
T-2 Toxin Induces Oxidative Stress at Low Doses via Atf3ΔZip2a/2b-Mediated Ubiquitination and Degradation of Nrf2

 ,
, 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
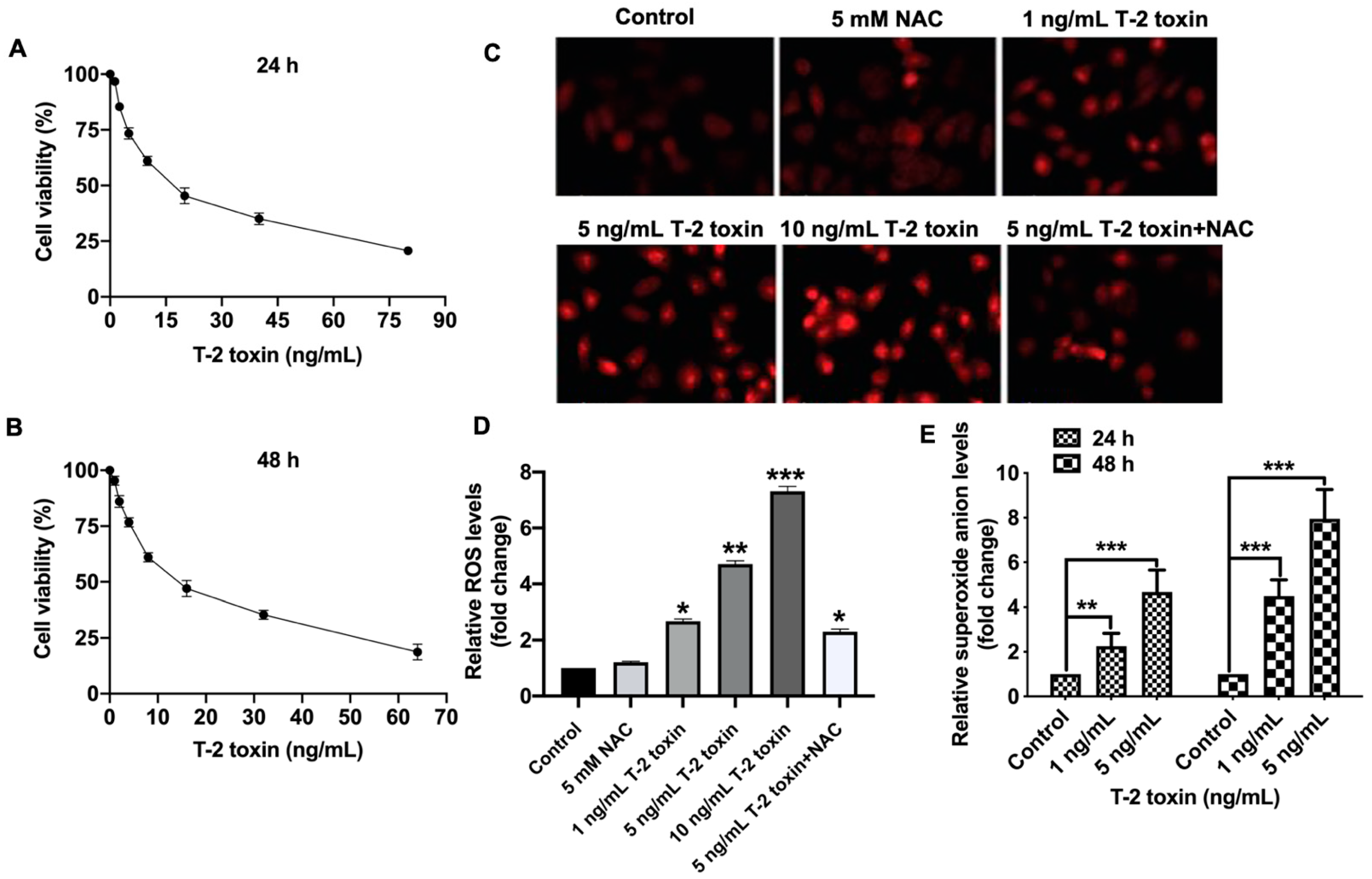
2.1. T-2 Toxin Promoted Ros Accumulation in Mcf-7 Cells at Low Doses
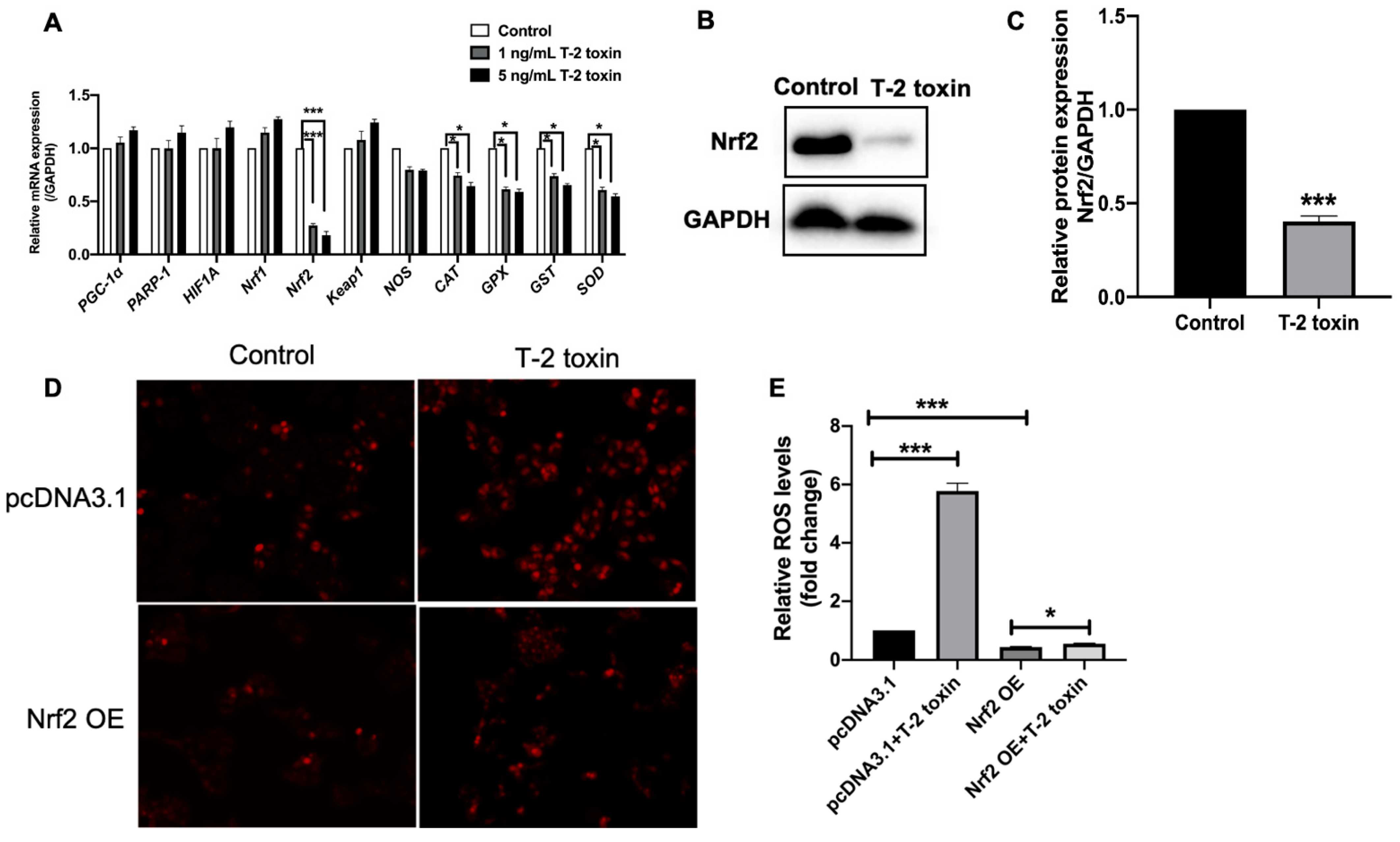
2.2. Nrf2 Was Critical for T-2 Toxin-Induced Ros Accumulation
2.3. T-2 Toxin Downregulated the Expression of Antioxidant Genes by Nrf2
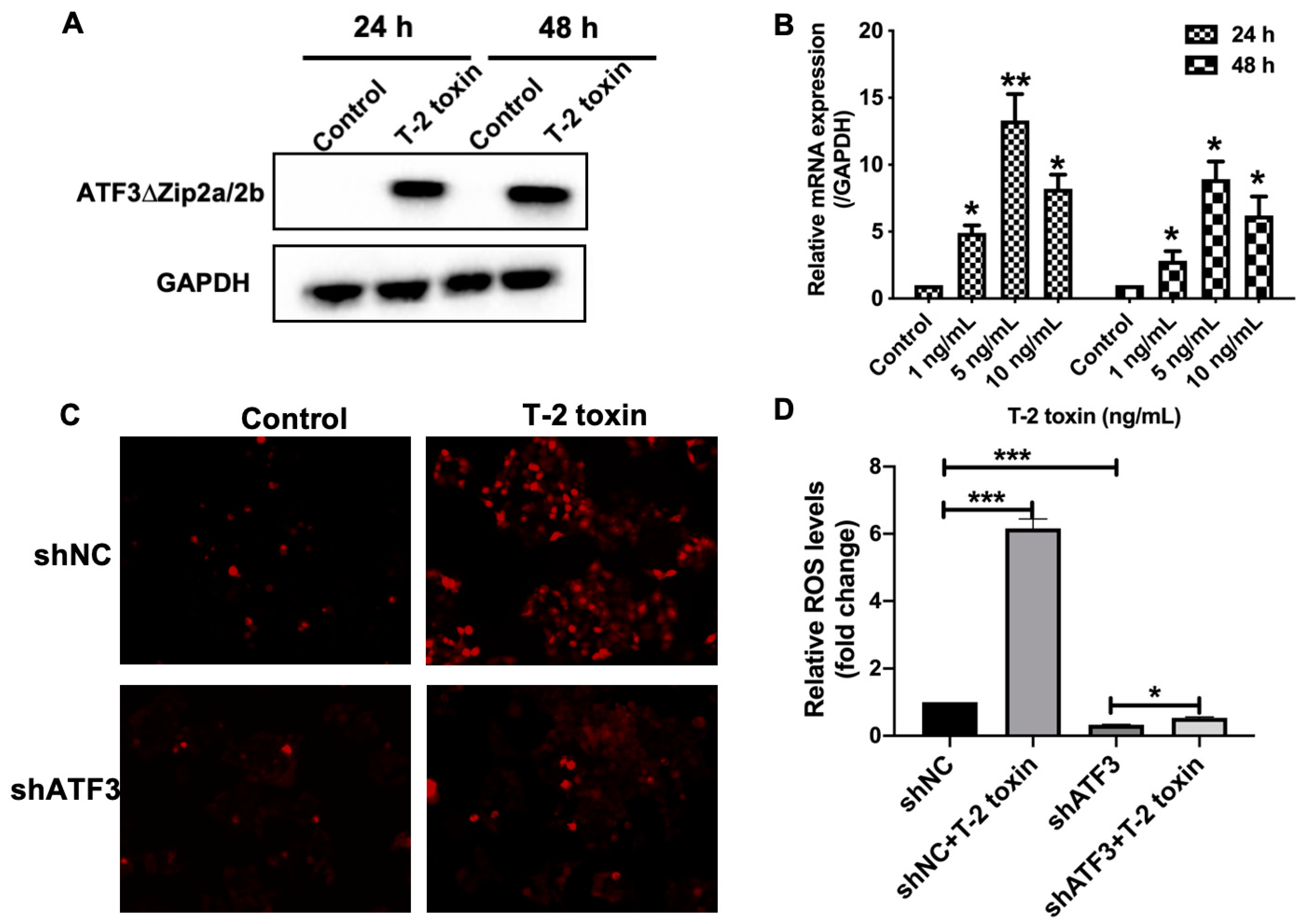
2.4. ATF3ΔZip2a/2b Was Involved in the T-2 Toxin-Induced Ros Accumulation
2.5. T-2 Toxin Reduced the Expression of Nrf2 and Its Targeted Antioxidant Genes via Inducing ATF3ΔZip2a/2b
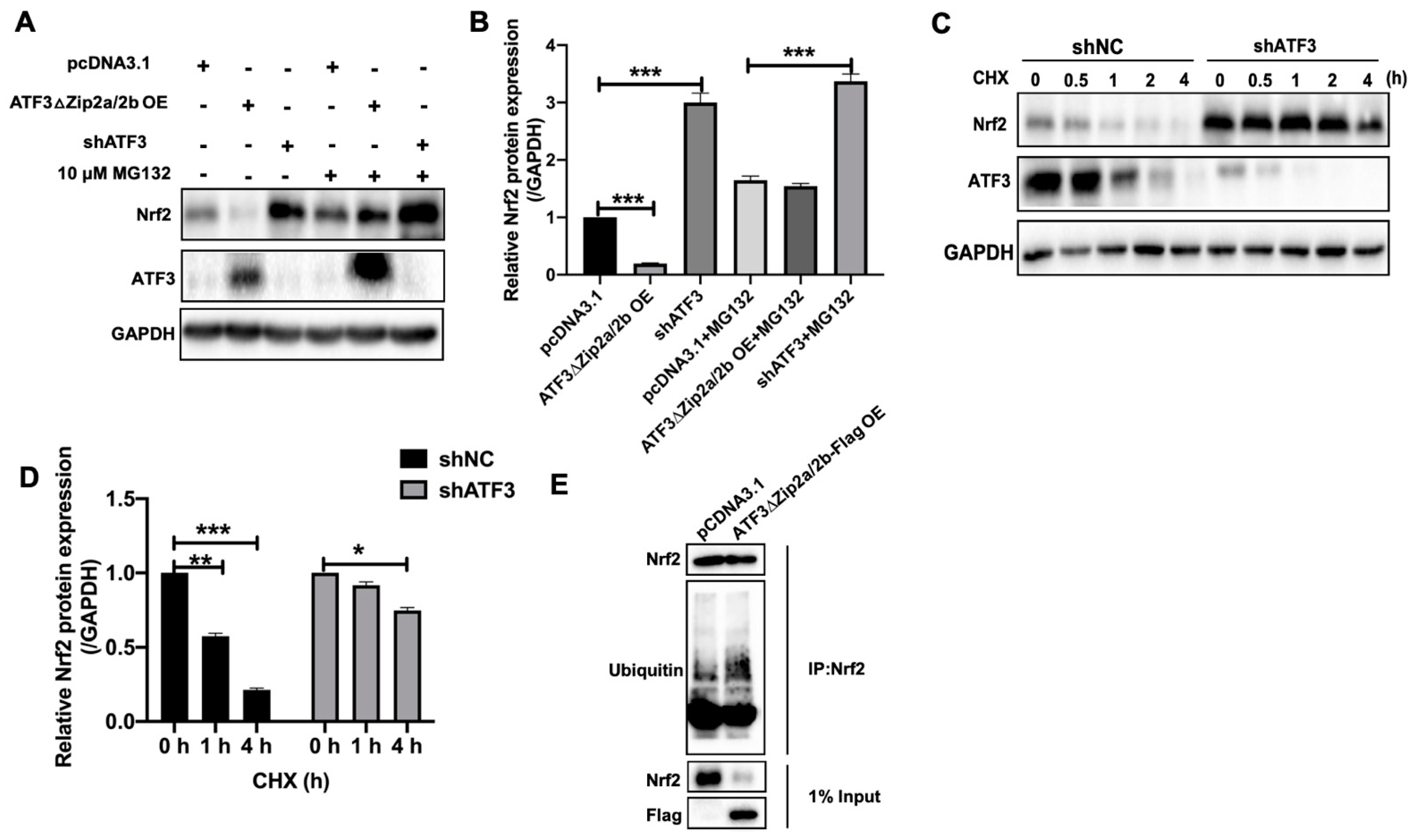
2.6. Overexpression of ATF3ΔZip2a/2b Promoted Ubiquitination and Degradation of Nrf2
3. Discussion
4. Materials and Methods
4.1. Cell Culture and Chemical Reagents
4.2. Plasmids Construction and Cell Transfection
4.3. Cell Viability Assay
4.4. Measurement of Intracellular Ros Levels
4.5. RNA Extraction and Quantitative RT-PCR
4.6. Western Blot
4.7. RNA Interference Assay
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Dai, C.; Xiao, X.; Sun, F.; Zhang, Y.; Hoyer, D.; Shen, J.; Tang, S.; Velkov, T. T-2 toxin neurotoxicity: Role of oxidative stress and mitochondrial dysfunction. Arch. Toxicol. 2019, 93, 3041–3056. [Google Scholar] [CrossRef]
- Wu, Q.; Dohnal, V.; Huang, L.; Kuča, K.; Yuan, Z. Metabolic pathways of trichothecenes. Drug Metab. Rev. 2010, 42, 250–267. [Google Scholar] [CrossRef]
- Doi, K.; Ishigami, N.; Sehata, S. T-2 toxin-induced toxicity in pregnant mice and rats. Int. J. Mol. Sci. 2008, 9, 2146–2158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinozuka, J.; Suzuki, M.; Noguchi, N.; Sugimoto, T.; Uetsuka, K.; Nakayama, H.; Doi, K. T-2 toxin-induced apoptosis in hematopoietic tissues of mice. Toxicol. Pathol. 1998, 26, 674–681. [Google Scholar] [CrossRef] [PubMed]
