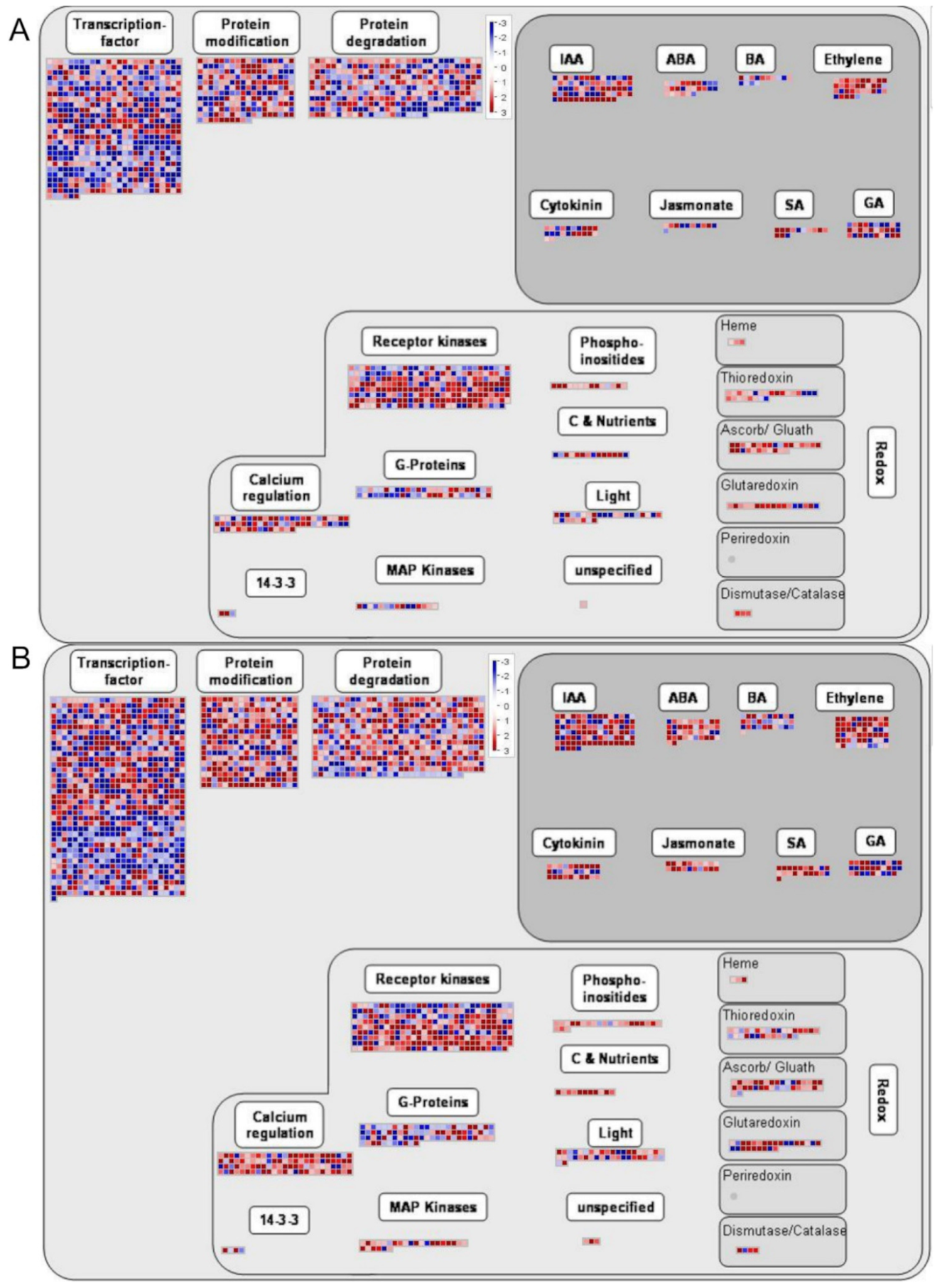
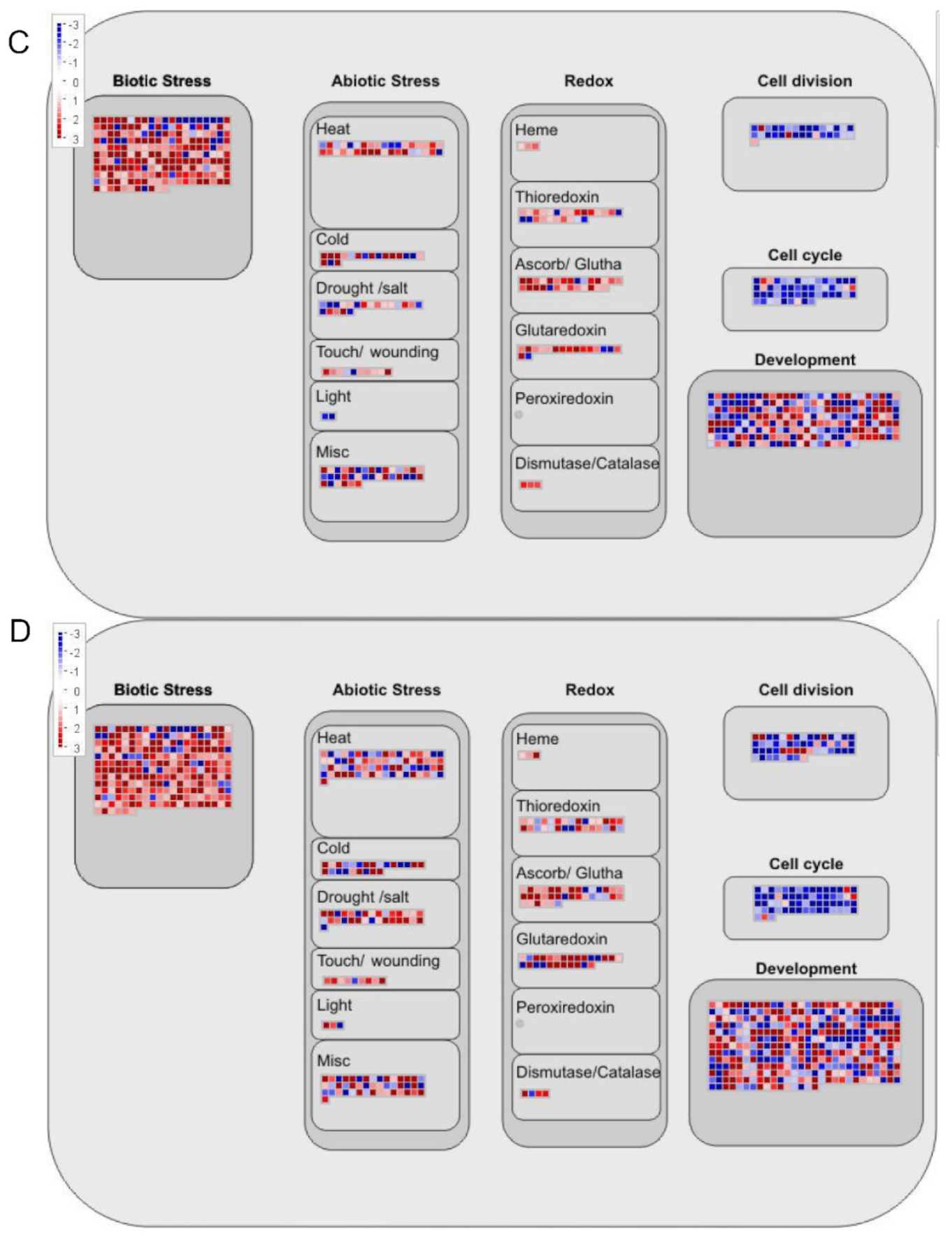
2.6. DEGs Bioregulation Analysis
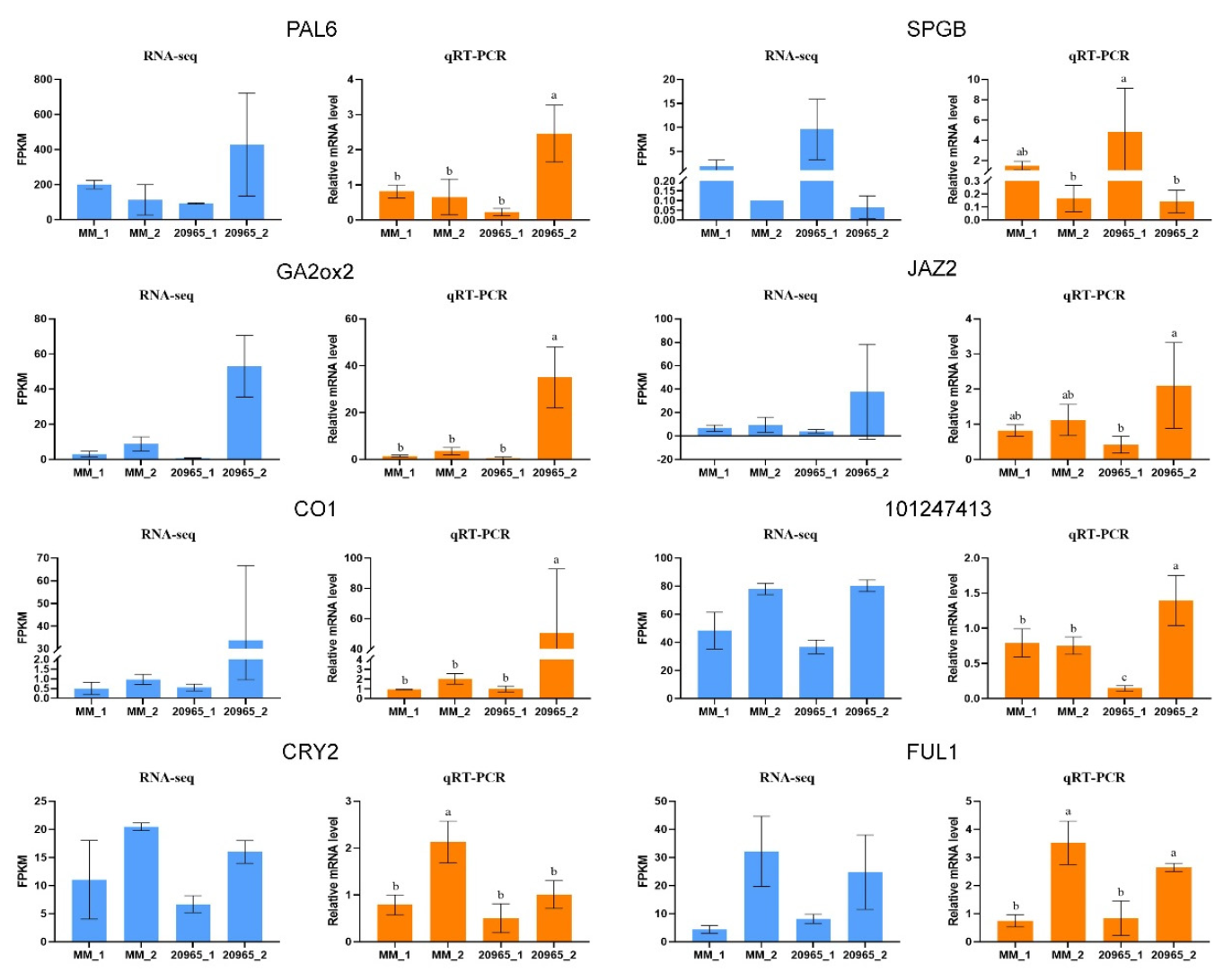
2.7. Validation of RNA-Seq Data by qRT-PCR
3. Discussion
4. Materials and Methods
4.1. Experimental Material
4.2. RNA-Seq Library Construction and Sequencing
4.3. Sequencing Reads and DEG Analysis
4.4. GO and KEGG Analysis
4.5. Weighted Gene Coexpression Network Analysis (WGCNA)
4.6. Analysis of MapMan Biological Functions of DEGs
4.7. Real-Time Quantitative PCR Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Zhang, J.; Wu, K.; Zeng, S.; da Silva, J.A.T.; Zhao, X.; Tian, C.E.; Xia, H.; Duan, J. Transcriptome analysis of cymbidium sinense and its application to the identification of genes associated with floral development. BMC Genom. 2013, 14, 279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, K.; Feng, S.; Pan, Y.; Zhong, J.; Chen, Y.; Yuan, C.; Li, H. Transcriptome analysis and identification of genes associated with floral transition and flower development in sugar apple. Front. Plant Sci. 2016, 7, 1695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, Y.L.; Tsay, Y.F. Influence of differing nitrate and nitrogen availability on flowering control in Arabidopsis. J. Exp. Bot. 2017, 68, 2603–2609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, J.; Tian, W.; Zhou, M.; Zhu, Q.; Du, W.; Jin, C. Improved plant nitrate status involves in flowering induction by extended photoperiod. Front. Plant Sci. 2021, 12, 629857. [Google Scholar] [CrossRef]
- Song, Y.H.; Shim, J.S.; Kinmonth-Schultz, H.A.; Imaizumi, T. Photoperiodic flowering: Time measurement mechanisms in leaves. Annu. Rev. Plant Biol. 2015, 66, 441–464. [Google Scholar] [CrossRef] [Green Version]
- Gaudinier, A.; Blackman, B.K. Evolutionary processes from the perspective of flowering time diversity. New Phytol. 2020, 225, 1883–1898. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Zhang, B.; Ma, N.; Liu, X.; Qin, M.; Zhang, Y.; Wang, K.; Guo, N.; Zuo, K.; Liu, X.; et al. Quantitative trait locus mapping and identification of candidate genes controlling flowering time in Brassica napus L. Front. Plant Sci. 2021, 11, 626205. [Google Scholar] [CrossRef]
- Jack, T. Molecular and genetic mechanisms of floral control. Plant Cell. 2004, 16, S1–S17. [Google Scholar] [CrossRef] [Green Version]
- Irish, V.F. The flowering of Arabidopsis flower development. Plant J. 2010, 61, 1014–1028. [Google Scholar] [CrossRef]
