
Emerging Approaches to Understanding Microvascular Endothelial Heterogeneity: A Roadmap for Developing Anti-Inflammatory Therapeutics


Abstract
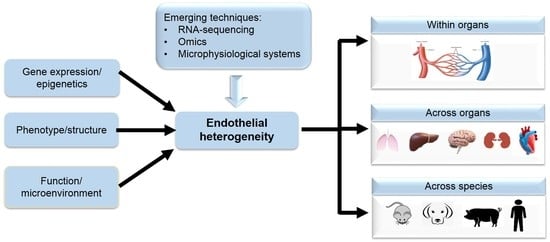
:
1. Introduction
Structure/Function of the Endothelium
2. Endothelium Structure and Heterogeneity
2.1. Endothelium Heterogeneity across Organs
2.2. Endothelium Heterogeneity across Species
3. Endothelial Cell Alterations in Acute Inflammation
3.1. Endothelium Activation in Sepsis

3.2. The Classical Leukocyte Recruitment Process in Sepsis
3.3. The Classical Rules of Leukocyte–Endothelial Interactions Do Not Apply Universally
3.4. Endothelium Activation in COVID-19
4. Emerging Techniques and Biomimetic Models to Study Endothelial Cell Function
4.1. RNA Sequencing
4.2. Proteomics
4.3. The Promise of Microphysiological Systems and Their Limitations


5. Leveraging Emerging Technologies to Enhance Translatability of Ec-Targeted Therapeutics

6. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| EC | Endothelial cells |
| VEGF | Vascular endothelial growth factor |
| VEGFR2 | VEGF receptor 2 |
| BBB | Blood–brain barrier |
| HLMVEC | Human lung microvascular endothelial cells |
| MLMVEC | Mouse lung microvascular endothelial cells |
| TEER | Transendothelial electrical resistance |
| ICAM-1 | Intercellular adhesion molecule-1 |
| VCAM-1 | Vascular cell adhesion molecule-1 |
| HUVEC | Human umbilical vein endothelial cells |
| PAMPs | Pathogen-associated molecular patterns |
| DAMPs | Damage-associated molecular patterns |
| PRR | Pattern recognition receptors |
| VE-cadherin | Vascular endothelial cadherin |
| DIC | Disseminated intravascular coagulation |
| NETs | Neutrophil extracellular traps |
| ROS | Reactive oxygen species |
| RNS | Reactive nitrogen species |
| MPO | Myeloperoxidase |
| MODS | Multiple organ dysfunction syndrome |
| JAM | Junctional adhesion molecules |
| PECAM | Platelet endothelial adhesion molecule |
| COVID-19 | Coronavirus disease of 2019 |
| SARS-CoV-2 | Severe acute respiratory syndrome coronavirus-2 |
| ACE-2 | Angiotensin converting enzyme 2 |
| ARDS | Acute respiratory distress syndrome |
| vWF | von Willebrand factor |
| 2DGE | 2D gel electrophoresis |
| LC-MS | Liquid chromatography in combination with mass spectrometry |
| MALDI | Matrix-assisted laser desorption/ionization |
| ESI | Electrospray ionization |
| MPS | Microphysiological systems |
| bMFA | Biomimetic microfluidic assay |
| PKCδ | Protein Kinase C-delta |
| B3C | Blood–brain barrier on-a-chip |
| EGFR | Epidermal growth factor receptor |
| bMTM | Biomimetic microfluidic tumor microenvironment |
References
- Jurisic, G.; Detmar, M. Lymphatic endothelium in health and disease. Cell Tissue Res. 2009, 335, 97–108. [Google Scholar] [CrossRef] [Green Version]
- Pepper, M.S.; Skobe, M. Lymphatic endothelium: Morphological, molecular and functional properties. J. Cell Biol. 2003, 163, 209. [Google Scholar] [CrossRef] [PubMed]
- Augustin, H.G.; Koh, G.Y. Organotypic vasculature: From descriptive heterogeneity to functional pathophysiology. Science 2017, 357, 12. [Google Scholar] [CrossRef] [Green Version]