- Pisoschi, A.M.; Pop, A. The role of antioxidants in the chemistry of oxidative stress: A review. Eur. J. Med. Chem. 2015, 97, 55–74. [Google Scholar] [CrossRef]
- Li, Y.; Wang, Z.; Beier, R.C.; Shen, J.; De Smet, S.; De Saeger, S.; Zhang, S. T-2 toxin, a trichothecene mycotoxin: Review of toxicity, metabolism, and analytical methods. J. Agric. Food Chem. 2011, 59, 3441–3453. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, Y.; Velkov, T.; Tang, S.; Dai, C. T-2 toxin–induced toxicity in neuroblastoma–2a cells involves the generation of reactive oxygen, mitochondrial dysfunction and inhibition of Nrf2/HO–1 pathway. Food Chem. Toxicol. 2018, 114, 88–97. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, M.; Jayaraj, R.; Bhaskar, A.S.; Rao, P.L. Oxidative stress induction by T-2 toxin causes DNA damage and triggers apoptosis via caspase pathway in human cervical cancer cells. Toxicology 2009, 262, 153–161. [Google Scholar] [CrossRef] [PubMed]
- Mate, J.M. Effects of antioxidant enzymes in the molecular control of reactive oxygen species toxicology. Toxicology 2000, 153, 83–104. [Google Scholar] [CrossRef]
- Udensi, U.K.; Tchounwou, P.B. Oxidative stress in prostate hyperplasia and carcinogenesis. J. Exp. Clin. Cancer Res. 2016, 35, 139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Tan, J.; Miao, Y.; Lei, P.; Zhang, Q. ROS and autophagy: Interactions and molecular regulatory mechanisms. Cell Mol. Neurobiol. 2015, 35, 615–621. [Google Scholar] [CrossRef]
- Hashad, I.M.; Abdel, R.M.F.; Abdel–Maksoud, S.M.; Amr, K.S.; Effat, L.K.; Shaban, G.M.; Gad, M.Z. C242T polymorphism of NADPH oxidase p22phox gene reduces the risk of coronary artery disease in a random sample of Egyptian population. Mol. Biol. Rep. 2014, 41, 2281–2286. [Google Scholar] [CrossRef]
- Heslop, C.L.; Tebbutt, S.J.; Podder, M.; Ruan, J.; Hill, J.S. Combined polymorphisms in oxidative stress genes predict coronary artery disease and oxidative stress in coronary angiography patients. Ann. Hum. Genet. 2012, 76, 435–447. [Google Scholar] [CrossRef]
