
Differences in MPS I and MPS II Disease Manifestations

 ,
,
Abstract
:1. Introduction
2. Accumulated Glycosaminoglycans
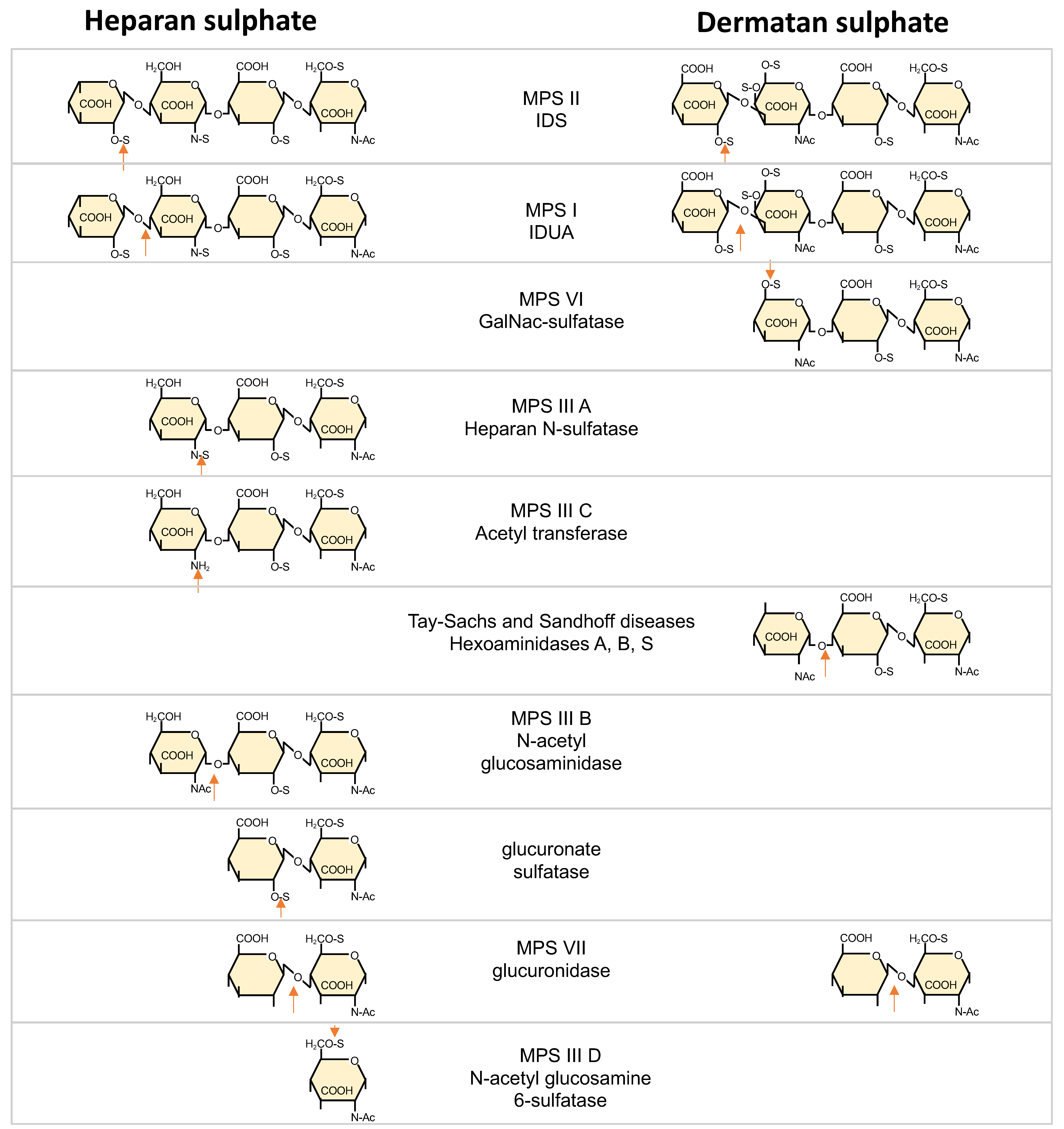
2.1. DS/HS Ratio
2.2. HS and DS Functions
2.3. Sulfation Levels
3. Disease-Specific Gene Expression
4. Disease-Specific Proteomics
5. Disease Manifestation
5.1. Systemic Manifestations: Skin, Kyphosis, Corneal Clouding, Valvular Heart Disease
5.2. Neurological Involvement: White Matter Abnormalities, Neurocognitive Functioning, Behavioral Manifestations, Seizures, Sleep Abnormalities
HS in Neurological Manifestations
6. Animal Models
7. Natural History
8. Treatment
8.1. Treatment in Animals
8.2. Experimental Therapies and Clinical Trials
8.2.1. ERT to the Brain
8.2.2. Shuttling of IDS Across the BBB
8.2.3. Gene Therapy: In Vivo
8.2.4. Genome Editing
8.2.5. Oligodendrocyte-Like Cells, DUOC-01, to Accelerate CNS Engraftment of Donor Cells
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Hunter, C. A Rare Disease in Two Brothers. Proc. R. Soc. Med. 1917, 10, 104–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hashimoto, A.; Kumagai, T.; Mineta, H. Hunter Syndrome Diagnosed by Otorhinolaryngologist. Case Rep. Otolaryngol. 2018, 2018, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.A.; Peracha, H.; Ballhausen, D.; Wiesbauer, A.; Gautschi, M.; Mason, R.W.; Giugliani, R.; Suzuki, Y.; Orii, K.E.; Orii, T.; et al. Epidemiology of mucopolysaccharidoses. Mol. Genet. Metab. 2017, 121, 227–240. [Google Scholar] [CrossRef]
- Shapiro, E.; Eisengart, J. The natural history of neurocognition in MPS disorders: A review. Mol. Genet. Metab. 2021, 133, 8–34. [Google Scholar] [CrossRef]
- Broomfield, A.; Davison, J.; Roberts, J.; Stewart, C.; Hensman, P.; Beesley, C.; Tylee, K.; Rust, S.; Schwahn, B.; Jameson, E.; et al. Ten years of enzyme replacement therapy in paediatric onset mucopolysaccharidosis II in England. Mol. Genet. Metab. 2020, 129, 98–105. [Google Scholar] [CrossRef] [PubMed]
- Mohamed, S.; He, Q.Q.; Singh, A.A.; Ferro, V. Mucopolysaccharidosis type II (Hunter syndrome): Clinical and biochemical aspects of the disease and approaches to its diagnosis and treatment. Adv. Carbohydr. Chem. Biochem. 2020, 77, 71–117. [Google Scholar] [CrossRef] [PubMed]
- Al Sawaf, S.; Mayatepek, E.; Hoffmann, B. Neurological findings in Hunter disease: Pathology and possible therapeutic effects reviewed. J. Inherit. Metab. Dis. 2008, 31, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Holt, J.B.; Poe, M.D.; Escolar, M.L. Natural progression of neurological disease in mucopolysaccharidosis type II. Pediatrics 2011, 127, e1258–e1265. [Google Scholar] [CrossRef]
- Neufeld, E.F.; Muenzer, I. The Metabolic & Molecular Basis of Inherited Disease, 8th ed.; Scriver, C.R., Beudet, A.L., Sly, W.S., Valle, D., Eds.; McGraw-Hill: New York, NY, USA, 2001; Volume III. [Google Scholar]
- Wraith, J.E.; Scarpa, M.; Beck, M.; Bodamer, O.A.; De Meirleir, L.; Guffon, N.; Meldgaard Lund, A.; Malm, G.; Van der Ploeg, A.T.; Zeman, J. Mucopolysaccharidosis type II (Hunter syndrome): A clinical review and recommendations for treatment in the era of enzyme replacement therapy. Eur. J. Pediatr. 2008, 167, 267–277. [Google Scholar] [CrossRef] [Green Version]
- Martin, R.; Beck, M.; Eng, C.; Giugliani, R.; Harmatz, P.; Muñoz, V.; Muenzer, J. Recognition and diagnosis of mucopolysaccharidosis II (Hunter syndrome). Pediatrics 2008, 121, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Young, I.D.; Harper, P.S. The Natural History of the Severe Form of Hunter’s Syndrome: A Study Based on 52 Cases. Dev. Med. Child Neurol. 1983, 25, 481–489. [Google Scholar] [CrossRef]
- Pastores, G.M.; Arn, P.; Beck, M.; Clarke, J.T.R.; Guffon, N.; Kaplan, P.; Muenzer, J.; Norato, D.Y.J.; Shapiro, E.; Thomas, J.; et al. The MPS I registry: Design, methodology, and early findings of a global disease registry for monitoring patients with Mucopolysaccharidosis Type I. Mol. Genet. Metab. 2007, 91, 37–47. [Google Scholar] [CrossRef]
- D’Aco, K.; Underhill, L.; Rangachari, L.; Arn, P.; Cox, G.F.; Giugliani, R.; Okuyama, T.; Wijburg, F.; Kaplan, P. Diagnosis and treatment trends in mucopolysaccharidosis I: Findings from the MPS I registry. Eur. J. Pediatr. 2012, 171, 911–919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tylki-Szymańska, A. Mucopolysaccharidosis type II, Hunter’s syndrome. Pediatr. Endocrinol. Rev. 2014, 12, 107–113. [Google Scholar] [PubMed]
- Scarpa, M.; Almássy, Z.; Beck, M.; Bodamer, O.; Bruce, I.A.; De Meirleir, L.; Guffon, N.; Guillén-Navarro, E.; Hensman, P.; Jones, S.; et al. Mucopolysaccharidosis type II: European recommendations for the diagnosis and multidisciplinary management of a rare disease. Orphanet J. Rare Dis. 2011, 6, 72. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendelsohn, N.J.; Harmatz, P.; Bodamer, O.; Burton, B.K.; Giugliani, R.; Jones, S.A.; Lampe, C.; Malm, G.; Steiner, R.D.; Parini, R. Importance of surgical history in diagnosing mucopolysaccharidosis type II (Hunter syndrome): Data from the Hunter Outcome Survey. Genet. Med. 2010, 12, 816–822. [Google Scholar] [CrossRef] [PubMed]
- Beck, M.; Arn, P.; Giugliani, R.; Muenzer, J.; Okuyama, T.; Taylor, J.; Fallet, S. The natural history of MPS I: Global perspectives from the MPS I Registry. Genet. Med. 2014, 16, 759–765. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leroy, J.G. Clinical Definition of the Hurler-Hunter Phenotypes. Am. J. Dis. Child. 1966, 112, 518–530. [Google Scholar] [CrossRef]
- Kiely, B.T.; Kohler, J.L.; Coletti, H.Y.; Poe, M.D.; Escolar, M.L. Early disease progression of Hurler syndrome. Orphanet J. Rare Dis. 2017, 12, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fesslová, V.; Corti, P.; Sersale, G.; Rovelli, A.; Russo, P.; Mannarino, S.; Butera, G.; Parini, R. The natural course and the impact of therapies of cardiac involvement in the mucopolysaccharidoses. Cardiol. Young 2009, 19, 170–178. [Google Scholar] [CrossRef] [PubMed]
- Muenzer, J.; Wraith, J.E.; Clarke, L.A. Mucopolysaccharidosis I: Management and treatment guidelines. Pediatrics 2009, 123, 19–29. [Google Scholar] [CrossRef]
- Wraith, J.E.; Beck, M.; Giugliani, R.; Clarke, J.; Martin, R.; Muenzer, J. Initial report from the Hunter Outcome Survey. Genet. Med. 2008, 10, 508–516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Link, B.; Lapagesse de Camargo Pinto, L.; Giugliani, R.; Wraith, J.E.; Guffon, N.; Eich, E.; Beck, M. Orthopedic manifestations in patients with muco polysaccharidosis type II (Hunter syndrome) enrolled in the Hunter Outcome Survey. Orthop. Rev. (Pavia) 2010, 2, 16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aldenhoven, M.; Wynn, R.F.; Orchard, P.J.; O’Meara, A.; Veys, P.; Fischer, A.; Valayannopoulos, V.; Neven, B.; Rovelli, A.; Prasad, V.K.; et al. Long-term outcome of Hurler syndrome patients after hematopoietic cell transplantation: An international multicenter study. Blood 2015, 125, 2164–2172. [Google Scholar] [CrossRef] [Green Version]
- White, K.K. Orthopaedic aspects of mucopolysaccharidoses. Rheumatology 2011, 50, 26–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, M.; Breyer, S.; Löbel, U.; Yarar, S.; Stücker, R.; Ullrich, K.; Müller, I.; Muschol, N. Musculoskeletal manifestations in mucopolysaccharidosis type i (Hurler syndrome) following hematopoietic stem cell transplantation. Orphanet J. Rare Dis. 2016, 11, 93. [Google Scholar] [CrossRef] [Green Version]
- Schwartz, I.V.D.; Ribeiro, M.G.; Mota, J.G.; Toralles, M.B.P.; Correia, P.; Horovitz, D.; Santos, E.S.; Monlleo, I.L.; Fett-Conte, A.C.; Sobrinho, R.P.O.; et al. A clinical study of 77 patients with mucopolysaccharidosis type II. Acta Paediatr. Int. J. Paediatr. 2007, 96, 63–70. [Google Scholar] [CrossRef]
- Thappa, D.; Singh, A.; Jaisankar, T.; Rao, R.; Ratnakar, C. Pebbling of the skin: A marker of Hunter’s syndrome. Pediatr. Dermatol. 1998, 15, 370–373. [Google Scholar] [CrossRef]
- Ochiai, T.; Suzuki, Y.; Kato, T.; Shichino, H.; Chin, M.; Mugishima, H.; Orii, T. Natural history of extensive Mongolian spots in mucopolysaccharidosis type II (Hunter syndrome): A survey among 52 Japanese patients. J. Eur. Acad. Dermatol. Venereol. 2007, 21, 1082–1085. [Google Scholar] [CrossRef] [PubMed]
- Broomfield, A.; Sims, J.; Mercer, J.; Hensman, P.; Ghosh, A.; Tylee, K.; Stepien, K.M.; Oldham, A.; Prathivadi Bhayankaram, N.; Wynn, R.; et al. The evolution of pulmonary function in childhood onset Mucopolysaccharidosis type I. Mol. Genet. Metab. 2020, 132, 94–99. [Google Scholar] [CrossRef]
- Keilmann, A.; Nakarat, T.; Bruce, I.A.; Molter, D.; Malm, G. Hearing loss in patients with mucopolysaccharidosis II: Data from HOS - The Hunter Outcome Survey. J. Inherit. Metab. Dis. 2012, 35, 343–353. [Google Scholar] [CrossRef]
- Kampmann, C.; Beck, M.; Morin, I.; Loehr, J.P. Prevalence and characterization of cardiac involvement in hunter syndrome. J. Pediatr. 2011, 159, 327–331.e2. [Google Scholar] [CrossRef]
- Rigante, D.; Segni, G. Cardiac Structural Involvement in Mucopolysaccharidoses. Cardiology 2002, 98, 18–20. [Google Scholar] [CrossRef]
- Dangel, J.H. Cardiovascular changes in children with mucopolysaccharide storage diseases and related disorders- clinical and echocardiographic findings in 64 patients. Eur. J. Pediatr. 1998, 157, 534–538. [Google Scholar] [CrossRef]
- Leal, G.N.; De Paula, A.C.; Leone, C.; Kim, C.A. Echocardiographic study of paediatric patients with mucopolysaccharidosis. Cardiol. Young 2010, 20, 254–261. [Google Scholar] [CrossRef]