- Marcu, R.; Choi, Y.J.; Xue, J.; Fortin, C.L.; Wang, Y.; Nagao, R.J.; Xu, J.; MacDonald, J.W.; Bammler, T.K.; Murry, C.E.; et al. Human Organ-Specific Endothelial Cell Heterogeneity. iScience 2018, 4, 20–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cahill, P.A.; Redmond, E.M. Vascular endothelium—Gatekeeper of vessel health. Atherosclerosis 2016, 248, 97–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aird, W.C. Endothelial cell heterogeneity. Cold Spring Harb. Perspect. Med. 2012, 2, a006429. [Google Scholar] [CrossRef] [PubMed]
- Rajendran, P.; Rengarajan, T.; Thangavel, J.; Nishigaki, Y.; Sakthisekaran, D.; Sethi, G.; Nishigaki, I. The vascular endothelium and human diseases. Int. J. Biol. Sci. 2013, 9, 1057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Chang, Y.; Wei, W. Endothelial dysfunction and inflammation: Immunity in rheumatoid arthritis. Mediat. Inflamm. 2016, 2016, 6813016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wijerathne, H.; Langston, J.; Yang, Q.; Sun, S.; Miyamoto, C.; Kilpatrick, L.E.; Kiani, M.F. Mechanisms of radiation-induced endothelium damage: Emerging models and technologies. Radiother. Oncol. 2021, 158, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Gan, X.; Curran, E.; Lamberti, G.; Krynska, B.; Kiani, M.F.; Wang, B. Targeted delivery of vascular endothelial growth factor improves stem cell therapy in a rat myocardial infarction model. Nanomed. Nanotechnol. Biol. Med. 2014, 10, 1711–1718. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Soroush, F.; Tong, Z.; Kiani, M.F.; Wang, B. Targeted multidrug delivery system to overcome chemoresistance in breast cancer. Int. J. Nanomed. 2017, 12, 671. [Google Scholar] [CrossRef] [Green Version]
- Muzykantov, V.R. Targeted drug delivery to endothelial adhesion molecules. ISRN Vasc. Med. 2013, 2013, 916254. [Google Scholar] [CrossRef] [Green Version]
- Brenner, J.S.; Greineder, C.; Shuvaev, V.; Muzykantov, V. Endothelial nanomedicine for the treatment of pulmonary disease. Expert Opin. Drug Deliv. 2015, 12, 239–261. [Google Scholar] [CrossRef] [PubMed]
- Glassman, P.M.; Myerson, J.W.; Ferguson, L.T.; Kiseleva, R.Y.; Shuvaev, V.V.; Brenner, J.S.; Muzykantov, V.R. Targeting drug delivery in the vascular system: Focus on endothelium. Adv. Drug Deliv. Rev. 2020, 157, 96–117. [Google Scholar] [CrossRef]
- Oliver, G.; Srinivasan, R.S. Endothelial cell plasticity: How to become and remain a lymphatic endothelial cell. Development 2010, 137, 363–372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, J.; Hirschi, K.K. Endothelial cell development and its application to regenerative medicine. Circ. Res. 2019, 125, 489–501. [Google Scholar] [CrossRef] [PubMed]
- Corada, M.; Morini, M.F.; Dejana, E. Signaling pathways in the specification of arteries and veins. Arterioscler. Thromb. Vasc. Biol. 2014, 34, 2372–2377. [Google Scholar] [CrossRef]
- Sriram, G.; Tan, J.Y.; Islam, I.; Rufaihah, A.J.; Cao, T. Efficient differentiation of human embryonic stem cells to arterial and venous endothelial cells under feeder-and serum-free conditions. Stem Cell Res. Ther. 2015, 6, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, Z.; Mo, Z. Regulation of endothelial cell differentiation in embryonic vascular development and its therapeutic potential in cardiovascular diseases. Life Sci. 2021, 119406. [Google Scholar] [CrossRef]