- Mathews, M.T.; Berk, B.C. PARP-1 inhibition prevents oxidative and nitrosative stress–induced endothelial cell death via transactivation of the VEGF receptor 2. Arterioscler. Thromb. Vasc. Biol. 2008, 28, 711–717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szabo, C. Roles of poly(ADP–ribose) polymerase activation in the pathogenesis of diabetes mellitus and its complications. Pharmacol. Res. 2005, 52, 60–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.G.; Wang, Z.Q.; Tong, W.M.; Shen, Y. PARP1 Val762Ala polymorphism reduces enzymatic activity. Biochem. Biophys. Res. Commun. 2007, 354, 122–126. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Liu, Y.; Zhou, Y.; Ruiz–Rodado, V.; Larion, M.; Xu, G.; Yang, C. Triptolide suppresses IDH1–mutated malignancy via Nrf2–driven glutathione metabolism. Proc. Natl. Acad. Sci. USA 2020, 117, 9964–9972. [Google Scholar] [CrossRef] [PubMed]
- Miura, S.; Yamaguchi, M.; Yoshino, H.; Nakai, Y.; Kashiwakura, I. Dose–dependent increase of Nrf2 target gene expression in mice exposed to ionizing radiation. Radiat. Res. 2019, 191, 176–188. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013, 53, 401–426. [Google Scholar] [CrossRef] [Green Version]
- Murakami, S.; Motohashi, H. Roles of Nrf2 in cell proliferation and differentiation. Free. Radic. Biol. Med. 2015, 88, 168–178. [Google Scholar] [CrossRef] [Green Version]
- Tonelli, C.; Chio, I.I.C.; Tuveson, D.A. Transcriptional regulation by Nrf2. Antioxid. Redox Signal. 2018, 29, 1727–1745. [Google Scholar] [CrossRef] [Green Version]
- Taguchi, K.; Yamamoto, M. The Keap1–Nrf2 system in cancer. Front. Oncol. 2017, 7, 85. [Google Scholar] [CrossRef]