- Srikanth, A.; Schmid, M. Regulation of flowering time: All roads lead to Rome. Cell. Mol. Life Sci. 2011, 68, 2013–2037. [Google Scholar] [CrossRef]
- Yuan, S.; Zhang, Z.W.; Zheng, C.; Zhao, Z.Y.; Wang, Y.; Feng, L.Y.; Niu, G.; Wang, C.Q.; Wang, J.H.; Feng, H.; et al. Arabidopsis cryptochrome 1 functions in nitrogen regulation of flowering. Proc. Natl. Acad. Sci. USA 2016, 113, 7661–7666. [Google Scholar] [CrossRef] [Green Version]
- Liu, C.; Zhang, Q.; Zhu, H.; Cai, C.; Li, S. Characterization of mungbean CONSTANS-LIKE genes and functional analysis of CONSTANS-LIKE 2 in the regulation of flowering time in Arabidopsis. Front. Plant Sci. 2021, 12, 608603. [Google Scholar] [CrossRef]
- Greenham, K.; McClung, C.R. Integrating circadian dynamics with physiological processes in plants. Nat. Rev. Genet. 2015, 16, 598–610. [Google Scholar] [CrossRef]
- Suarez-Lopez, P.; Wheatley, K.; Robson, F.; Onouchi, H.; Valverde, F.; Coupland, G. CONSTANS mediates between the circadian clock and the control of flowering in Arabidopsis. Nature 2001, 410, 1116–1120. [Google Scholar] [CrossRef]
- Andrés, F.; Coupland, G. The genetic basis of flowering responses to seasonal cues. Nat. Rev. Genet. 2012, 13, 627–639. [Google Scholar] [CrossRef]
- Wang, L.; Sun, S.; Wu, T.; Liu, L.; Sun, X.; Cai, Y.; Li, J.; Jia, H.; Yuan, S.; Chen, L.; et al. Natural variation and CRISPR/Cas9-mediated mutation in GmPRR37 affect photoperiodic flowering and contribute to regional adaptation of soybean. Plant Biotechnol. J. 2020, 18, 1869–1881. [Google Scholar] [CrossRef] [Green Version]
- An, H.; Roussot, C.; Suarez-Lopez, P.; Corbesier, L.; Vincent, C.; Piñeiro, M.; Hepworth, S.; Mouradov, A.; Justin, S.; Turnbull, C.; et al. CONSTANS acts in the phloem to regulate a systemic signal that induces photoperiodic flowering of Arabidopsis. Development 2004, 131, 3615–3626. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.H.; Ito, S.; Imaizumi, T. Flowering time regulation: Photoperiod- and temperature-sensing in leaves. Trends Plant Sci. 2013, 18, 575–583. [Google Scholar] [CrossRef] [Green Version]
- Gangappa, S.N.; Botto, J.F. The BBX family of plant transcription factors. Trends Plant Sci. 2014, 19, 460–470. [Google Scholar] [CrossRef]
- Wellmer, F.; Riechmann, J.L. Gene networks controlling the initiation of flower development. Trends Genet. 2010, 26, 519–527. [Google Scholar] [CrossRef]