- Tsuji-Tamura, K.; Ogawa, M. Morphology regulation in vascular endothelial cells. Inflamm. Regen. 2018, 38, 1–13. [Google Scholar] [CrossRef]
- Chi, J.-T.; Chang, H.Y.; Haraldsen, G.; Jahnsen, F.L.; Troyanskaya, O.G.; Chang, D.S.; Wang, Z.; Rockson, S.G.; Van De Rijn, M.; Botstein, D. Endothelial cell diversity revealed by global expression profiling. Proc. Natl. Acad. Sci. USA 2003, 100, 10623–10628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aird, W.C. Phenotypic heterogeneity of the endothelium: I. Structure, function, and mechanisms. Circ. Res. 2007, 100, 158–173. [Google Scholar] [CrossRef]
- Young, E.W.; Simmons, C.A. Macro-and microscale fluid flow systems for endothelial cell biology. Lab Chip 2010, 10, 143–160. [Google Scholar] [CrossRef] [PubMed]
- Predescu, S.A.; Predescu, D.N.; Palade, G.E. Endothelial transcytotic machinery involves supramolecular protein–lipid complexes. Mol. Biol. Cell 2001, 12, 1019–1033. [Google Scholar] [CrossRef] [Green Version]
- Vogel, S.M.; Malik, A.B. Cytoskeletal dynamics and lung fluid balance. Compr. Physiol. 2012, 2, 449–478. [Google Scholar]
- Komarova, Y.; Malik, A.B. Regulation of endothelial permeability via paracellular and transcellular transport pathways. Annu. Rev. Physiol. 2010, 72, 463–493. [Google Scholar] [CrossRef]
- Sukriti, S.; Tauseef, M.; Yazbeck, P.; Mehta, D. Mechanisms regulating endothelial permeability. Pulm. Circ. 2014, 4, 535–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benson, K.; Cramer, S.; Galla, H.-J. Impedance-based cell monitoring: Barrier properties and beyond. Fluids Barriers CNS 2013, 10, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.; Soroush, F.; Sun, S.; Liverani, E.; Langston, J.C.; Yang, Q.; Kilpatrick, L.E.; Kiani, M.F. Protein kinase C-delta inhibition protects blood-brain barrier from sepsis-induced vascular damage. J. Neuroinflamm. 2018, 15, 309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tse, D.; Stan, R.V. Morphological Heterogeneity of Endothelium; Seminars in thrombosis and hemostasis, 2010; Thieme Medical Publishers: New York, NY, USA, 2010; pp. 236–245. [Google Scholar]
- Bearer, E.L.; Orci, L. Endothelial fenestral diaphragms: A quick-freeze, deep-etch study. J. Cell Biol. 1985, 100, 418–428. [Google Scholar] [CrossRef] [Green Version]
- Stan, R.V.; Tse, D.; Deharvengt, S.J.; Smits, N.C.; Xu, Y.; Luciano, M.R.; McGarry, C.L.; Buitendijk, M.; Nemani, K.V.; Elgueta, R. The diaphragms of fenestrated endothelia: Gatekeepers of vascular permeability and blood composition. Dev. Cell 2012, 23, 1203–1218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stan, R.-V.; Kubitza, M.; Palade, G.E. PV-1 is a component of the fenestral and stomatal diaphragms in fenestrated endothelia. Proc. Natl. Acad. Sci. USA 1999, 96, 13203–13207. [Google Scholar] [CrossRef] [Green Version]
- Shetty, S.; Lalor, P.F.; Adams, D.H. Liver sinusoidal endothelial cells—Gatekeepers of hepatic immunity. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Weinbaum, S.; Tarbell, J.M.; Damiano, E.R. The structure and function of the endothelial glycocalyx layer. Annu. Rev. Biomed. Eng. 2007, 9, 121–167. [Google Scholar] [CrossRef] [PubMed]
- Reiterer, M.; Branco, C.M. Endothelial cells and organ function: Applications and implications of understanding unique and reciprocal remodelling. FEBS J. 2020, 287, 1088–1100. [Google Scholar] [CrossRef] [PubMed]