- Kawai, Y.; Garduno, L.; Theodore, M.; Yang, J.; Arinze, I.J. Acetylation–deacetylation of the transcription factor Nrf2 (nuclear factor erythroid 2–related factor 2) regulates its transcriptional activity and nucleocytoplasmic localization. J. Biol. Chem. 2011, 286, 7629–7640. [Google Scholar] [CrossRef] [Green Version]
- Zhou, H.; Li, N.; Yuan, Y.; Jin, Y.G.; Guo, H.; Deng, W.; Tang, Q. Activating transcription factor 3 in cardiovascular diseases: A potential therapeutic target. Basic Res. Cardiol. 2018, 113, 37. [Google Scholar] [CrossRef]
- Zhao, W.; Sun, M.; Li, S.; Chen, Z.; Geng, D. Transcription factor ATF3 mediates the radioresistance of breast cancer. J. Cell Mol. Med. 2018, 22, 4664–4675. [Google Scholar] [CrossRef] [Green Version]
- Brown, S.L.; Sekhar, K.R.; Rachakonda, G.; Sasi, S.; Freeman, M.L. Activating transcription factor 3 is a novel repressor of the nuclear factor erythroid–derived 2–related factor 2 (Nrf2)–regulated stress pathway. Cancer Res. 2008, 68, 364–368. [Google Scholar] [CrossRef] [Green Version]
- Nyunt, T.; Britton, M.; Wanichthanarak, K.; Budamagunta, M.; Voss, J.C.; Wilson, D.W.; Aung, H.H. Mitochondrial oxidative stress–induced transcript variants of ATF3 mediate lipotoxic brain microvascular injury. Free Radic. Biol. Med. 2019, 143, 25–46. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Kim, G.L.; Kim, N.Y.; Kim, S.J.; Ghosh, P.; Rhee, D.K. ATF3 stimulates IL–17A by regulating intracellular Ca2+/ROS–dependent IL–1beta activation during streptococcus pneumoniae infection. Front. Immunol. 2018, 9, 1954. [Google Scholar] [CrossRef] [PubMed]
- Bueno, M.; Brands, J.; Voltz, L.; Fiedler, K.; Mays, B.; Croix, C.S.; Mallampalli, R.K.; Rojas, M.; Mora, A.L. ATF3 represses PINK1 gene transcription in lung epithelial cells to control mitochondrial homeostasis. Aging Cell 2018, 17, e12720. [Google Scholar] [CrossRef] [PubMed]