- Finnegan, E.J.; Robertson, M.; Helliwell, C.A. Resetting FLOWERING LOCUS C expression after vernalization is just activation in the early embryo by a different name. Front. Plant Sci. 2021, 11, 620155. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.; Galvão, V.C.; Zhang, Y.C.; Horrer, D.; Zhang, T.Q.; Hao, Y.H.; Feng, Y.Q.; Wang, S.; Schmid, M.; Wang, J.W. Gibberellin regulates the Arabidopsis floral transition through miR15-targeted SQUAMOSA promoter bindin-like transcription factors. Plant Cell 2012, 24, 3320–3332. [Google Scholar] [CrossRef] [Green Version]
- Silva, G.F.F.; Silva, E.M.; Correa, J.P.O.; Vicente, M.H.; Jiang, N.; Notini, M.M.; Junior, A.C.; Jesus, F.A.D.; Castilho, P.; Carrera, E.; et al. Tomato floral induction and flower development are orchestrated by the interplay between gibberellin and two unrelated microRNA-controlled modules. New Phytol. 2019, 221, 1328–1344. [Google Scholar] [CrossRef] [Green Version]
- Molinero-Rosales, N.; Latorre, A.; Jamilena, M.; Lozano, R. SINGLE FLOWER TRUSS regulates the transition and maintenance of flowering in tomato. Planta 2004, 218, 427–434. [Google Scholar] [CrossRef]
- Pnueli, L.; Carmel-Goren, L.; Hareven, D.; Gutfinger, T.; Alvarez, J.; Ganal, M.; Zamir, D.; Lifschitz, E. The SELF-PRUNING gene of tomato regulates vegetative to reproductive switching of sympodial meristems and is the ortholog of CEN and TFL1. Development 1998, 125, 1979–1989. [Google Scholar] [CrossRef]
- Lifschitz, E.; Ayre, B.G.; Eshed, Y. Florigen and anti-florigen-a systemic mechanism for coordinating growth and termination in flowering plants. Front. Plant Sci. 2014, 5, 465. [Google Scholar] [CrossRef] [Green Version]
- Jiang, K.; Liberatore, K.L.; Park, S.J.; Alvarez, J.P.; Lippman, Z.B. Tomato yield heterosis is triggered by a dosage sensitivity of the florigen pathway that fine-tunes shoot architecture. PLoS Genet. 2013, 9, e1004043. [Google Scholar] [CrossRef] [Green Version]
- Dielen, V.; Quinet, M.; Chao, J.; Batoko, H.; Havelange, A.; Kinet, J.M. UNIFLORA, a pivotal gene that regulates floral transition and meristem identity in tomato (Lycopersicon esculentum). New Phytol. 2004, 161, 393–400. [Google Scholar] [CrossRef]
- Quinet, M.; Dielen, V.; Batoko, H.; Boutry, M.; Havelange, A.; Kinet, J.M. Genetic interactions in the control of flowering time and reproductive structure development in tomato (Solanum lycopersicum). New Phytol. 2006, 170, 701–710. [Google Scholar] [CrossRef]