- Jiménez-Arredondo, R.E.; Brambila-Tapia, A.J.L.; Mercado-Silva, F.M.; Ortiz-Aranda, M.; Benites-Godinez, V.; Olmos-García-de-ALBA, G.; Figuera, L.E. Association between brain structural anomalies, electroencephalogram and history of seizures in Mucopolysaccharidosis type II (Hunter syndrome). Neurol. Sci. 2017, 38, 445–450. [Google Scholar] [CrossRef]
- Eisengart, J.; Rudser, K.; Tolar, J.; Orchared, P.; Kivisto, T.; Ziegler, R.S.; Whitley, C.; Shapiro, E. Enzyme replacement is associated with better cognitive outcomes after transplant in Hurler syndrome. J. Pedatr. 2013, 162, 375–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shapiro, E.G.; Nestrasil, I.; Rudser, K.; Delaney, K.; Kovac, V.; Ahmed, A.; Yund, B.; Orchard, P.J.; Eisengart, J.; Niklason, G.R.; et al. Neurocognition across the spectrum of mucopolysaccharidosis type I: Age, severity, and treatment. Mol. Genet. Metab. 2015, 116, 61–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clarke, L. Mucopolysaccharidosis Type I. In GeneReviews; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Mirzaa, G., Eds.; University of Washington: Seattle, WA, USA, 2002. [Google Scholar]
- Young, I.D.; Harper, P.S. Psychosocial problems in Hunter’s syndrome. Child. Care. Health Dev. 1981, 7, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Holley, R.J.; Deligny, A.; Wei, W.; Watson, H.A.; Niñonuevo, M.R.; Dagälv, A.; Leary, J.A.; Bigger, B.W.; Kjellén, L.; Merry, C.L.R. Mucopolysaccharidosis type I, unique structure of accumulated heparan sulfate and increased N-sulfotransferase activity in mice lacking α-L-iduronidase. J. Biol. Chem. 2011, 286, 37515–37524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, H.Y.; Lo, Y.T.; Wang, T.J.; Huang, S.F.; Tu, R.Y.; Chen, T.L.; Lin, S.P.; Chuang, C.K. Normalization of glycosaminoglycan-derived disaccharides detected by tandem mass spectrometry assay for the diagnosis of mucopolysaccharidosis. Sci. Rep. 2019, 9, 10755. [Google Scholar] [CrossRef] [Green Version]
- Langereis, E.J.; van Vlies, N.; Church, H.J.; Geskus, R.B.; Hollak, C.E.M.; Jones, S.A.; Kulik, W.; van Lenthe, H.; Mercer, J.; Schreider, L.; et al. Biomarker responses correlate with antibody status in mucopolysaccharidosis type I patients on long-term enzyme replacement therapy. Mol. Genet. Metab. 2015, 114, 129–137. [Google Scholar] [CrossRef]
- Zhang, H.; Wood, T.; Young, S.; Millington, D. A straightforward, quantitative ultra-performance liquid chromatography-tandem mass spectrometric method for heparan sulfate, dermatan sulfate and chondroitin sulfate in urine: An improved clinical screening test for the mucopolysaccharidoses. Mol. Genet. Metab. 2015, 114, 123–128. [Google Scholar] [CrossRef] [PubMed]
- Auray-Blais, C.; Bhérer, P.; Gagnon, R.; Young, S.P.; Zhang, H.H.; An, Y.; Clarke, J.T.R.; Millington, D.S. Efficient analysis of urinary glycosaminoglycans by LC-MS/MS in mucopolysaccharidoses type I, II and VI. Mol. Genet. Metab. 2011, 102, 49–56. [Google Scholar] [CrossRef]
- Tanaka, N.; Kida, S.; Kinoshita, M.; Morimoto, H.; Shibasaki, T.; Tachibana, K.; Yamamoto, R. Evaluation of cerebrospinal fluid heparan sulfate as a biomarker of neuropathology in a murine model of mucopolysaccharidosis type II using high-sensitivity LC/MS/MS. Mol. Genet. Metab. 2018, 125, 53–58. [Google Scholar] [CrossRef]
- Menkovic, I.; Lavoie, P.; Boutin, M.; Auray-Blais, C. Distribution of heparan sulfate and dermatan sulfate in mucopolysaccharidosis type II mouse tissues pre- and post-enzyme-replacement therapy determined by UPLC-MS/MS. Bioanalysis 2019, 11, 727–740. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Dickson, P.I.; Stiles, A.R.; Chen, A.H.; Le, S.Q.; McCaw, P.; Beasley, J.; Millington, D.S.; Young, S.P. Comparison of dermatan sulfate and heparan sulfate concentrations in serum, cerebrospinal fluid and urine in patients with mucopolysaccharidosis type I receiving intravenous and intrathecal enzyme replacement therapy. Clin. Chim. Acta 2020, 508, 179–184. [Google Scholar] [CrossRef] [PubMed]
- Andrade, F.; Aldámiz-Echevarría, L.; Llarena, M.; Couce, M.L. Sanfilippo syndrome: Overall review. Pediatr. Int. 2015, 57, 331–338. [Google Scholar] [CrossRef] [PubMed]
- Harmatz, P.R.; Shediac, R. Mucopolysaccharidosis VI: Pathophysiology, diagnosis and treatment. Front. Biosci. Landmark 2017, 22, 385–406. [Google Scholar] [CrossRef] [Green Version]
- Lin, H.Y.; Lee, C.L.; Lo, Y.T.; Wang, T.J.; Huang, S.F.; Chen, T.L.; Wang, Y.S.; Niu, D.M.; Chuang, C.K.; Lin, S.P. The relationships between urinary glycosaminoglycan levels and phenotypes of mucopolysaccharidoses. Mol. Genet. Genomic Med. 2018, 6, 982–992. [Google Scholar] [CrossRef]
- Tomatsu, S.; Gutierrez, M.A.; Ishimaru, T.; Peña, O.M.; Montaño, A.M.; Maeda, H.; Velez-Castrillon, S.; Nishioka, T.; Fachel, A.A.; Cooper, A.; et al. Heparan sulfate levels in mucopolysaccharidoses and mucolipidoses. J. Inherit. Metab. Dis. 2005, 28, 743–757. [Google Scholar] [CrossRef]
- Coppa, G.V.; Gabrielli, O.; Zampini, L.; Maccari, F.; Mantovani, V.; Galeazzi, T.; Santoro, L.; Padella, L.; Marchesiello, R.L.; Galeotti, F.; et al. Mental retardation in mucopolysaccharidoses correlates with high molecular weight urinary heparan sulphate derived glucosamine. Metab. Brain Dis. 2015, 30, 1343–1348. [Google Scholar] [CrossRef] [PubMed]
- Mashima, R.; Sakai, E.; Tanaka, M.; Kosuga, M.; Okuyama, T. The levels of urinary glycosaminoglycans of patients with attenuated and severe type of mucopolysaccharidosis II determined by liquid chromatography-tandem mass spectrometry. Mol. Genet. Metab. Reports 2016, 7, 87–91. [Google Scholar] [CrossRef]
- Hendriksz, C.J.; Muenzer, J.; Burton, B.K.; Pan, L.; Wang, N.; Naimy, H.; Pano, A.; Barbier, A.J. A cerebrospinal fluid collection study in pediatric and adult patients with hunter syndrome. J. Inborn Errors Metab. Screen. 2015, 2015, 1–5. [Google Scholar] [CrossRef]
- Bishop, J.R.; Schuksz, M.; Esko, J.D. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature 2007, 446, 1030–1037. [Google Scholar] [CrossRef] [PubMed]
- Billings, P.C.; Pacifici, M. Interactions of signaling proteins, growth factors and other proteins with heparan sulfate: Mechanisms and mysteries. Connect. Tissue Res. 2015, 56, 272–280. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, Y.; Adams, D.H.; Shaw, S. Proteoglycans on endothelial cells present adhesion-inducing cytokines to leukocytes. Immunol. Today 1993, 14, 111–115. [Google Scholar] [CrossRef]
- Celie, J.W.A.M.; Beelen, R.H.J.; Van Den Born, J. Heparan sulfate proteoglycans in extravasation: Assisting leukocyte guidance. Front. Biosci. 2009, 14, 4932–4949. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bigger, B.W.; Begley, D.J.; Virgintino, D.; Pshezhetsky, A.V. Anatomical changes and pathophysiology of the brain in mucopolysaccharidosis disorders. Mol. Genet. Metab. 2018, 125, 322–331. [Google Scholar] [CrossRef]
- De Pasquale, V.; Pavone, L.M. Heparan sulfate proteoglycans: The sweet side of development turns sour in mucopolysaccharidoses. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 1865, 165539. [Google Scholar] [CrossRef]
- Trowbridge, J.M.; Gallo, R.L. Dermatan sulfate: New functions from an old glycosaminoglycan. Glycobiology 2002, 12, 117–125. [Google Scholar] [CrossRef]
- Fischer, J. Tilorone-induced lysosomal storage of glycosaminoglycans in cultured corneal fibroblasts: Biochemical and physicochemical investigations. Biochem. J. 1995, 312, 215–222. [Google Scholar] [CrossRef] [Green Version]
- Prokopek, M. The tilorone-induced mucopolysaccharidosis in rats. Biochemical investigations. Biochem. Pharmacol. 1991, 42, 2187–2191. [Google Scholar] [CrossRef]
- Lullmann-Rauch, R. Keratopathy in rats after treatment with tilorone. Graefe ’ s Arch. Ophthalmol. 1986, 224, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Hein, L.; Lüllmann-Rauch, R. Mucopolysaccharidosis and lipidosis in rats treated with tilorone analogues. Toxicology 1989, 58, 145–154. [Google Scholar] [CrossRef]
- Hochuli, M.; Wüthrich, K.; Steinmann, B. Two-dimensional NMR spectroscopy of urinary glycosaminoglycans from patients with different mucopolysaccharidoses. NMR Biomed. 2003, 16, 224–236. [Google Scholar] [CrossRef]
- Fuller, M.; Chau, A.; Nowak, R.C.; Hopwood, J.J.; Meikle, P.J. A defect in exodegradative pathways provides insight into endodegradation of heparan and dermatan sulfates. Glycobiology 2006, 16, 318–325. [Google Scholar] [CrossRef] [Green Version]
- Ramage, P.; Cummingham, W. Comparative structural studies of urinary glycosaminoglycans in the Hurler and Hunter Syndromes. Biochim. Biophys. Acta 1975, 411, 325–333. [Google Scholar] [CrossRef]
- Lawrence, R.; Brown, J.R.; Al-Mafraji, K.; Lamanna, W.C.; Beitel, J.R.; Boons, G.; Esko, J.D.; Crawford, B.E. Disease-specific non-reducing end carbohydrate biomarkers for mucopolysaccharidoses. Nat. Chem. Biol. 2012, 8, 197–204. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, F.L.; Holley, R.J.; Langford-Smith, K.J.; Badrinath, S.; Liao, A.; Langford-Smith, A.; Cooper, J.D.; Jones, S.A.; Wraith, J.E.; Wynn, R.F.; et al. Neuropathology in mouse models of mucopolysaccharidosis type I, IIIA and IIIB. PLoS ONE 2012, 7, e35787. [Google Scholar] [CrossRef] [PubMed]
- Gleitz, H.F.; Liao, A.Y.; Cook, J.R.; Rowlston, S.F.; Forte, G.M.; D’Souza, Z.; O’Leary, C.; Holley, R.J.; Bigger, B.W. Brain-targeted stem cell gene therapy corrects mucopolysaccharidosis type II via multiple mechanisms. EMBO Mol. Med. 2018, 10, e8730. [Google Scholar] [CrossRef]