- Jyrkkanen, H.K.; Kansanen, E.; Inkala, M.; Kivela, A.M.; Hurttila, H.; Heinonen, S.E.; Goldsteins, G.; Jauhiainen, S.; Tiainen, S.; Makkonen, H.; et al. Nrf2 regulates antioxidant gene expression evoked by oxidized phospholipids in endothelial cells and murine arteries in vivo. Circ. Res. 2008, 103, e1–e9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoetzenecker, W.; Echtenacher, B.; Guenova, E.; Hoetzenecker, K.; Woelbing, F.; Bruck, J.; Teske, A.; Valtcheva, N.; Fuchs, K.; Kneilling, M.; et al. ROS–induced ATF3 causes susceptibility to secondary infections during sepsis–associated immunosuppression. Nat. Med. 2011, 18, 128–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, L.; Mu, P.; Huang, B.; Li, H.; Mu, H.; Deng, Y. EGR1 is essential for deoxynivalenol–induced G2/M cell cycle arrest in HepG2 cells via the ATF3DeltaZip2a/2b–EGR1–p21 pathway. Toxicol. Lett. 2018, 299, 95–103. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Wang, X.; Xiao, S.; Bu, X.; Lin, Z.; Qi, C.; Qin, J.; Chen, L. T-2 toxin in the diet suppresses growth and induces immunotoxicity in juvenile Chinese mitten crab (Eriocheir sinensis). Fish. Shellfish Immunol. 2020, 97, 593–601. [Google Scholar] [CrossRef]
- Wu, Q.; Wang, X.; Nepovimova, E.; Miron, A.; Liu, Q.; Wang, Y.; Su, D.; Yang, H.; Li, L.; Kuca, K. Trichothecenes: Immunomodulatory effects, mechanisms, and anti–cancer potential. Arch. Toxicol. 2017, 91, 3737–3785. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yang, C.; Yuan, Z.; Yi, J.; Wu, J. T-2 toxin exposure induces apoptosis in TM3 cells by inhibiting mammalian target of rapamycin/serine/threonine protein kinase(mTORC2/AKT) to promote Ca2+ production. Int. J. Mol. Sci. 2018, 19, 3360. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Z.; Matias, F.B.; Yi, J.E.; Wu, J. T-2 toxin-induced cytotoxicity and damage on TM3 Leydig cells. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2016, 181–182, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Huang, W.; Xiao, H.; Xie, Y.; Yuan, Z.; Yi, J.; Chen, J.; Tu, D.; Tian, Y. Procyanidins B2 reverses the T-2 toxin–induced mitochondrial apoptosis in TM3 Leydig cells. J. Funct. Foods 2018, 45, 118–128. [Google Scholar] [CrossRef]