- Quinet, M.; Kinet, J.M.; Lutts, S. Flowering response of the uniflora:blind:self-pruning and jointless:uniflora:self-pruning tomato (Solanum lycopersicum) triple mutants. Physiol. Plant 2011, 141, 166–176. [Google Scholar] [CrossRef]
- Cao, K.; Cui, L.; Zhou, X.; Ye, L.; Zou, Z.; Deng, S. Four Tomato FLOWERING LOCUS T-Like proteins act antagonistically to regulate floral initiation. Front. Plant Sci. 2016, 6, 1213. [Google Scholar] [CrossRef] [PubMed]
- Soyk, S.; Müller, N.A.; Park, S.J.; Schmalenbach, I.; Jiang, K.; Hayama, R.; Zhang, L.; Eck, J.V.; Jiménez-Gómez, J.M.; Lippman, Z.B. Variation in the flowering gene SELF PRUNING 5G promotes day-neutrality and early yield in tomato. Nat. Genet. 2017, 49, 162–168. [Google Scholar] [CrossRef] [PubMed]
- MacAlister, C.A.; Park, S.J.; Jiang, K.; Marcel, F.; Bendahmane, A.; Izkovich, Y.; Eshed, Y.; Lippman, Z.B. Synchronization of the flowering transition by the tomato TERMINATING FLOWER gene. Nat. Genet. 2012, 44, 1393–1398. [Google Scholar] [CrossRef] [PubMed]
- Fornara, F.; de Montaigu, A.; Coupland, G. SnapShot: Control of flowering in Arabidopsis. Cell 2010, 141, 550–550.e2. [Google Scholar] [CrossRef] [Green Version]
- Lyu, T.; Cao, J. Cys2/His2 Zinc-Finger proteins in transcriptional regulation of flower development. Int. J. Mol. Sci. 2018, 19, 2589. [Google Scholar] [CrossRef] [Green Version]
- Waheed, S.; Zeng, L. The critical role of miRNAs in regulation of flowering time and flower development. Genes 2020, 11, 319. [Google Scholar] [CrossRef] [Green Version]
- Dodd, A.N.; Salathia, N.; Hall, A.; Kevei, E.; Toth, R.; Nagy, F.; Hibberd, J.M.; Millar, A.J.; Webb, A.A. Plant circadian clocks increase photosynthesis, growth, survival, and competitive advantage. Science 2005, 309, 630–633. [Google Scholar] [CrossRef] [Green Version]
- Wijnen, H.; Young, M.W. Interplay of circadian clocks and metabolic rhythms. Annu. Rev. Genet. 2006, 40, 409–448. [Google Scholar] [CrossRef]
- Matsushika, A.; Makino, S.; Kojima, M.; Mizuno, T. Circadian waves of expression of the APRR1/TOC1 family of pseudo-response regulators in Arabidopsis thaliana: Insight into the plant circadian clock. Plant Cell Physiol. 2000, 41, 1002–1012. [Google Scholar] [CrossRef] [Green Version]
- Farre’, E.M.; Harmer, S.L.; Harmon, F.G.; Yanovsky, M.J.; Kay, S.A. Overlapping and distinct roles of PRR7 and PRR9 in the Arabidopsis circadian clock. Curr. Biol. 2005, 15, 47–54. [Google Scholar] [CrossRef] [Green Version]