- Ruan, C.-C.; Gao, P.-J. Role of complement-related inflammation and vascular dysfunction in hypertension. Hypertension 2019, 73, 965–971. [Google Scholar] [CrossRef]
- Sartain, S.E.; Turner, N.A.; Moake, J.L. Brain microvascular endothelial cells exhibit lower activation of the alternative complement pathway than glomerular microvascular endothelial cells. J. Biol. Chem. 2018, 293, 7195–7208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kerr, H.; Richards, A. Complement-mediated injury and protection of endothelium: Lessons from atypical haemolytic uraemic syndrome. Immunobiology 2012, 217, 195–203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosshart, S.P.; Herz, J.; Vassallo, B.G.; Hunter, A.; Wall, M.K.; Badger, J.H.; McCulloch, J.A.; Anastasakis, D.G.; Sarshad, A.A.; Leonardi, I.; et al. Laboratory mice born to wild mice have natural microbiota and model human immune responses. Science 2019, 365. [Google Scholar] [CrossRef]
- Cavaillon, J.M.; Singer, M.; Skirecki, T. Sepsis therapies: Learning from 30 years of failure of translational research to propose new leads. EMBO Mol. Med. 2020, 12, e10128. [Google Scholar] [CrossRef] [PubMed]
- Brubaker, D.K.; Lauffenburger, D.A. Translating preclinical models to humans. Science 2020, 367, 742–743. [Google Scholar] [CrossRef] [PubMed]
- Shultz, L.D.; Keck, J.; Burzenski, L.; Jangalwe, S.; Vaidya, S.; Greiner, D.L.; Brehm, M.A. Humanized mouse models of immunological diseases and precision medicine. Mamm. Genome 2019, 30, 123–142. [Google Scholar] [CrossRef] [PubMed]
- Kalla, D.; Kind, A.; Schnieke, A. Genetically engineered pigs to study cancer. Int. J. Mol. Sci. 2020, 21, 488. [Google Scholar] [CrossRef] [PubMed]
- Perlman, H.; Budinger, G.R.; Ward, P.A. Humanizing the mouse: In defense of murine models of critical illness. Am. J. Respir. Crit. Care Med. 2013, 187, 898–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Seok, J.; Warren, H.S.; Cuenca, A.G.; Mindrinos, M.N.; Baker, H.V.; Xu, W.; Richards, D.R.; McDonald-Smith, G.P.; Gao, H.; Hennessy, L.; et al. Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc. Natl. Acad. Sci. USA 2013, 110, 3507–3512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soroush, F.; Tang, Y.; Mustafa, O.; Sun, S.; Yang, Q.; Kilpatrick, L.E.; Kiani, M.F. Neutrophil-endothelial interactions of murine cells is not a good predictor of their interactions in human cells. FASEB J. 2020, 34, 2691–2702. [Google Scholar] [CrossRef] [Green Version]
- Jang, K.J.; Otieno, M.A.; Ronxhi, J.; Lim, H.K.; Ewart, L.; Kodella, K.R.; Petropolis, D.B.; Kulkarni, G.; Rubins, J.E.; Conegliano, D.; et al. Reproducing human and cross-species drug toxicities using a Liver-Chip. Sci. Transl. Med. 2019, 11. [Google Scholar] [CrossRef]
- Miller, S.; Walker, S.; Arthur, J.; Lewin, M.; Pickard, K.; Nicol, F.; Howie, A.; Beckett, G. Selenoprotein expression in endothelial cells from different human vasculature and species. Biochim. Biophys. Acta BBA Mol. Basis Dis. 2002, 1588, 85–93. [Google Scholar] [CrossRef] [Green Version]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.-D.; Coopersmith, C.M. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef] [PubMed]