- Luo, J.F.; Shen, X.Y.; Lio, C.K.; Dai, Y.; Cheng, C.S.; Liu, J.X.; Yao, Y.; Yu, Y.; Xie, Y.; Luo, P.; et al. Activation of Nrf2/HO–1 pathway by nardochinoid c inhibits inflammation and oxidative stress in lipopolysaccharide–stimulated macrophages. Front. Pharmacol. 2018, 9, 911. [Google Scholar] [CrossRef]
- Li, L.; Dong, H.; Song, E.; Xu, X.; Liu, L.; Song, Y. Nrf2/ARE pathway activation, HO–1 and NQO1 induction by polychlorinated biphenyl quinone is associated with reactive oxygen species and PI3K/AKT signaling. Chem. Biol. Interact. 2014, 209, 56–67. [Google Scholar] [CrossRef]
- He, F.; Antonucci, L.; Yamachika, S.; Zhang, Z.; Taniguchi, K.; Umemura, A.; Hatzivassiliou, G.; Roose-Girma, M.; Reina-Campos, M.; Duran, A.; et al. NRF2 activates growth factor genes and downstream AKT signaling to induce mouse and human hepatomegaly. J. Hepatol. 2020, 72, 1182–1195. [Google Scholar] [CrossRef] [Green Version]
- Chen, Z.; Zhong, H.; Wei, J.; Lin, S.; Zong, Z.; Gong, F.; Huang, X.; Sun, J.; Li, P.; Lin, H.; et al. Inhibition of Nrf2/HO–1 signaling leads to increased activation of the NLRP3 inflammasome in osteoarthritis. Arthritis Res. Ther. 2019, 21, 300. [Google Scholar] [CrossRef] [Green Version]
- Ma, Q. Transcriptional responses to oxidative stress: Pathological and toxicological implications. Pharmacol. Ther. 2010, 125, 376–393. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Gu, X.; Gao, J.; Wang, Z.; Liu, G.; Barkema, H.W.; Han, B. Chlorogenic acid promotes the Nrf2/HO–1 anti–oxidative pathway by activating p21(Waf1/Cip1) to resist dexamethasone–induced apoptosis in osteoblastic cells. Free Radic. Biol. Med. 2019, 137, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Komatsu, M.; Kurokawa, H.; Waguri, S.; Taguchi, K.; Kobayashi, A.; Ichimura, Y.; Sou, Y.; Ueno, I.; Sakamoto, A.; Tong, K.I.; et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat. Cell Biol. 2010, 12, 213–223. [Google Scholar] [CrossRef] [PubMed]