- Salomé, P.A.; McClung, C.R. PSEUDO-RESPONSE REGULATOR 7 and 9 are partially redundant genes essential for the temperature responsiveness of the Arabidopsis circadian clock. Plant Cell 2005, 17, 791–803. [Google Scholar] [CrossRef] [Green Version]
- Quail, P.H. Phytochrome photosensory signalling networks. Nat. Rev. Mol. Cell Biol. 2002, 3, 85–93. [Google Scholar] [CrossRef]
- Yu, J.W.; Rubio, V.; Lee, N.Y.; Bai, S.; Lee, S.Y.; Kim, S.S.; Liu, L.; Zhang, Y.; Irigoyen, M.L.; Sullivan, J.A.; et al. COP1 and ELF3 control circadian function and photoperiodic flowering by regulating GI stability. Mol. Cell 2008, 32, 617–630. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Huang, X.; Deng, X.W. The Photomorphogenic Central Repressor COP1: Conservation and functional diversification during evolution. Plant Commun. 2020, 1, 100044. [Google Scholar] [CrossRef]
- Kim, W.Y.; Hicks, K.A.; Somers, D.E. Independent roles for EARLY FLOWERING 3 and ZEITLUPE in the ccntrol of circadian timing, hypocotyl length, and flowering time. Plant Physiol. 2005, 139, 1557–1569. [Google Scholar] [CrossRef] [Green Version]
- Dally, N.; Eckel, M.; Batschauer, A.; Höft, N.; Jung, C. Two CONSTANS-LIKE genes jointly control flowering time in beet. Sci Rep. 2018, 8, 16120. [Google Scholar] [CrossRef] [Green Version]
- Rashid, A.; Deyholos, M.K. PELPK1 (At5g09530) contains a unique pentapeptide repeat and is a positive regulator of germination in Arabidopsis thaliana. Plant Cell Rep. 2011, 30, 1735–1745. [Google Scholar] [CrossRef]
- Johanson, U.; West, J.; Lister, C.; Michaels, S.; Amasino, R.; Dean, C. Molecular analysis of FRIGIDA, a major determinant of natural variation in Arabidopsis flowering time. Science 2000, 290, 344–347. [Google Scholar] [CrossRef]
- Jiang, D.; Gu, X.; He, Y. Establishment of the winter-annual growth habit via FRIGIDA-mediated histone methylation at FLOWERING LOCUS C in Arabidopsis. Plant Cell 2009, 21, 1733–1746. [Google Scholar] [CrossRef] [Green Version]
- Xu, C.; Park, S.J.; Eck, J.V.; Lippman, Z.B. Control of inflorescence architecture in tomato by BTB/POZ transcriptional regulators. Genes Dev. 2016, 30, 2048–2061. [Google Scholar] [CrossRef] [Green Version]
- Andrés, F.; Romera-Branchat, M.; Martínez-Gallegos, R.; Patel, V.; Schneeberger, K.; Jang, S.; Altmüller, J.; Nürnberg, P.; Coupland, G. Floral induction in Arabidopsis by FLOWERING LOCUS T requires direct repression of BLADE-ON-PETIOLE genes by the homeodomain protein PENNYWISE. Plant Physiol. 2015, 169, 2187–2199. [Google Scholar] [CrossRef]