- Wegrzyn, G.; Jakóbkiewicz-Banecka, J.; Narajczyk, M.; Wiśniewski, A.; Piotrowska, E.; Gabig-Cimińska, M.; Kloska, A.; Słomińska-Wojewódzka, M.; Korzon-Burakowska, A.; Wegrzyn, A. Why are behaviors of children suffering from various neuronopathic types of mucopolysaccharidoses different? Med. Hypotheses 2010, 75, 605–609. [Google Scholar] [CrossRef] [Green Version]
- Herndon, M.E.; Stipp, C.S.; Lander, A.D. Interactions of neural glycosaminoglycans and proteoglycans with protein ligands: Assessment of selectivity, heterogeneity and the participation of core proteins in binding. Glycobiology 1999, 9, 143–155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsuo, I.; Kimura-Yoshida, C. Extracellular distribution of diffusible growth factors controlled by heparan sulfate proteoglycans during mammalian embryogenesis. Philos. Trans. R. Soc. B Biol. Sci. 2014, 369, 20130545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mertens, G.; Van Der Schueren, B.; Van Den Berghe, H.; David, G. Heparan sulfate expression in polarized epithelial cells: The apical sorting of glypican (GPI-anchored proteoglycan) is inversely related to its heparan sulfate content. J. Cell Biol. 1996, 132, 487–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afratis, N.; Gialeli, C.; Nikitovic, D.; Tsegenidis, T.; Karousou, E.; Theocharis, A.D.; Pavão, M.S.; Tzanakakis, G.N.; Karamanos, N.K. Glycosaminoglycans: Key players in cancer cell biology and treatment. FEBS J. 2012, 279, 1177–1197. [Google Scholar] [CrossRef]
- Ohtsubo, K.; Marth, J.D. Glycosylation in Cellular Mechanisms of Health and Disease. Cell 2006, 126, 855–867. [Google Scholar] [CrossRef] [Green Version]
- Watson, H.A.; Holley, R.J.; Langford-Smith, K.J.; Wilkinson, F.L.; Van Kuppevelt, T.H.; Wynn, R.F.; Wraith, J.E.; Merry, C.L.R.; Bigger, B.W. Heparan sulfate inhibits hematopoietic stem and progenitor cell migration and engraftment in mucopolysaccharidosis I. J. Biol. Chem. 2014, 289, 36194–36203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallagher, J.T. Multiprotein signalling complexes: Regional assembly on heparan sulphate. Biochem. Soc. Trans. 2006, 34, 438–441. [Google Scholar] [CrossRef]
- Allen, B.L.; Filla, M.S.; Rapraeger, A.C. Role of heparan sulfate as a tissue-specific regulator of FGF-4 and FGF receptor recognition. J. Cell Biol. 2001, 155, 845–857. [Google Scholar] [CrossRef] [Green Version]
- Brokowska, J.; Pierzynowska, K.; Gaffke, L.; Rintz, E.; Węgrzyn, G. Expression of genes involved in apoptosis is dysregulated in mucopolysaccharidoses as revealed by pilot transcriptomic analyses. Cell Biol. Int. 2021, 45, 549–557. [Google Scholar] [CrossRef] [PubMed]
- Gaffke, L.; Pierzynowska, K.; Podlacha, M.; Hoinkis, D.; Rintz, E.; Brokowska, J.; Cyske, Z.; Wegrzyn, G. Underestimated aspect of mucopolysaccharidosis pathogenesis: Global changes in cellular processes revealed by transcriptomic studies. Int. J. Mol. Sci. 2020, 21, 1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salvalaio, M.; D’Avanzo, F.; Rigon, L.; Zanetti, A.; D’Angelo, M.; Valle, G.; Scarpa, M.; Tomanin, R. Brain RNA-seq profiling of the mucopolysaccharidosis type II mouse model. Int. J. Mol. Sci. 2017, 18, 1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heywood, W.E.; Camuzeaux, S.; Doykov, I.; Patel, N.; Preece, R.L.; Footitt, E.; Cleary, M.; Clayton, P.; Grunewald, S.; Abulhoul, L.; et al. Proteomic Discovery and Development of a Multiplexed Targeted MRM-LC-MS/MS Assay for Urine Biomarkers of Extracellular Matrix Disruption in Mucopolysaccharidoses I, II, and VI. Anal. Chem. 2015, 87, 12238–12244. [Google Scholar] [CrossRef]
- Yuan, X.; Meng, Y.; Chen, C.; Liang, S.; Ma, Y.; Jiang, W.; Duan, J.; Wang, C. Proteomic approaches in the discovery of potential urinary biomarkers of mucopolysaccharidosis type II. Clin. Chim. Acta 2019, 499, 34–40. [Google Scholar] [CrossRef]
- Ou, L.; Przybilla, M.; Whitley, C. Proteomic analysis of muccopolysaccharidosis I mouse brain with two-dimensional polyacrylamide gel electrophoresis. Mol. Genet. Metab. 2017, 120, 101–110. [Google Scholar] [CrossRef] [Green Version]
- Baldo, G.; Lorenzini, D.M.; Santos, D.S.; Mayer, F.Q.; Vitry, S.; Bigou, S.; Heard, J.M.; Matte, U.; Giugliani, R. Shotgun proteomics reveals possible mechanisms for cognitive impairment in Mucopolysaccharidosis I mice. Mol. Genet. Metab. 2015, 114, 138–145. [Google Scholar] [CrossRef]
- Cardona, C.; Benincore, E.; Pimentel, N.; Reyes, L.H.; Patarroyo, C.; Rodríguez-López, A.; Rufián, M.M.; Barrera, L.A.; Alméciga-Díaz, C.J. Identification of the iduronate-2-sulfatase proteome in wild-type mouse brain. Heliyon 2019, 5, e01667. [Google Scholar] [CrossRef] [Green Version]
- Lonergan, C.; Payne, A.; Wilson, W.; Patterson, J.; Englisch III, J. What syndrome is this? Pediatr. Dermatol. 2005, 22, 266–267. [Google Scholar] [CrossRef]
- Prystowsky, S.D.; Maumenee, I.H.; Freeman, R.G.; Herndon, J.H.; Jo Harrod, M. A Cutaneous Marker in the Hunter Syndrome: A Report of Four Cases. Arch. Dermatol. 1977, 113, 602–605. [Google Scholar] [CrossRef]
- Freeman, R.G. A Pathological Basis for the Cutaneous Papules of Mucopolysaccharidosis II (The Hunter Syndrome). J. Cutan. Pathol. 1977, 4, 318–328. [Google Scholar] [CrossRef]
- Demitsu, T.; Kakurait, M.; Okubo, Y.; Shibayama, C.; Kikuchi, Y.; Mori, Y.; Sukegawa, K.; Mizuguchi, M. Skin eruption as the presenting sign of Hunter syndrome IIB. Clin. Exp. Dermatol. 1999, 24, 179–182. [Google Scholar] [CrossRef] [PubMed]
- Warner, T.F.; Wrone, D.A.; Williams, E.C.; Cripps, D.J.; Mundhenke, C.F.A. Heparan sulphate proteoglycan in scleromyxedema promotes FGF-2 activity. Pathol Res. Pr. 2002, 198, 701–707. [Google Scholar] [CrossRef] [PubMed]
- Cordova, A. The Mongolian Spot. Clin. Pediatr. 1981, 20, 714–719. [Google Scholar] [CrossRef] [PubMed]
- Quantock, A.J.; Young, R.D. Development of the corneal stroma, and the collagen-proteoglycan associations that help define its structure and function. Dev. Dyn. 2008, 237, 2607–2621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bredrup, C.; Knappskog, P.M.; Majewski, J.; Rødabi, E.; Boman, H. Congenital stromal dystrophy of the cornea caused by a mutation in the decorin gene. Investig. Ophthalmol. Vis. Sci. 2005, 46, 420–426. [Google Scholar] [CrossRef] [PubMed]
- Rødahl, E.; Van Ginderdeuren, R.; Knappskog, P.M.; Bredrup, C.; Boman, H. A Second Decorin Frame Shift Mutation in a Family With Congenital Stromal Corneal Dystrophy. Am. J. Ophthalmol. 2006, 142, 520–521. [Google Scholar] [CrossRef]
- Rühland, C.; Schönherr, E.; Robenek, H.; Hansen, U.; Iozzo, R.V.; Bruckner, P.; Seidler, D.G. The glycosaminoglycan chain of decorin plays an important role in collagen fibril formation at the early stages of fibrillogenesis. FEBS J. 2007, 274, 4246–4255. [Google Scholar] [CrossRef]
- Mollard, R.; Telegan, P.; Haskins, M.; Aguirre, G. Corneal endothelium in mucopolysaccharide storage disorders. Morphologic studies in animal models. Cornea 1996, 15, 25–34. [Google Scholar] [CrossRef]
- Fahnehjelm, K.T.; Ashworth, J.L.; Pitz, S.; Olsson, M.; Törnquist, A.L.; Lindahl, P.; Summers, C.G. Clinical guidelines for diagnosing and managing ocular manifestations in children with mucopolysaccharidosis. Acta Ophthalmol. 2012, 90, 595–602. [Google Scholar] [CrossRef]
- Alroy, J.; Haskins, M.; Birk, D.E. Altered corneal stromal matrix organization is associated with mucopolysaccharidosis I, III and VI. Exp. Eye Res. 1999, 68, 523–530. [Google Scholar] [CrossRef]
- Yuan, C.; Bothun, E.; Hardten, D.; Tolar, J.; McLoon, L. A novel explanation of corneal clouding in a bone marrow transplant-treated patient with Hurler syndrome. Exp. Eye Res. 2016, 148, 83–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levin, T.L.; Berdon, W.E.; Lachman, R.S.; Anyane-Yeboa, K.; Ruzal-Shapiro, C.; Roye, D.P. Lumbar gibbus in storage diseases and bone dysplasias. Pediatr. Radiol. 1997, 27, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Janecke, A.R.; Li, B.; Boehm, M.; Krabichler, B.; Rohrbach, M.; Müller, T.; Fuchs, I.; Golas, G.; Katagiri, Y.; Ziegler, S.G.; et al. The phenotype of the musculocontractural type of Ehlers-Danlos syndrome due to CHST14 mutations. Am. J. Med. Genet. Part A 2016, 107A, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Uehara, M.; Kosho, T.; Yamamoto, N.; Takahashi, H.E.; Shimakura, T.; Nakayama, J.; Kato, H.; Takahashi, J. Spinal manifestations in 12 patients with musculocontractural Ehlers-Danlos syndrome caused by CHST14/D4ST1 deficiency (mcEDS-CHST14). Am. J. Med. Genet. Part A 2018, 176, 2331–2341. [Google Scholar] [CrossRef]
- Boffi, L.; Russo, P.; Limongelli, G. Early diagnosis and management of cardiac manifestations in mucopolysaccharidoses: A practical guide for paediatric and adult cardiologists. Ital. J. Pediatr. 2018, 44, 122. [Google Scholar] [CrossRef] [Green Version]
- Braunlin, E.A.; Harmatz, P.R.; Scarpa, M.; Furlanetto, B.; Kampmann, C.; Loehr, J.P.; Ponder, K.P.; Roberts, W.C.; Rosenfeld, H.M.; Giugliani, R. Cardiac disease in patients with mucopolysaccharidosis: Presentation, diagnosis and management. J. Inherit. Metab. Dis. 2011, 34, 1183–1197. [Google Scholar] [CrossRef] [Green Version]
- Azevedo, A.C.M.M.; Schwartz, I.V.; Kalakun, L.; Brustolin, S.; Burin, M.G.; Beheregaray, A.P.C.; Leistner, S.; Giugliani, C.; Rosa, M.; Barrios, P.; et al. Clinical and biochemical study of 28 patients with mucopolysaccharides type VI. Clin. Genet. 2004, 66, 208–213. [Google Scholar] [CrossRef] [PubMed]