- Limonciel, A.; Jennings, P. A review of the evidence that ochratoxin A is an Nrf2 inhibitor: Implications for nephrotoxicity and renal carcinogenicity. Toxins 2014, 6, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.Y.; Park, K.J.; Hwang, J.Y.; Kim, G.H.; Lee, D.; Lee, Y.J.; HyunSong, E.; Yoo, M.; Kim, B.; HoSuh, Y.; et al. Activating transcription factor 3 is a target molecule linking hepatic steatosis to impaired glucose homeostasis. J. Hepatol. 2017, 67, 349–359. [Google Scholar] [CrossRef] [Green Version]
- Liang, Y.; Jiang, Y.; Jin, X.; Chen, P.; Heng, Y.; Cai, L.; Zhang, W.; Li, L.; Jia, L. Neddylation inhibition activates the protective autophagy through NF–kappaB–catalase–ATF3 Axis in human esophageal cancer cells. Cell Commun. Signal. 2020, 18, 72. [Google Scholar] [CrossRef]
- Rao, J.; Qian, X.; Li, G.; Pan, X.; Zhang, C.; Zhang, F.; Zhai, Y.; Wang, X. ATF3–mediated NRF2/HO–1 signaling regulates TLR4 innate immune responses in mouse liver ischemia/reperfusion injury. Am. J. Transplant. 2015, 15, 76–87. [Google Scholar] [CrossRef] [Green Version]
- Bakin, A.V.; Stourman, N.V.; Sekhar, K.R.; Rinehart, C.; Yan, X.; Meredith, M.J.; Arteaga, C.L.; Freemanb, M.L. Smad3–ATF3 signaling mediates TGF–beta suppression of genes encoding Phase II detoxifying proteins. Free Radic. Biol. Med. 2005, 38, 375–387. [Google Scholar] [CrossRef]
- Shan, Y.; Akram, A.; Amatullah, H.; Zhou, D.Y.; Gali, P.L.; Maron–Gutierrez, T.; González-López, A.; Zhou, L.; Rocco, P.R.; Hwang, D.; et al. ATF3 protects pulmonary resident cells from acute and ventilator–induced lung injury by preventing Nrf2 degradation. Antioxid. Redox Signal. 2015, 22, 651–668. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.H.; Jeong, J.Y.; Surh, Y.J.; Kim, K.W. Expression of stress–response ATF3 is mediated by Nrf2 in astrocytes. Nucleic Acids Res. 2010, 38, 48–59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalliokoski, T.; Kramer, C.; Vulpetti, A.; Gedeck, P. Comparability of mixed IC50 data a statistical analysis. PLoS ONE 2013, 8, e61007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bustin, S.A.; Benes, V.; Nolan, T.; Pfaffl, M.W. Quantitative real–time RT–PCR—A perspective. J. Mol. Endocrinol. 2005, 34, 597–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real–time quantitative PCR and the 2(–Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.L.; Zhang, H.Y.; Guo, Z.Y.; Ying, W.T.; Qian, X.H.; Wang, J.L. Comparison of three methods of protein extraction from Dermatophagoides pteronyssinus for two–dimensional electrophoresis. Chin. Med. Sci. J. 2009, 24, 64–68. [Google Scholar] [CrossRef]
- Rao, D.D.; Vorhies, J.S.; Senzer, N.; Nemunaitis, J. siRNA vs. shRNA: Similarities and differences. Adv. Drug Deliv. Rev. 2009, 61, 746–759. [Google Scholar] [CrossRef] [PubMed]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, X.; Mu, P.; Zhu, L.; Mao, X.; Chen, S.; Zhong, H.; Deng, Y. T-2 Toxin Induces Oxidative Stress at Low Doses via Atf3ΔZip2a/2b-Mediated Ubiquitination and Degradation of Nrf2. Int. J. Mol. Sci. 2021, 22, 7936. https://doi.org/10.3390/ijms22157936
Chen X, Mu P, Zhu L, Mao X, Chen S, Zhong H, Deng Y. T-2 Toxin Induces Oxidative Stress at Low Doses via Atf3ΔZip2a/2b-Mediated Ubiquitination and Degradation of Nrf2. International Journal of Molecular Sciences. 2021; 22(15):7936. https://doi.org/10.3390/ijms22157936
Chicago/Turabian StyleChen, Xiaoxuan, Peiqiang Mu, Lang Zhu, Xiaoxiao Mao, Shuang Chen, Huali Zhong, and Yiqun Deng. 2021. "T-2 Toxin Induces Oxidative Stress at Low Doses via Atf3ΔZip2a/2b-Mediated Ubiquitination and Degradation of Nrf2" International Journal of Molecular Sciences 22, no. 15: 7936. https://doi.org/10.3390/ijms22157936