- Shang, X.L.; Xie, R.R.; Tian, H.; Wang, Q.L.; Guo, F.Q. Putative zeatin O-glucosyltransferase OscZOG1 regulates root and shoot development and formation of agronomic traits in rice. J. Integr. Plant Biol. 2016, 58, 627–641. [Google Scholar] [CrossRef]
- Dong, B.; Deng, Y.; Wang, H.; Gao, R.; Stephen, K.; Chen, S.; Jiang, J.; Chen, F. Gibberellic acid signaling is required to induce flowering of chrysanthemums grown under both short and long days. Int. J. Mol. Sci. 2017, 18, 1259. [Google Scholar] [CrossRef] [Green Version]
- Mutasa-Göttgens, E.; Hedden, P. Gibberellin as a factor in floral regulatory networks. J. Exp. Bot. 2009, 60, 1979–1989. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, T.P. The molecular mechanism and evolution of the GA-GID1-DELLA signaling module in plants. Curr. Biol. 2011, 21, R338–R345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, X.; Chen, G.; Tang, B.; Zhang, J.; Zhou, S.; Hu, Z. The Jasmonate ZIM-domain protein gene SlJAZ2 regulates plant morphology and accelerates flower initiation in Solanum lycopersicum plants. Plant Sci. 2018, 267, 65–73. [Google Scholar] [CrossRef]
- Guan, Y.; Ding, L.; Jiang, J.; Shentu, Y.; Zhao, W.; Zhang, X.; Song, A.; Chen, S.; Chen, F. Overexpression of the CmJAZ1-like gene delays flowering in Chrysanthemum morifolium. Hortic. Res. 2021, 8, 87. [Google Scholar] [CrossRef] [PubMed]
- Heisler, M.G.; Atkinson, A.; Bylstra, Y.H.; Walsh, R.; Smyth, D.R. SPATULA, a gene that controls development of carpel margin tissues in Arabidopsis, encodes a bHLH protein. Development 2001, 128, 1089–1098. [Google Scholar] [CrossRef]
- Masiero, S.; Colombo, L.; Grini, P.E.; Schnittger, A.; Kater, M.M. The emerging importance of type I MADS box transcription factors for plant reproduction. Plant Cell 2011, 23, 865–872. [Google Scholar] [CrossRef] [Green Version]
- Niwa, T.; Suzuki, T.; Takebayashi, Y.; Takebayashi, R.; Higashiyama, T.; Sakakibara, H.; Ishiguro, S. Jasmonic acid facilitates flower opening and floral organ development through the upregulated expression of SlMYB21 transcription factor in tomato. Biosci. Biotechnol. Biochem. 2018, 82, 292–303. [Google Scholar] [CrossRef] [Green Version]
- Cock, P.J.; Fields, C.J.; Goto, N.; Heuer, M.L.; Rice, P.M. The Sanger FASTQ file format for sequences with quality scores, and the Solexa/Illumina FASTQ variants. Nucleic Acids Res. 2010, 38, 1767–1771. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357–360. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef] [Green Version]