- Scarpa, M.; Barone, R.; Fiumara, A.; Astarita, L.; Parenti, G.; Rampazzo, A.; Sala, S.; Sorge, G.; Parini, R. Mucopolysaccharidosis VI: The Italian experience. Eur. J. Pediatr. 2009, 168, 1203–1206. [Google Scholar] [CrossRef]
- Latif, N.; Sarathchandra, P.; Taylor, P.; Antoniw, J.; Yacoub, M. Localization and pattern of expression of extracellular matrix components in human heart valves. J. Hear. Valve Dis. 2005, 14, 218–227. [Google Scholar]
- Dekaban, A.S.; Constantopoulos, G. Mucopolysaccharidosis types I, II, IIIA and V - Pathological and biochemical abnormalities in the neural and mesenchymal elements of the brain. Acta Neuropathol. 1977, 39, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Constantopoulos, G.; Iqbal, K.; Dekaban, A.S. Mucopolysaccharidosis Types IH, IS, II and IIIA: Glycosaminoglycans and Lipids of Isolated Brain Cells and Other Fractions from Autopsied Tissues. J. Neurochem. 1980, 34, 1399–1411. [Google Scholar] [CrossRef] [PubMed]
- Lach, B.; Haust, M. Nodular lesions of choroid plexus in Hurler disease. Fetal Pediatr. Pathol. 2011, 30, 189–198. [Google Scholar] [CrossRef]
- Shapiro, E.; Nestrasil, I.; Delaney, K.A.; Rudser, K.; Kovac, V.; Nair, N.; Richard, C.I.; Haslett, P.; Whitley, C.B. A prospective natural history study of Mucopolysaccharidosis Type IIIA. J. Pediatr. 2016, 170, 278–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neufeld, E.; Muenzer, J. The Mucopolysaccharidoses, 8th ed.; Scriver, C., Beaudet, A., Sly, W., Valle, D., Childs, B., Kinzler, K., Vogelstein, B., Eds.; McGraw-Hill: New York, NY, USA, 2001. [Google Scholar]
- Whitley, C.B.; Cleary, M.; Eugen Mengel, K.; Harmatz, P.; Shapiro, E.; Nestrasil, I.; Haslett, P.; Whiteman, D.; Alexanderian, D. Observational Prospective Natural History of Patients with Sanfilippo Syndrome Type B. J. Pediatr. 2018, 197, 198–206.e2. [Google Scholar] [CrossRef] [Green Version]
- Nestrasil, I.; Vedolin, L. Quantitative neuroimaging in mucopolysaccharidoses clinical trials. Mol. Genet. Metab. 2017, 122, 17–24. [Google Scholar] [CrossRef] [PubMed]
- King, K.E.; Rudser, K.D.; Nestrasil, I.; Kovac, V.; Delaney, K.A.; Wozniak, J.R.; Mueller, B.A.; Lim, K.O.; Eisengart, J.B.; Mamak, E.G.; et al. Attention and corpus callosum volumes in individuals with mucopolysaccharidosis type I. Neurology 2019, 92, E2321–E2328. [Google Scholar] [CrossRef] [PubMed]
- Yund, B.; Rudser, K.; Ahmed, A.; Kovac, V.; Nestrasil, I.; Raiman, J.; Mamak, E.; Harmatz, P.; Steiner, R.; Lau, H.; et al. Cognitive, medical, and neuroimaging characteristics of attenuated mucopolysaccharidosis type II. Mol. Genet. Metab. 2015, 114, 170–177. [Google Scholar] [CrossRef] [Green Version]
- Shapiro, E.; Guler, O.E.; Rudser, K.; Delaney, K.; Bjoraker, K.; Whitley, C.; Tolar, J.; Orchard, P.; Provenzale, J.; Thomas, K.M. An exploratory study of brain function and structure in mucopolysaccharidosis type I: Long term observations following hematopoietic cell transplantation (HCT). Mol. Genet. Metab. 2012, 107, 116–121. [Google Scholar] [CrossRef] [Green Version]
- Cross, E.M.; Hare, D.J. Behavioural phenotypes of the mucopolysaccharide disorders: A systematic literature review of cognitive, motor, social, linguistic and behavioural presentation in the MPS disorders. J. Inherit. Metab. Dis. 2013, 36, 189–200. [Google Scholar] [CrossRef]
- Shapiro, E.G.; Whitley, C.B.; Eisengart, J.B. Beneath the floor: Re-analysis of neurodevelopmental outcomes in untreated Hurler syndrome. Orphanet J. Rare Dis. 2018, 13, 76. [Google Scholar] [CrossRef]
- Ahmed, A.; Shapiro, E.; Rudser, K.; Kunin-Batson, A.; King, K.; Whitley, C.B. Association of somatic burden of disease with age and neuropsychological measures in attenuated mucopolysaccharidosis types I, II and VI. Mol. Genet. Metab. Reports 2016, 7, 27–31. [Google Scholar] [CrossRef]
- Ahmed, A.; Whitley, C.B.; Cooksley, R.; Rudser, K.; Cagle, S.; Ali, N.; Delaney, K.; Yund, B.; Shapiro, E. Neurocognitive and neuropsychiatric phenotypes associated with the mutation L238Q of the alpha-L-iduronidase gene in Hurler-Scheie syndrome. Mol. Genet. Metab. 2014, 111, 123–127. [Google Scholar] [CrossRef] [Green Version]
- Bax, M.C.O.; Colville, G.A. Behaviour in mucopolysaccharide disorders. Arch. Dis. Child. 1995, 73, 77–81. [Google Scholar] [CrossRef] [Green Version]
- Shapiro, E.G.; Escolar, M.L.; Delaney, K.A.; Mitchell, J.J. Assessments of neurocognitive and behavioral function in the mucopolysaccharidoses. Mol. Genet. Metab. 2017, 122, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Potegal, M.; Yund, B.; Rudser, K.; Ahmed, A.; Delaney, K.; Nestrasil, I.; Whitley, C.B.; Shapiro, E.G. Mucopolysaccharidosis Type IIIA presents as a variant of Klüver-Bucy syndrome. J. Clin. Exp. Neuropsychol. 2013, 35, 608–616. [Google Scholar] [CrossRef] [PubMed]
- King, K.; Shapiro, E.; Rudser, K.; Whitley, C. Emotional-behavioral functioning in individuals with MPS I: A longitudinal approach. Mol. Genet. Metab. 2016, 2, S67–S68. [Google Scholar] [CrossRef]
- Staba, S.L.; Escolar, M.L.; Poe, M.; Kim, Y.; Martin, P.L.; Szabolcs, P.; Allison-Thacker, J.; Wood, S.; Wenger, D.A.; Rubinstein, P.; et al. Cord-blood transplants from unrelated donors in patients with Hurler’s syndrome. N. Engl. J. Med. 2004, 350, 1960–1969. [Google Scholar] [CrossRef] [Green Version]
- Seto, T.; Kono, K.; Morimoto, K.; Inoue, Y.; Shintaku, H.; Hattori, H.; Matsuoka, O.; Yamano, T.; Tanaka, A. Brain magnetic resonance imaging in 23 patients with mucopolysaccharidoses and the effect of bone marrow transplantation. Ann. Neurol. 2001, 50, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Guffon, N.; Pettazzoni, M.; Pangaud, N.; Garin, C.; Lina-Granade, G.; Plault, C.; Mottolese, C.; Froissart, R.; Fouilhoux, A. Long term disease burden post-transplantation: Three decades of observations in 25 Hurler patients successfully treated with hematopoietic stem cell transplantation (HSCT). Orphanet J. Rare Dis. 2021, 16, 60. [Google Scholar] [CrossRef]
- Grant, N. Evaluating strategies to manage and endure challenging behaviors in mucopolysaccharidoses. Orphanet J. Rare Dis. 2021, 16, 165. [Google Scholar] [CrossRef] [PubMed]
- Holt, J.; Poe, M.D.; Escolar, M.L. Early clinical markers of central nervous system involvement in mucopolysaccharidosis type II. J. Pediatr. 2011, 159, 320–326.e2. [Google Scholar] [CrossRef] [PubMed]
- Eisengart, J.B.; King, K.E.; Shapiro, E.G.; Whitley, C.B.; Muenzer, J. The nature and impact of neurobehavioral symptoms in neuronopathic Hunter syndrome. Mol. Genet. Metab. Reports 2020, 22, 100549. [Google Scholar] [CrossRef]
- Roberts, J.; Stewart, C.; Kearney, S. Management of the behavioural manifestations of hunter syndrome. Br. J. Nurs. 2016, 25, 22–30. [Google Scholar] [CrossRef]
- Scarpa, M.; Lourenço, C.M.; Amartino, H. Epilepsy in mucopolysaccharidosis disorders. Mol. Genet. Metab. 2017, 122, 55–61. [Google Scholar] [CrossRef]
- Kuzenkova, L.; Podkletnova, T.; Namazova-Baranova, L.; Gevorkya, A.; Vashakmadze, N.; Zhurkova, N.; Nechaeva, N. Particular features of neurological symptoms with children suffering from MPS syndrome type II. Mol. Genet. Metab. 2013, 108, S56–S57. [Google Scholar] [CrossRef]
- Young, I.D.; Harper, P.S. Mild form of Hunter’s syndrome: Clinical delineation based on 31 cases. Arch. Dis. Child. 1982, 57, 828–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inatani, M.; Irie, F.; Plump, A.S.; Tessier-Lavigne, M.; Yamaguchi, Y. Mammalian Brain Morphogenesis and Midline Axon Guidance Require Heparan Sulfate. Science 2003, 302, 1044–1046. [Google Scholar] [CrossRef]
- Poulain, F.E.; Joseph Yost, H. Heparan sulfate proteoglycans: A sugar code for vertebrate development? Development 2015, 142, 3456–3467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pan, C.; Nelson, M.S.; Reyes, M.; Koodie, L.; Brazil, J.J.; Stephenson, E.J.; Zhao, R.C.; Peters, C.; Selleck, S.B.; Stringer, S.E.; et al. Functional abnormalities of heparan sulfate in mucopolysaccharidosis-I are associated with defective biologic activity of FGF-2 on human multipotent progenitor cells. Blood 2005, 106, 1956–1964. [Google Scholar] [CrossRef] [Green Version]
- Fuller, M.; Brooks, D.A.; Evangelista, M.; Hein, L.K.; Hopwood, J.J.; Meikle, P.J. Prediction of neuropathology in mucopolysaccharidosis I patients. Mol. Genet. Metab. 2005, 84, 18–24. [Google Scholar] [CrossRef]
- Jastrebova, N.; Vanwildemeersch, M.; Lindahl, U.; Spillmann, D. Heparan sulfate domain organization and sulfation modulate FGF-induced cell signaling. J. Biol. Chem. 2010, 285, 26842–26851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simonaro, C.M.; Ge, Y.; Eliyahu, E.; He, X.; Jepsen, K.J.; Schuchman, E.H. Involvement of the Toll-like receptor 4 pathway and use of TNF-α antagonists for treatment of the mucopolysaccharidoses. Proc. Natl. Acad. Sci. USA 2010, 107, 222–227. [Google Scholar] [CrossRef] [Green Version]
- Goodall, K.J.; Poon, I.K.H.; Phipps, S.; Hulett, M.D. Soluble heparan sulfate fragments generated by heparanase trigger the release of pro-inflammatory cytokines through TLR-4. PLoS ONE 2014, 9, e109596. [Google Scholar] [CrossRef]
- O’Callaghan, P.; Zhang, X.; Li, J.P. Heparan Sulfate Proteoglycans as Relays of Neuroinflammation. J. Histochem. Cytochem. 2018, 66, 305–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lloyd-Evans, E.; Haslett, L.J. The lysosomal storage disease continuum with ageing-related neurodegenerative disease. Ageing Res. Rev. 2016, 32, 104–121. [Google Scholar] [CrossRef]
- Snow, A.D.; Mar, H.; Nochlin, D.; Sekiguchi, R.T.; Kimata, K.; Koike, Y.; Wight, T.N. Early accumulation of heparan sulfate in neurons and in the beta-amyloid protein-containing lesions of Alzheimer’s disease and Down’s syndrome. Am. J. Pathol. 1990, 137, 1253–1270. [Google Scholar] [CrossRef] [Green Version]
- Holmes, B.B.; DeVos, S.L.; Kfoury, N.; Li, M.; Jacks, R.; Yanamandra, K.; Ouidja, M.O.; Brodsky, F.M.; Marasa, J.; Bagchi, D.P.; et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc. Natl. Acad. Sci. USA 2013, 110, e3138–e3147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maïza, A.; Chantepie, S.; Vera, C.; Fifre, A.; Huynh, M.B.; Stettler, O.; Ouidja, M.O.; Papy-Garcia, D. The role of heparan sulfates in protein aggregation and their potential impact on neurodegeneration. FEBS Lett. 2018, 592, 3806–3818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walkley, S.U. Secondary accumulation of gangliosides in lysosomal storage disorders. Semin. Cell Dev. Biol. 2004, 15, 433–444. [Google Scholar] [CrossRef]
- Walkley, S.U.; Vanier, M. Secondary lipid accumulation in lysosomal disease. Biochim. Biophys. Acta 2009, 1793, 726–736. [Google Scholar] [CrossRef] [Green Version]
- Viana, G.M.; Priestman, D.A.; Platt, F.M.; Khan, S.; Tomatsu, S.; Pshezhetsky, A.V. Brain Pathology in Mucopolysaccharidoses (MPS) Patients with Neurological Forms. J. Clin. Med. 2020, 9, 396. [Google Scholar] [CrossRef] [Green Version]
- McGlynn, R.; Dobrenis, K.; Walkley, S.U. Differential subcellular localization of cholesterol, gangliosides, and glycosaminoglycans in murine models of mucopolysaccharide storage disorders. J. Comp. Neurol. 2004, 480, 415–426. [Google Scholar] [CrossRef]
- Hampe, C.S.; Polgreen, L.E.; Lund, T.C.; McIvor, R.S. Dysostosis Multiplex in Human Mucopolysaccharidosis Type 1 H and in Animal Models of the Disease. Pediatr. Endocrinol. Rev. 2020, 17, 317–326. [Google Scholar] [CrossRef]
- Wilkerson, M.; Lewis, D.; Marks, S.; Prieur, D. Syndrome, Clinical and Morphologic Features of Mucopolysaccharidosis Type II in a Dog: Naturally Occurring Model of Hunter Syndrome. Vet. Pathol. 1998, 35, 230–233. [Google Scholar] [CrossRef] [Green Version]
- Muenzer, J.; Lamsa, J.C.; Garcia, A.; Dacosta, J.; Garcia, J.; Treco, D.A. Enzyme replacement therapy in mucopolysaccharidosis type II (Hunter syndrome): A preliminary report. Acta Paediatr. Suppl. 2002, 91, 98–99. [Google Scholar] [CrossRef]
- Hinderer, C.; Katz, N.; Louboutin, J.P.; Bell, P.; Yu, H.; Nayal, M.; Kozarsky, K.; O’Brien, W.T.; Goode, T.; Wilson, J.M. Delivery of an Adeno-associated virus vector into cerebrospinal fluid attenuates central nervous system disease in mucopolysaccharidosis type II mice. Hum. Gene Ther. 2016, 27, 906–915. [Google Scholar] [CrossRef]
- Higuchi, T.; Shimizu, H.; Fukuda, T.; Kawagoe, S.; Matsumoto, J.; Shimada, Y.; Kobayashi, H.; Ida, H.; Ohashi, T.; Morimoto, H.; et al. Enzyme replacement therapy (ERT) procedure for mucopolysaccharidosis type II (MPS II) by intraventricular administration (IVA) in murine MPS II. Mol. Genet. Metab. 2012, 107, 122–128. [Google Scholar] [CrossRef]
- Jung, S.C.; Park, E.S.; Choi, E.N.; Kim, C.H.; Kim, S.J.; Jin, D.K. Characterization of a novel mucopolysaccharidosis type II mouse model and recombinant AAV2/8 vector-mediated gene therapy. Mol. Cells 2010, 30, 13–18. [Google Scholar] [CrossRef]
- Hong, S.H.; Chu, H.; Kim, K.R.; Ko, M.H.; Kwon, S.Y.; Moon, I.J.; Chung, W.H.; Cho, Y.S.; Kim, C.H.; Suh, M.W.; et al. Auditory characteristics and therapeutic effects of enzyme replacement in mouse model of the mucopolysaccharidosis (MPS) II. Am. J. Med. Genet. Part A 2012, 158 A, 2131–2138. [Google Scholar] [CrossRef]
- Garcia, A.R.; Pan, J.; Lamsa, J.C.; Muenzer, J. The characterization of a murine model of mucopolysaccharidosis II (Hunter syndrome). J. Inherit. Metab. Dis. 2007, 30, 924–934. [Google Scholar] [CrossRef]
- Friso, A.; Tomanin, R.; Alba, S.; Gasparotto, N.; Puicher, E.P.; Fusco, M.; Hortelano, G.; Muenzer, J.; Marin, O.; Zacchello, F.; et al. Reduction of GAG storage in MPS II mouse model following implantation on encapsulated recombinant myoblasts. J. Gene Med. 2005, 7, 1482–1491. [Google Scholar] [CrossRef]
- Cardone, M.; Polito, V.A.; Pepe, S.; Mann, L.; D’Azzo, A.; Auricchio, A.; Ballabio, A.; Cosma, M.P. Correction of Hunter syndrome in the MPSII mouse model by AAV2/8-mediated gene delivery. Hum. Mol. Genet. 2006, 15, 1225–1236. [Google Scholar] [CrossRef]
- Wakabayashi, T.; Shimada, Y.; Akiyama, K.; Higuchi, T.; Fukuda, T.; Kobayashi, H.; Eto, Y.; Ida, H.; Ohashi, T. Hematopoietic Stem Cell Gene Therapy Corrects Neuropathic Phenotype in Murine Model of Mucopolysaccharidosis Type II. Hum. Gene Ther. 2015, 26, 357–366. [Google Scholar] [CrossRef]
- Polito, V.A.; Abbondante, S.; Polishchuk, R.S.; Nusco, E.; Salvia, R.; Cosma, M.P. Correction of CNS defects in the MPSII mouse model via systemic enzyme replacement therapy. Hum. Mol. Genet. 2010, 19, 4871–4885. [Google Scholar] [CrossRef]
- Motas, S.; Haurigot, V.; Garcia, M.; Marcó, S.; Ribera, A.; Roca, C.; Sánchez, X.; Sánchez, V.; Molas, M.; Bertolin, J.; et al. CNS-directed gene therapy for the treatment of neurologic and somatic mucopolysaccharidosis type II (Hunter syndrome). JCI Insight 2016, 1, e86696. [Google Scholar] [CrossRef]
- Provoost, L.; Siracusa, C.; Stefanovski, D.; Che, Y.; Li, M.; Casal, M. Cognitive abilities of dogs with mucopolysaccharidosis I: Learning and memory. Animals 2020, 10, 397. [Google Scholar] [CrossRef] [Green Version]
- Shull, R.; Munger, R.; Spellacy, E.; Hall, C.; Constantopoulos, G.C.; Neufeld, E.F. Canine alpha-L-Iduronidase Deficiency A Model of Mucopolysaccharidosis I. Am. J. Pathol. 1982, 109, 244–248. [Google Scholar] [PubMed]
- Spellacy, E.; Shull, R.M.; Constantopoulos, G.; Neufeld, E.F. A canine model of human α-L-iduronidase deficiency. Proc. Natl. Acad. Sci. USA 1983, 80, 6091–6095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, D.; Shukla, C.; Liu, X.; Schoeb, T.R.; Clarke, L.A.; Bedwell, D.M.; Keeling, K.M. Characterization of an MPS I-H knock-in mouse that carries a nonsense mutation analogous to the human IDUA-W402X mutation. Mol. Genet. Metab. 2010, 99, 62–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oestreich, A.K.; Garcia, M.R.; Yao, X.; Pfeiffer, F.M.; Nobakhti, S.; Shefelbine, S.J.; Wang, Y.; Brodeur, A.C.; Phillips, C.L. Characterization of the MPS I-H knock-in mouse reveals increased femoral biomechanical integrity with compromised material strength and altered bone geometry. Mol. Genet. Metab. Rep. 2015, 5, 3–11. [Google Scholar] [CrossRef]
- Galimberti, C.; Madeo, A.; Di Rocco, M.; Fiumara, A. Mucopolysaccharidoses: Early diagnostic signs in infants and children. Ital. J. Pediatr. 2018, 44, 133. [Google Scholar] [CrossRef]
- Aldenhoven, M.; Boelens, J.; de Koning, T.J. The Clinical Outcome of Hurler Syndrome after Stem Cell Transplantation. Biol. Blood Marrow Transplant. 2008, 14, 485–498. [Google Scholar] [CrossRef] [Green Version]
- Barone, R.; Pellico, A.; Pittalà, A.; Gasperini, S. Neurobehavioral phenotypes of neuronopathic mucopolysaccharidoses. Ital. J. Pediatr. 2018, 44, 121. [Google Scholar] [CrossRef] [Green Version]
- Shapiro, E.G.; Lockman, L.A.; Balthazor, M.; Krivit, W. Neuropsychological outcomes of several storage diseases with and without bone marrow transplantation. J. Inherit. Metab. Dis. 1995, 18, 413–429. [Google Scholar] [CrossRef]
- Seo, J.H.; Okuyama, T.; Shapiro, E.; Fukuhara, Y.; Kosuga, M. Natural history of cognitive development in neuronopathic mucopolysaccharidosis type II (Hunter syndrome): Contribution of genotype to cognitive developmental course. Mol. Genet. Metab. Rep. 2020, 24, 100630. [Google Scholar] [CrossRef] [PubMed]
- Parini, R.; Jones, S.A.; Harmatz, P.R.; Giugliani, R.; Mendelsohn, N.J. The natural history of growth in patients with Hunter syndrome: Data from the Hunter Outcome Survey (HOS). Mol. Genet. Metab. 2016, 117, 438–446. [Google Scholar] [CrossRef]
- Colville, G.A.; Bax, M.A. Early presentation in the mucopolysaccharide disorders. Child. Care. Health Dev. 1996, 22, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Pan, D.; Sciascia, A.; Vorhees, C.V.; Williams, M.T. Progression of multiple behavioral deficits with various ages of onset in a murine model of Hurler syndrome. Brain Res. 2008, 1188, 241–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldo, G.; Mayer, F.Q.; Martinelli, B.Z.; de Carvalho, T.G.; Meyer, F.S.; de Oliveira, P.G.; Meurer, L.; Tavares, Â.; Matte, U.; Giugliani, R. Enzyme replacement therapy started at birth improves outcome in difficult-to-treat organs in mucopolysaccharidosis I mice. Mol. Genet. Metab. 2013, 109, 33–40. [Google Scholar] [CrossRef]
- Clarke, L.A.; Russell, C.S.; Pownall, S.; Warrington, C.L.; Borowski, A.; Dimmick, J.E.; Toone, J.; Jirik, F.R. Murine mucopolysaccharidosis type I: Targeted disruption of the murine alpha-L-iduronidase gene. Hum. Mol. Genet. 1997, 6, 503–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gleitz, H.F.E.; O’Leary, C.; Holley, R.J.; Bigger, B.W. Identification of age-dependent motor and neuropsychological behavioural abnormalities in a mouse model of mucopolysaccharidosis type II. PLoS ONE 2017, 12, e0172435. [Google Scholar] [CrossRef] [PubMed]