- Tokimatsu, T.; Yamanishi, Y. KEGG for linking genomes to life and the environment. Nucleic Acids Res. 2008, 36, D480–D484. [Google Scholar] [CrossRef]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Raw Reads (M) | Clean Reads (M) | Clean Bases (Gb) | Q20 (%) | Q30 (%) | Clean Reads Ratio (%) | Total Mapping (%) | Uniquely Mapping (%) |
|---|---|---|---|---|---|---|---|---|
| MM_1A | 45.57 | 44.22 | 6.63 | 97.9 | 94.07 | 97.04 | 94.44 | 92.71 |
| MM_1B | 47.33 | 45.43 | 6.81 | 97.89 | 94.08 | 95.98 | 93.54 | 91.97 |
| MM_1C | 45.57 | 43.92 | 6.59 | 97.97 | 94.34 | 96.37 | 93.85 | 92.19 |
| MM_2A | 45.57 | 43.95 | 6.59 | 97.87 | 94.03 | 96.44 | 92.9 | 91.05 |
| MM_2B | 45.57 | 44.32 | 6.65 | 97.91 | 94.13 | 97.24 | 93.12 | 91.54 |
| MM_2C | 45.57 | 44.41 | 6.66 | 97.68 | 93.5 | 97.44 | 94.57 | 93.01 |
| 20965_1A | 45.57 | 43.95 | 6.59 | 97.27 | 92.66 | 96.43 | 93.62 | 91.94 |
| 20965_1B | 47.33 | 45.02 | 6.75 | 97.45 | 93.17 | 95.13 | 93.14 | 91.47 |
| 20965_1C | 45.57 | 44.35 | 6.65 | 97.3 | 92.75 | 97.31 | 94.21 | 92.55 |
| 20965_2A | 45.57 | 44.23 | 6.63 | 97.39 | 92.99 | 97.06 | 92.86 | 90.6 |
| 20965_2B | 45.57 | 44.34 | 6.65 | 97.28 | 92.68 | 97.28 | 93.73 | 92.03 |
| 20965_2C | 45.57 | 44.03 | 6.60 | 97.33 | 92.82 | 96.6 | 93.42 | 91.39 |
| Comparison Group | Down | Up | Total |
|---|---|---|---|
| MM_1-vs-MM_2 | 2828 | 3152 | 5980 |
| 20965_1-vs-20965_2 | 3825 | 4954 | 8779 |
| MM_1-vs-20965_1 | 565 | 214 | 779 |
| MM_2-vs-20965_2 | 486 | 859 | 1345 |
| Flowering Pathway | Gene ID | Gene Symbol | Log2 Fold-Change | |
|---|---|---|---|---|
| MM_1-vs-MM_2 | 20965_1-vs-20965_2 | |||
| Photoperiod | 543547 | COP1 | 0.30 | 1.15 |
| Photoperiod | 101259391 | COP1-like | 2.58 | 3.74 |
| Photoperiod | 101263075 | COP1-interacting | 0.53 | 1.78 |
| Photoperiod | 543596 | CRY2 | 1.01 | 1.41 |
| Photoperiod | 778253 | CO1 | 1.02 | 6.16 |
| Photoperiod | 100191137 | CO | 1.42 | 2.35 |
| Photoperiod | 101245415 | CO3 | 1.29 | 1.73 |
| Photoperiod | 100134891 | EZ1 | 0.59 | 1.15 |
| Photoperiod | 101245336 | FT | 1.95 | 2.26 |
| Photoperiod | 109119816 | PELPK1-like | 2.72 | 5.29 |
| Photoperiod | 101256688 | ELF4-like | 0.75 | 1.13 |
| Photoperiod | 101265040 | ELF3-like | 0.67 | 1.47 |
| Photoperiod | 101252363 | BEE2 | 3.03 | 4.23 |
| Photoperiod | 101252729 | bHLH63 | 2.06 | 4.05 |
| Photoperiod | 101265165 | bHLH130 | 2.30 | 2.32 |
| Photoperiod | 101255722 | WNK5 | 2.55 | 4.49 |
| Photoperiod | 101258346 | GI-like | 1.34 | 0.42 |