- Schachern, P.A.; Shea, D.A.; Paparella, M.M. Mucopolysaccharidosis I-H (Hurler’s syndrome) and human temporal bone histopathology. Ann. Otol. Rhinol. Laryngol. 1984, 93, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Gomez-Ospina, N.; Scharenberg, S.G.; Mostrel, N.; Bak, R.O.; Mantri, S.; Quadros, R.M.; Gurumurthy, C.B.; Lee, C.; Bao, G.; Suarez, C.J.; et al. Human genome-edited hematopoietic stem cells phenotypically correct Mucopolysaccharidosis type I. Nat. Commun. 2019, 10, 4045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hampe, C.S.; Wesley, J.; Lund, T.C.; Orchard, P.J.; Polgreen, L.E.; Eisengart, J.B.; McLoon, L.K.; Cureoglu, S.; Schachern, P.; McIvor, R.S. Mucopolysaccharidosis type I: Current treatments, limitations and prospects for improvement. Biomolecules 2021, 11, 189. [Google Scholar] [CrossRef]
- Parini, R.; Deodato, F.; Di Rocco, M.; Lanino, E.; Locatelli, F.; Messina, C.; Rovelli, A.; Scarpa, M. Open issues in Mucopolysaccharidosis type I-Hurler. Orphanet J. Rare Dis. 2017, 12, 112. [Google Scholar] [CrossRef]
- Chung, Y.K.; Sohn, Y.B.; Sohn, J.M.; Lee, J.J.Y.; Chang, M.S.; Kwun, Y.; Kim, C.H.; Lee, J.J.Y.; Yook, Y.J.; Ko, A.-R.; et al. A biochemical and physicochemical comparison of two recombinant enzymes used for enzyme replacement therapies of hunter syndrome. Glycoconj. J. 2014, 31, 309–315. [Google Scholar] [CrossRef]
- Burton, B.K.; Jego, V.; Mikl, J.; Jones, S.A. Survival in idursulfase-treated and untreated patients with mucopolysaccharidosis type II: Data from the Hunter Outcome Survey (HOS). J. Inherit. Metab. Dis. 2017, 40, 867–874. [Google Scholar] [CrossRef] [Green Version]
- Wikman-Jorgensen, P.E.; López Amorós, A.; Peris García, J.; Esteve Atienzar, P.J.; Cañizares Navarro, R.; Asensio Tomás, M.L.; Seguí Ripoll, J.M.; Bonet, D.; Esteban-Giner, M.J.; Robert, J.; et al. Enzyme replacement therapy for the treatment of Hunter disease: A systematic review with narrative synthesis and meta-analysis. Mol. Genet. Metab. 2020, 131, 206–210. [Google Scholar] [CrossRef]
- Whiteman, D.; Kimura, A. Development of idursulfase therapy for mucopolysaccharidosis type II (Hunter syndrome): The past, the present and the future. Drug Des. Devel. Ther. 2017, 11, 2467–2480. [Google Scholar] [CrossRef] [Green Version]
- Burrow, T.A.; Hopkin, R.J.; Leslie, N.D.; Tinkle, B.T.; Grabowski, G.A. Enzyme reconstitution/replacement therapy for lysosomal storage diseases. Curr. Opin. Pediatr. 2007, 19, 628–635. [Google Scholar] [CrossRef] [PubMed]
- Sohn, Y.B.; Cho, S.Y.; Park, S.W.; Kim, S.J.; Ko, A.-R.; Kwon, E.-K.; Han, S.J.; Jin, D.-K. Phase I/II clinical trial of enzyme replacement therapy with idursulfase beta in patients with mucopolysaccharidosis II (Hunter syndrome). Orphanet J. Rare Dis 2013, 8, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhalla, A.; Ravi, R.; Fang, M.; Arguello, A.; Davis, S.S.; Chiu, C.L.; Blumenfeld, J.R.; Nguyen, H.N.; Earr, T.K.; Wang, J.; et al. Characterization of fluid biomarkers reveals lysosome dysfunction and neurodegeneration in neuronopathic MPS II patients. Int. J. Mol. Sci. 2020, 21, 5188. [Google Scholar] [CrossRef] [PubMed]
- Parini, R.; Deodato, F. Intravenous enzyme replacement therapy in mucopolysaccharidoses: Clinical effectiveness and limitations. Int. J. Mol. Sci. 2020, 21, 2975. [Google Scholar] [CrossRef] [PubMed]
- Barth, A.L.; de Magalhães, T.S.P.C.; Reis, A.B.R.; de Oliveira, M.L.; Scalco, F.B.; Cavalcanti, N.C.; Silva, D.S.E.; Torres, D.A.; Costa, A.A.P.; Bonfim, C.; et al. Early hematopoietic stem cell transplantation in a patient with severe mucopolysaccharidosis II: A 7 years follow-up. Mol. Genet. Metab. Rep. 2017, 12, 62–68. [Google Scholar] [CrossRef]
- Tajima, G.; Sakura, N.; Kosuga, M.; Okuyama, T.; Kobayashi, M. Effects of idursulfase enzyme replacement therapy for Mucopolysaccharidosis type II when started in early infancy: Comparison in two siblings. Mol. Genet. Metab. 2013, 108, 172–177. [Google Scholar] [CrossRef]
- Muenzer, J.; Gucsavas-Calikoglu, M.; McCandless, S.E.; Schuetz, T.J.; Kimura, A. A phase I/II clinical trial of enzyme replacement therapy in mucopolysaccharidosis II (Hunter syndrome). Mol. Genet. Metab. 2007, 90, 329–337. [Google Scholar] [CrossRef]
- Muenzer, J.; Bodamer, O.; Burton, B.; Clarke, L.; Frenking, G.S.; Giugliani, R.; Jones, S.; Rojas, M.V.M.; Scarpa, M.; Beck, M.; et al. The role of enzyme replacement therapy in severe Hunter syndrome-an expert panel consensus. Eur. J. Pediatr. 2012, 171, 181–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muenzer, J.; Beck, M.; Eng, C.M.; Giugliani, R.; Harmatz, P.; Martin, R.; Ramaswami, U.; Vellodi, A.; Wraith, J.E.; Cleary, M.; et al. Long-term, open-labeled extension study of idursulfase in the treatment of Hunter syndrome. Genet. Med. 2011, 13, 95–101. [Google Scholar] [CrossRef] [Green Version]
- Muhlebach, M.S.; Wooten, W.; Muenzer, J. Respiratory Manifestations in Mucopolysaccharidoses. Paediatr. Respir. Rev. 2011, 12, 133–138. [Google Scholar] [CrossRef]
- Parini, R.; Rigoldi, M.; Tedesco, L.; Boffi, L.; Brambilla, A.; Bertoletti, S.; Boncimino, A.; Del Longo, A.; De Lorenzo, P.; Gaini, R.; et al. Enzymatic replacement therapy for Hunter disease: Up to 9 years experience with 17 patients. Mol. Genet. Metab. Rep. 2015, 3, 65–74. [Google Scholar] [CrossRef]
- Lampe, C.; Bosserhoff, A.-K.; Burton, B.K.; Giugliani, R.; de Souza, C.F.; Bittar, C.; Muschol, N.; Olson, R.; Mendelsohn, N.J. Long-term experience with enzyme replacement therapy (ERT) in MPS II patients with a severe phenotype: An international case series. J. Inherit. Metab. Dis. 2014, 37, 823–829. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Luan, Z.; Jiang, H.; Fang, J.; Qin, M.; Lee, V.; Chen, J. Allogeneic Hematopoietic Stem Cell Transplantation in Thirty-Four Pediatric Cases of Mucopolysaccharidosis—A Ten-Year Report from the China Children Transplant Group. Biol. Blood Marrow Transplant. 2016, 22, 2104–2108. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, Y.; Taylor, M.; Orii, K.; Fukao, T.; Orii, T.; Tomatsu, S. Assessment of activity of daily life in mucopolysaccharidosis type II patients with hematopoietic stem cell transplantation. Diagnostics 2020, 10, 46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKinnis, E.J.R.; Sulzbacher, S.; Rutledge, J.C.; Sanders, J.; Scott, C.R. Bone marrow transplantation in Hunter syndrome. J. Pediatr. 1996, 129, 145–148. [Google Scholar] [CrossRef]
- Guffon, N.; Bertrand, Y.; Forest, I.; Fouilhoux, A.; Froissart, R. Bone Marrow Transplantation in Children with Hunter Syndrome: Outcome after 7 to 17 Years. J. Pediatr. 2009, 154, 733–737. [Google Scholar] [CrossRef]
- Annibali, R.; Caponi, L.; Morganti, A.; Manna, M.; Gabrielli, O.; Ficcadenti, A. Hunter syndrome (Mucopolysaccharidosis type II), severe phenotype: Long term follow-up on patients undergone to hematopoietic stem cell transplantation. Minerva Pediatr. 2013, 65, 487–496. [Google Scholar] [PubMed]
- Jones, S.A.; Parini, R.; Harmatz, P.; Giugliani, R.; Fang, J.; Mendelsohn, N.J. The effect of idursulfase on growth in patients with Hunter syndrome: Data from the Hunter Outcome Survey (HOS). Mol. Genet. Metab. 2013, 109, 41–48. [Google Scholar] [CrossRef]
- Patel, P.; Suzuki, Y.; Tanaka, A.; Yabe, H.; Kato, S.; Shimada, T.; Mason, R.W.; Orii, K.E.; Fukao, T.; Orii, T.; et al. Impact of enzyme replacement therapy and hematopoietic stem cell therapy on growth in patients with Hunter syndrome. Mol. Genet. Metab. Rep. 2014, 1, 184–196. [Google Scholar] [CrossRef]
- Coppa, G.V.; Gabrielli, O.; Zampini, L.; Pierani, P.; Giorgi, P.; Jezequel, A.; Orlandi, F.; Miniero, R.; Busca, A.; De Tuca, T. Bone marrow transplantation in Hunter syndrome (mucopolysaccharidosis type II): Two-year follow-up of the first Italian patient and review of the literature. Pediatr. Med. Chir. 1995, 17, 227–235. [Google Scholar] [PubMed]
- Imaizumi, M.; Gushi, K.; Kurobane, I.; Inoue, S.; Suzuki, J.; Koizumi, Y.; Suzuki, H.; Sato, A.; Gotoh, Y.-I.; Haginoya, K.; et al. Long-term effects of bone marrow transplantation for inborn errors. Acta Paediatr. Jpn. 1994, 36, 30–36. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, A.; Okuyama, T.; Suzuki, Y.; Sakai, N.; Takakura, H.; Sawada, T.; Tanaka, T.; Otomo, T.; Ohashi, T.; Ishige-Wada, M.; et al. Long-term efficacy of hematopoietic stem cell transplantation on brain involvement in patients with mucopolysaccharidosis type II: A nationwide survey in Japan. Mol. Genet. Metab. 2012, 107, 513–520. [Google Scholar] [CrossRef]
- Bergstrom, S.; Quinn, J.; Greenstein, R.; Ascensao, J. Long-term follow-up of a patient transplanted for Hunter’s disease type IIB: A case report and literature review. Bone Marrow Transpl. 1994, 14, 653–658. [Google Scholar]
- Selvanathan, A.; Ellaway, C.; Wilson, C.; Owens, P.; Shaw, P.; Bhattacharya, K. Effectiveness of Early Hematopoietic Stem Cell Transplantation in Preventing Neurocognitive Decline in Mucopolysaccharidosis Type II: A Case Series. JIMD Rep. 2018, 41, 81–89. [Google Scholar] [CrossRef]
- Warkentin, P.; Dixon, M.; Schafer, I.; Strandjord, S.; Coccia, P. Bone marrow transplantation in Hunter syndrome: A preliminary report. Birth Defects Orig Artic Ser 1986, 22, 31–39. [Google Scholar]
- Li, P.; Thompson, J.N.; Hug, G.; Huffman, P.; Chuck, G. Biochemical and Molecular Analysis in a Patient with the Severe Form of Hunter Syndrome after Bone Marrow Transplantation. Am. J. Med. Genet. Semin. Med. Genet. 1996, 64, 531–535. [Google Scholar] [CrossRef]
- Kubaski, F.; Yabe, H.; Suzuki, Y.; Seto, T.; Hamazaki, T.; Mason, R.W.; Xie, L.; Onsten, T.G.H.; Leistner-Segal, S.; Giugliani, R.; et al. Hematopoietic Stem Cell Transplantation for Patients with Mucopolysaccharidosis II. Biol. Blood Marrow Transplant. 2017, 23, 1795–1803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Noh, H.; Lee, J.I. Current and potential therapeutic strategies for mucopolysaccharidoses. J. Clin. Pharm. Ther. 2014, 39, 215–224. [Google Scholar] [CrossRef]
- Marin, L.; Gutierrez-Solana, L.; Fernandez, A. Hunter syndrome: Resolution of extensive typical skin lesions after 9 months of enzyme replacement therapy with idursulfase. Pediatr. Dermatol. 2012, 29, 369–371. [Google Scholar] [CrossRef]
- Ito, K.; Ochiai, T.; Suzuki, H.; Chin, M.; Shichino, H.; Mugishima, H. The effect of haematopoietic stem cell transplant on papules with “pebbly” appearance in Hunter’s syndrome. Br. J. Dermatol. 2004, 151, 207–211. [Google Scholar] [CrossRef] [PubMed]
- Vellodi, A.; Young, E.; Cooper, A.; Lidchi, V.; Winchester, B.; Wraith, J.E. Long-term follow-up following bone marrow transplantation for Hunter disease. J. Inherit. Metab. Dis. 1999, 22, 638–648. [Google Scholar] [CrossRef]
- Tanjuakio, J.; Suzuki, Y.; Patel, P.; Yasuda, E.; Tanaka, A.; Yabe, H.; Mason, R.W.; Montaño, A.M.; Orii, K.E.; Orii, K.O.; et al. Activities of Daily Living in patients with Hunter syndrome: Impact of enzyme replacement therapy and hematopoietic stem cell transplantation. Mol. Genet. Metab. 2015, 114, 161–169. [Google Scholar] [CrossRef] [Green Version]
- Hobbs, J.; Hugh-Jones, K.; Barrett, A.; Byrom, N.; James, D.C.O.; Lucas, C.F. Reversal of clinical features of Hurler’s disease and biochemical improvement after treatment by bone-marrow transplantation. Lancet 1981, 2, 709–712. [Google Scholar] [CrossRef]
- Peters, C.; Balthazor, M.; Shapiro, E.G.; King, R.J.; Kollman, C.; Hegland, J.D.; Henslee-Downey, J.; Trigg, M.E.; Cowan, M.J.; Sanders, J.; et al. Outcome of unrelated donor bone marrow transplantation in 40 children with Hurler syndrome. Blood 1996, 87, 4894–4902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muenzer, J. Overview of the mucopolysaccharidoses. Rheumatology 2011, 50, 4–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araya, K.; Sakai, N.; Mohri, I.; Kagitani-Shimono, K.; Okinaga, T.; Hashii, Y.; Ohta, H.; Nakamichi, I.; Aozasa, K.; Taniike, M.; et al. Localized donor cells in brain of a Hunter disease patient after cord blood stem cell transplantation. Mol. Genet. Metab. 2009, 98, 255–263. [Google Scholar] [CrossRef] [PubMed]
- Vellodi, A. Bone marrow transplantation for lysosomal storage disorders. Expert Rev. Endocrinol. Metab. 2006, 1, 425–438. [Google Scholar] [CrossRef]
- Taylor, M.; Khan, S.; Stapleton, M.; Wang, J.; Chen, J.; Wynn, R.; Yabe, H.; Chinen, Y.; Boelens, J.J.; Robert, W.; et al. Hematopoietic stem cell transplantation for mucopolysaccharidoses; past, present, and future Madeleine. Biol. Blood Marrow Transpl. 2019, 25, 226–246. [Google Scholar] [CrossRef]
- Garcia, A.R.; DaCosta, J.M.; Pan, J.; Muenzer, J.; Lamsa, J.C. Preclinical dose ranging studies for enzyme replacement therapy with idursulfase in a knock-out mouse model of MPS II. Mol. Genet. Metab. 2007, 91, 183–190. [Google Scholar] [CrossRef]
- Laoharawee, K.; Podetz-Pedersen, K.M.; Nguyen, T.T.; Evenstar, L.B.; Kitto, K.F.; Nan, Z.; Fairbanks, C.A.; Low, W.C.; Kozarsky, K.F.; McIvor, R.S. Prevention of Neurocognitive Deficiency in Mucopolysaccharidosis Type II Mice by Central Nervous System-Directed, AAV9-Mediated Iduronate Sulfatase Gene Transfer. Hum. Gene Ther. 2017, 28, 626–638. [Google Scholar] [CrossRef] [PubMed]
- Polito, V.A.; Cosma, M.P. IDS Crossing of the Blood-Brain Barrier Corrects CNS Defects in MPSII Mice. Am. J. Hum. Genet. 2009, 85, 296–301. [Google Scholar] [CrossRef] [Green Version]
- Ahn, S.Y.; Chang, Y.S.; Sung, D.K.; Ko, A.R.; Kim, C.H.; Yoo, D.K.; Lim, K.H.; Sohn, Y.B.; Jin, D.K.; Park, W.S. High-dose enzyme replacement therapy attenuates cerebroventriculomegaly in a mouse model of mucopolysaccharidosis type II. J. Hum. Genet. 2013, 58, 728–733. [Google Scholar] [CrossRef]
- Wada, M.; Shimada, Y.; Iizuka, S.; Ishii, N.; Hiraki, H.; Tachibana, T.; Maeda, K.; Saito, M.; Arakawa, S.; Ishimoto, T.; et al. Ex Vivo Gene Therapy Treats Bone Complications of Mucopolysaccharidosis Type II Mouse Models through Bone Remodeling Reactivation. Mol. Ther. Methods Clin. Dev. 2020, 19, 261–274. [Google Scholar] [CrossRef]
- Froissart, R.; Da Silva, I.M.; Maire, I. Mucopolysaccharidosis type II: An update on mutation spectrum. Acta Paediatr. Int. J. Paediatr. 2007, 96, 71–77. [Google Scholar] [CrossRef]
- Dvorakova, L.; Vlaskova, H.; Sarajlija, A.; Ramadza, D.; Poupetova, H.; Hruba, E.; Hlavata, A.; Bzuduch, V.; Peskova, K.; Storkanova, G.; et al. Genotype-phenotype correlation in 44 Czech, Slovak, Croatian and Serbian patients with mucopolysaccharidosis type II. Clin. Genet. 2017, 91, 787–796. [Google Scholar] [CrossRef] [PubMed]
- Semyachkina, A.N.; Voskoboeva, E.Y.; Nikolaeva, E.A.; Zakharova, E.Y. Analysis of long-term observations of the large group of Russian patients with Hunter syndrome (mucopolysaccharidosis type II). BMC Med. Genomics 2021, 14, 71. [Google Scholar] [CrossRef]
- Alcántara-Ortigoza, M.A.; García-de Teresa, B.; González-del Angel, A.; Berumen, J.; Guardado-Estrada, M.; Fernández-Hernández, L.; Navarrete-Martínez, J.I.; Maza-Morales, M.; Rius-Domínguez, R. Wide allelic heterogeneity with predominance of large IDS gene complex rearrangements in a sample of Mexican patients with Hunter syndrome. Clin. Genet. 2016, 89, 574–583. [Google Scholar] [CrossRef] [PubMed]
- Brusius-Facchin, A.C.; Schwartz, I.V.D.; Zimmer, C.; Ribeiro, M.G.; Acosta, A.X.; Horovitz, D.; Monlleó, I.L.; Fontes, M.I.B.; Fett-Conte, A.; Sobrinho, R.P.O.; et al. Mucopolysaccharidosis type II: Identification of 30 novel mutations among Latin American patients. Mol. Genet. Metab. 2014, 111, 133–138. [Google Scholar] [CrossRef]
- Kosuga, M.; Mashima, R.; Hirakiyama, A.; Fuji, N.; Kumagai, T.; Seo, J.H.; Nikaido, M.; Saito, S.; Ohno, K.; Sakuraba, H.; et al. Molecular diagnosis of 65 families with mucopolysaccharidosis type II (Hunter syndrome) characterized by 16 novel mutations in the IDS gene: Genetic, pathological, and structural studies on iduronate-2-sulfatase. Mol. Genet. Metab. 2016, 118, 190–197. [Google Scholar] [CrossRef] [PubMed]
- Alkhzouz, C.; Lazea, C.; Bucerzan, S.; Nascu, I.; Kiss, E.; Denes, C.; Grigorescu-Sido, P. Clinical and Genetic Characteristics of Romanian Patients with Mucopolysaccharidosis Type II. JIMD Rep. 2017, 33, 19–25. [Google Scholar] [CrossRef] [PubMed]
- Calias, P.; Papisov, M.; Pan, J.; Savioli, N.; Belov, V.; Huang, Y.; Lotterhand, J.; Alessandrini, M.; Liu, N.; Fischman, A.J.; et al. CNS penetration of intrathecal-lumbar idursulfase in the monkey, dog and mouse: Implications for neurological outcomes of lysosomal storage disorder. PLoS ONE 2012, 7, e30341. [Google Scholar] [CrossRef]
- Sohn, Y.B.; Ko, A.R.; Seong, M.R.; Lee, S.; Kim, M.R.; Cho, S.Y.; Kim, J.S.; Sakaguchi, M.; Nakazawa, T.; Kosuga, M.; et al. The efficacy of intracerebroventricular idursulfase-beta enzyme replacement therapy in mucopolysaccharidosis II murine model: Heparan sulfate in cerebrospinal fluid as a clinical biomarker of neuropathology. J. Inherit. Metab. Dis. 2018, 41, 1235–1246. [Google Scholar] [CrossRef]
- Sohn, Y.B.; Lee, J.; Cho, S.Y.; Kim, S.J.; Ko, A.R.; Nam, M.H.; Jin, D.K. Improvement of CNS Defects Via Continuous Intrathecal Enzyme Replacement by Osmotic Pump in Mucopolysaccharidosis Type II Mice. Am. J. Med. Genet. Part A 2013, 161, 1036–1043. [Google Scholar] [CrossRef]
- Muenzer, J.; Hendriksz, C.J.; Fan, Z.; Vijayaraghavan, S.; Perry, V.; Santra, S.; Solanki, G.A.; Mascelli, M.A.; Pan, L.; Wang, N.; et al. A phase I/II study of intrathecal idursulfase-IT in children with severe mucopolysaccharidosis II. Genet. Med. 2016, 18, 73–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muenzer, J.; Hendriksz, C.J.; Stein, M.B.; Fan, Z.; Kearney, S.; Horton, J.; Vijayaraghavan, S.; Santra, S.; Solanki, G.A.; Pan, L.; et al. A long-term extension study evaluating intrathecal idursulfase-IT in children with Hunter syndrome and cognitive impairment. Mol. Genet. Metab. 2016, 99–100. [Google Scholar] [CrossRef]
- Giugliani, R.; Dalla Corte, A.; Poswar, F.; Vanzella, C.; Horovitz, D.; Riegel, M.; Baldo, G.; Vairo, F. Intrathecal/Intracerebroventricular enzyme replacement therapy for the mucopolysaccharidoses: Efficacy, safety, and prospects. Expert Opin. Orphan Drugs 2018, 6, 403–411. [Google Scholar] [CrossRef]
- Seo, J.H.; Kosuga, M.; Hamazaki, T.; Shintaku, H.; Okuyama, T. Impact of intracerebroventricular enzyme replacement therapy in patients with neuronopathic mucopolysaccharidosis type II. Mol. Ther. Methods Clin. Dev. 2021, 21, 67–75. [Google Scholar] [CrossRef] [PubMed]
- Sonoda, H.; Morimoto, H.; Yoden, E.; Koshimura, Y.; Kinoshita, M.; Golovina, G.; Takagi, H.; Yamamoto, R.; Minami, K.; Mizoguchi, A.; et al. A Blood-Brain-Barrier-Penetrating Anti-human Transferrin Receptor Antibody Fusion Protein for Neuronopathic Mucopolysaccharidosis II. Mol. Ther. 2018, 26, 1366–1374. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.H.; Boado, R.J.; Lu, J.Z.; Hui, E.K.W.; Pardridge, W.M. Brain-penetrating IgG-iduronate 2-sulfatase fusion protein for the mouse. Drug Metab. Dispos. 2012, 40, 329–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morimoto, H.; Kida, S.; Yoden, E.; Kinoshita, M.; Tanaka, N.; Yamamoto, R.; Koshimura, Y.; Takagi, H.; Takahashi, K.; Hirato, T.; et al. Clearance of heparan sulfate in the brain prevents neurodegeneration and neurocognitive impairment in MPS II mice. Mol. Ther. 2021, 29, 1853–1861. [Google Scholar] [CrossRef] [PubMed]