| Vernalization | 101244491 | AGL42-like | 1.26 | 3.29 |
| Vernalization | 101249083 | AGL104 | 8.63 | 8.31 |
| Gibberellin | 544211 | 20ox-2 | 1.52 | 1.54 |
| Gibberellin | 100037505 | GA2ox2 | 1.53 | 6.07 |
| Gibberellin | 100134889 | GA2ox4 | 2.92 | 3.92 |
| Gibberellin | 543839 | FUL1 | 2.90 | 1.76 |
| Gibberellin | 543887 | FUL2 | 1.25 | 1.98 |
| Gibberellin | 101247719 | LOC101247719 | 1.36 | 1.68 |
| Gibberellin | 101249868 | SOC1-like | 0.47 | 1.59 |
| Gibberellin | 101253625 | GID1ac | 1.59 | 0.60 |
| Gibberellin | 100736493 | GID1b-1 | 2.04 | 0.32 |
| Gibberellin | 101258522 | MYB21 | 7.64 | 11.52 |
| Gibberellin | 101265384 | GAI | 3.55 | 1.71 |
| Age | 100191138 | AP2a | 1.39 | 1.84 |
| Age | 101244632 | AP2-like | −2.04 | −3.56 |
| Temperature | 101245946 | actin6 | −1.00 | −1.55 |
| Temperature | 101246885 | actin7 | −1.38 | −1.72 |
| Temperature | 101266766 | SVP | −0.66 | −1.70 |
| Temperature | 101252303 | PIF4 | 1.31 | 1.39 |
| Autonomy | 101246029 | SFT | 3.52 | 2.21 |
| Autonomy | 101252492 | FRI | −0.84 | −1.14 |
| Autonomy | 101251416 | LOC101251416 | 6.15 | 5.36 |
| 544020 | PRK2 | 10.21 | 11.53 | |
| 544021 | PRK1 | 11.03 | 12.13 | |
| 101243937 | LOC101243937 | 6.84 | 6.88 | |
| 101245709 | BOP3 | −0.04 | −1.99 | |
| 101246001 | BOP2 | −1.46 | −1.27 | |
| 101247407 | LOC101247407 | 1.18 | 1.89 | |
| 101246927 | TAA1 | 4.30 | 6.68 | |
| 101252256 | AIM1 | 1.11 | 2.38 | |
| 101251501 | GPAT6 | 0.24 | 3.20 | |
| 100134911 | JAZ2 | 0.58 | 3.38 | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, H.; Yang, Y.; Zhang, Y.; Zhao, T.; Jiang, J.; Li, J.; Xu, X.; Yang, H. Transcriptome Analysis of Flower Development and Mining of Genes Related to Flowering Time in Tomato (Solanum lycopersicum). Int. J. Mol. Sci. 2021, 22, 8128. https://doi.org/10.3390/ijms22158128
Wang H, Yang Y, Zhang Y, Zhao T, Jiang J, Li J, Xu X, Yang H. Transcriptome Analysis of Flower Development and Mining of Genes Related to Flowering Time in Tomato (Solanum lycopersicum). International Journal of Molecular Sciences. 2021; 22(15):8128. https://doi.org/10.3390/ijms22158128
Chicago/Turabian StyleWang, Hexuan, Yahui Yang, Yiyao Zhang, Tingting Zhao, Jingbin Jiang, Jingfu Li, Xiangyang Xu, and Huanhuan Yang. 2021. "Transcriptome Analysis of Flower Development and Mining of Genes Related to Flowering Time in Tomato (Solanum lycopersicum)" International Journal of Molecular Sciences 22, no. 15: 8128. https://doi.org/10.3390/ijms22158128