- Okuyama, T.; Eto, Y.; Sakai, N.; Minami, K.; Yamamoto, T.; Sonoda, H.; Yamaoka, M.; Tachibana, K.; Hirato, T.; Sato, Y. Iduronate-2-Sulfatase with Anti-human Transferrin Receptor Antibody for Neuropathic Mucopolysaccharidosis II: A Phase 1/2 Trial. Mol. Ther. 2019, 27, 456–464. [Google Scholar] [CrossRef] [Green Version]
- Okuyama, T.; Eto, Y.; Sakai, N.; Nakamura, K.; Yamamoto, T.; Yamaoka, M.; Ikeda, T.; So, S.; Tanizawa, K.; Sonoda, H.; et al. A Phase 2/3 Trial of Pabinafusp Alfa, IDS Fused with Anti-Human Transferrin Receptor Antibody, Targeting Neurodegeneration in MPS-II. Mol. Ther. 2021, 29, 671–679. [Google Scholar] [CrossRef] [PubMed]
- Giugliani, R.; Martins, A.M.; So, S.; Yamamoto, T.; Yamaoka, M.; Ikeda, T.; Tanizawa, K.; Sonoda, H.; Schmidt, M.; Sato, Y. Iduronate-2-sulfatase fused with anti-human transferrin receptor antibody, pabinafusp alfa, for treatment of neuronopathic and non-neuronopathic mucopolysaccharidosis II: Report of a phase 2 trial in Brazil. Mol. Ther. 2021, 29, 2378–2386. [Google Scholar] [CrossRef]
- Boado, R.J.; Hui, E.K.-W.; Lu, J.Z.; Pardridge, W.M. Insulin Receptor Antibody-Iduronate 2-Sulfatase Fusion Protein: Pharmacokinetics, Anti-Drug Antibody, and Safety Pharmacology in Rhesus Monkeys. Biotechnol. Bioeng. 2014, 111, 2317–2325. [Google Scholar] [CrossRef] [Green Version]
- Boado, R.J.; Hui, E.K.W.; Lu, J.Z.; Sumbria, R.K.; Pardridge, W.M. Blood-brain barrier molecular trojan horse enables imaging of brain uptake of radioiodinated recombinant protein in the rhesus monkey. Bioconjug. Chem. 2013, 24, 1741–1749. [Google Scholar] [CrossRef]
- Foust, K.; Nurre, E.; Montgomery, C.; Hernandez, A.; Chan, C.; Kaspar, B. Intravascular AAV9 preferentially targets neonatal-neurons and adult-astrocytes in CNS. Nat. Biotechnol. 2009, 27, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fu, H.; Dirosario, J.; Killedar, S.; Zaraspe, K.; McCarty, D.M. Correction of neurological disease of mucopolysaccharidosis IIIB in adult mice by rAAV9 trans-blood-brain barrier gene delivery. Mol. Ther. 2011, 19, 1025–1033. [Google Scholar] [CrossRef]
- Ruzo, A.; Marcó, S.; García, M.; Villacampa, P.; Ribera, A.; Ayuso, E.; Maggioni, L.; Mingozzi, F.; Haurigot, V.; Bosch, F. Correction of pathological accumulation of glycosaminoglycans in central nervous system and peripheral tissues of MPSIIIA mice through systemic AAV9 gene transfer. Hum. Gene Ther. 2012, 23, 1237–1246. [Google Scholar] [CrossRef]
- Sharma, R.; Anguela, X.M.; Doyon, Y.; Wechsler, T.; DeKelver, R.C.; Sproul, S.; Paschon, D.E.; Miller, J.C.; Davidson, R.J.; Shivak, D.; et al. In vivo genome editing of the albumin locus as a platform for protein replacement therapy. Blood 2015, 126, 1777–1784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheridan, C. Sangamo’s landmark genome editing trial gets mixed reception. Nat. Biotechnol. 2018, 36, 907–908. [Google Scholar] [CrossRef] [PubMed]
- Krivit, W.; Sung, J.; Shapiro, E.G.; Lockman, L.A. Microglia: The effector cell for reconstitution of the central nervous system following bone marrow transplantation for lysosomal and peroxisomal storage diseases. Cell Transpl. 1995, 4, 385–392. [Google Scholar] [CrossRef]
- Capotondo, A.; Milazzo, R.; Politi, L.S.; Quattrini, A.; Palini, A.; Plati, T.; Merella, S.; Nonis, A.; Di Serio, C.; Montini, E.; et al. Brain conditioning is instrumental for successful microglia reconstitution following hematopoietic stem cell transplantation. Proc. Natl. Acad. Sci. USA 2012, 109, 15018–15023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeager, A.M.; Shinohara, M.; Shinn, C. Hematopoietic cell transplantation after administration of high-dose busulfan in murine globoid cell leukodystrophy (the twitcher mouse). Pediatr. Res. 1991, 29, 302–305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, D.W.; Abkowitz, J.L. Kinetics of central nervous system microglial and macrophage engraftment: Analysis using a transgenic bone marrow transplantation model. Blood 1997, 90, 986–993. [Google Scholar] [CrossRef] [Green Version]
- Saha, A.; Buntz, S.; Scotland, P.; Xu, L.; Noeldner, P.; Patel, S.; Wollish, A.; Gunaratne, A.; Gentry, T.; Troy, J.; et al. A cord blood monocyte–derived cell therapy product accelerates brain remyelination. JCI Insight 2016, 1, e86667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurtzberg, J.; Buntz, S.; Gentry, T.; Noeldner, P.; Ozamiz, A.; Rusche, B.; Stroms, R.; Wollish, A.; Wenger, D.; Balber, A. Preclinical characterization of DUOC-01, a cell therapy product derived from banked umbilical cord blood for use as an adjuvant to umbilical cord blood transplantation for treatment of inherited metabolic diseases. Cytotherapy 2015, 17, 803–815. [Google Scholar] [CrossRef] [PubMed] [Green Version]



{kind=link}
{kind=link}
| Manifestation | MPS II (Hunter Syndrome) | References | MPS IH (Hurler Syndrome) | References |
|---|---|---|---|---|
| Umbilical hernia | 70–95% | [12,19] | 60–75% | [19,20,21,22] |
| Inguinal hernia | 70–95% | [12,19] | 60–75% | [19,20,21,22] |
| Hepatosplenomegaly | 60–90% | [10,12,19,23] | 70–85% | [19,20] |
| Skeletal manifestations | 80% | [24] | 80% | [22,25] |
| Kyphosis | 34% | [10,12,18,19,23,24] | 70–90% | [26] |
| Odontoid hypoplasia | rare | [10] | 65% | [27] |
| Joint stiffness | 75–90% | [10,19,23,24] | 93% | [19,20,26] |
| Poor growth | 79% | [12,19,24] | 100% | [19,22,24] |
| Epidermal symptoms (thickened skin with pebble formation, persistent Mongolian spots) | 13–17% | [19,28,29,30] | rare | |
| Coarse facial features | 95% | [10,23] | 86–97% | [13] |
| Upper respiratory issues | 100% | [12,19] | 80–100% | [19,20,22,25] |
| Lower respiratory issues | 80–90% | [12,24] | 80–90% | [21,22,31] |
| Loss of hearing | 70–95% | [8,12,28,32] | 76–100% | [20,25] |
| Valvular heart disease | 50–60% | [10,23,28,33,34] | 40–100% | [13,20,21,22,25,35,36] |
| Corneal clouding | rare | [19] | 71–88% | [19,20,21,22,25] |
| Seizures | 60% | [12,37] | rare | [38] |
| Cognitive impairment | 100% | [12,23,28] | 100% | [22,39,40] |
| Behavioral disturbances | 30–45% | [8,12,19,23,28,41] | rare | [19] |
| Diarrhea | 60% | [12,19,23,28] | rare | [19] |
| MPS IH | MPS IIA | |
|---|---|---|
| Cognitive development | Progressive cognitive decline beginning at age 6–15 months [18,39,176,177,178] | Normal until age 3–4 years, followed by plateau and rapid decline [4,179] |
| Coarse facial features | 6–12 months [18] | 2–4 years [23] |
| Hearing loss | 6–12 months [20] | 4 years [32] |
| Cardiac valve disease | 1 year [18] | 6 years [23] |
| Kyphosis | 1 year [18] | 6 years [23,24] |
| Axial growth | Normal until age 3 years | Normal to accelerated until age 5–6 years [23,180] |
| Average age at diagnosis | 1 year [18] | 3–4 years [23,24,180,181] |
| MPS I | MPS II | |
|---|---|---|
| Behavior (hypoactivity) | 2–4 months [182,183] | 6–10 months [160,164,166,168] |
| Zygomatic arch width | Increased at 4 weeks [184] | Increased at 2–3 months [164,185] |
| Hearing loss | 2 months [186] | 17 weeks [163] |
| Elevated GAG levels in urine and tissue | 4 weeks [173,184,187] | 4–6 weeks [162,164,165,167,185] |
| Manifestation | ERT | HSCT |
|---|---|---|
| Hepatosplenomegaly | Improved [192,199,200,201,202,203,204,205] | Improved [206,207] Improved (case studies n < 10) [208,209,210] |
| Skeletal manifestations | No change [204] | No change (case study n = 1) [208] Improved (case study n = 1) [198] |
| Poor growth | Minimal effect [199,211] | Improved [212] Improved (case study n = 1) [198] |
| Coarse facial features | Improved [201] | Improved [206] Improved (case studies n < 10) [208,210,213,214] |
| Upper respiratory function | Improved [205] | Improved [206,207] |
| Lower respiratory function | Improved [191,199,201,203] | NA |
| Heart hypertrophy | Improved [191,194,199] | Improved [215] |
| Valvular heart disease | Prevention (when administered very early) [199] No change [204] | Stabilization/improved [206,215] Improved (case studies n < 10) [209,216,217] |
| Joint stiffness | No change [199,200] Improved (shoulders) [195,205] | No change [206] No change (case studies n < 10) [214,216,218,219] Improved [207,220] Improved (case studies n < 10) [208,209,213] |
| Endurance | Improved [192,194,195,201] | Improved [217] |
| Skin, thickened with pebble | Improved [199,221,222] | Improved (case studies n < 10)[214,216,218,223] |
| Cognitive impairment | No change [195,201] | Improved [217] Improved (case studies <10) [198,217] Worsening (case studies n < 10) [208,209,210,219,224] |
| Apnea | Improved/stabilized apnea (obstructive) [182] | Improved apnea (not clarified whether obstructive or central) [207] |
| Diarrhea | Improved/stabilized [205] | NA |
| Activity of daily living (ADL) | NA | Improved [207,215,225] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hampe, C.S.; Yund, B.D.; Orchard, P.J.; Lund, T.C.; Wesley, J.; McIvor, R.S. Differences in MPS I and MPS II Disease Manifestations. Int. J. Mol. Sci. 2021, 22, 7888. https://doi.org/10.3390/ijms22157888
Hampe CS, Yund BD, Orchard PJ, Lund TC, Wesley J, McIvor RS. Differences in MPS I and MPS II Disease Manifestations. International Journal of Molecular Sciences. 2021; 22(15):7888. https://doi.org/10.3390/ijms22157888
Chicago/Turabian StyleHampe, Christiane S., Brianna D. Yund, Paul J. Orchard, Troy C. Lund, Jacob Wesley, and R. Scott McIvor. 2021. "Differences in MPS I and MPS II Disease Manifestations" International Journal of Molecular Sciences 22, no. 15: 7888. https://doi.org/10.3390/ijms22157888