- Ince, C.; Mayeux, P.R.; Nguyen, T.; Gomez, H.; Kellum, J.A.; Ospina-Tascon, G.A.; Hernandez, G.; Murray, P.; De Backer, D.; Workgrp, A.X. The endothelium in sepsis. Shock 2016, 45, 259–270. [Google Scholar] [CrossRef] [Green Version]
- Joffre, J.; Hellman, J.; Ince, C.; Ait-Oufella, H. Endothelial Responses in Sepsis. Am. J. Respir. Crit. Care Med. 2020, 202, 361–370. [Google Scholar] [CrossRef]
- Brown, K.A.; Brain, S.D.; Pearson, J.D.; Edgeworth, J.D.; Lewis, S.M.; Treacher, D.F. Neutrophils in development of multiple organ failure in sepsis. Lancet 2006, 368, 157–169. [Google Scholar] [CrossRef]
- Goldenberg, N.M.; Steinberg, B.E.; Slutsky, A.S.; Lee, W.L. Broken Barriers: A New Take on Sepsis Pathogenesis. Sci. Transl. Med. 2011, 3, 88ps25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maniatis, N.A.; Orfanos, S.E. The endothelium in acute lung injury/acute respiratory distress syndrome. Curr. Opin. Crit. Care 2008, 14, 22–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ley, K.; Reutershan, J. Leucocyte-endothelial interactions in health and disease. Handb. Exp. Pharmacol. 2006, 176 Pt 2, 97–133. [Google Scholar]
- Ley, K.; Laudanna, C.; Cybulsky, M.I.; Nourshargh, S. Getting to the site of inflammation: The leukocyte adhesion cascade updated. Nat. Rev. Immunol. 2007, 7, 678–689. [Google Scholar] [CrossRef] [PubMed]
- Seynhaeve, A.L.B.; Vermeulen, C.E.; Eggermont, A.M.M.; ten Hagen, T.L.M. Cytokines and vascular permeability. Cell Biochem. Biophys. 2006, 44, 157–169. [Google Scholar] [CrossRef]
- Wachtel, M.; Bolliger, M.F.; Ishihara, H.; Frei, K.; Bluethmann, H.; Gloor, S.M. Down-regulation of occludin expression in astrocytes by tumour necrosis factor (TNF) is mediated via TNF type-1 receptor and nuclear factor-kappa B activation. J. Neurochem. 2001, 78, 155–162. [Google Scholar] [CrossRef]
- Mankertz, J.; Tavalali, S.; Schmitz, H.; Mankertz, A.; Riecken, E.O.; Fromm, M.; Schulzke, J.D. Expression from the human occludin promoter is affected by tumor necrosis factor alpha and interferon gamma. J. Cell Sci. 2000, 113, 2085–2090. [Google Scholar] [CrossRef] [PubMed]
- Adam, A. Regulation of Endothelial Adherens Junctions by Tyrosine Phosphorylation. Mediat. Inflamm. 2015, 2015, 272858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Millán, J.; Cain, R.J.; Reglero-Real, N.; Bigarella, C.; Marcos-Ramiro, B.; Fernández-Martín, L.; Correas, I.; Ridley, A.J. Adherens junctions connect stress fibres between adjacent endothelial cells. BMC Biol. 2010, 8, 11. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Zhang, R.; Qu, H.; Wu, J.; Li, L.; Tang, Y. Endothelial microparticles activate endothelial cells to facilitate the inflammatory response. Mol. Med. Rep. 2017, 15, 1291–1296. [Google Scholar] [CrossRef] [Green Version]
- Drake-Holland, A.J.; Noble, M.I.M. Update on the Important New Drug Target in Cardiovascular Medicine—The Vascular Glycocalyx. Cardiovasc. Hematol. Disord. Drug Targets 2012, 12, 76–81. [Google Scholar] [CrossRef]
- Broekhuizen, L.N.; Mooij, H.L.; Kastelein, J.J.P.; Stroes, E.S.G.; Vink, H.; Nieuwdorp, M. Endothelial glycocalyx as potential diagnostic and therapeutic target in cardiovascular disease. Curr. Opin. Lipidol. 2009, 20, 57–62. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Yang, X.; Chatterjee, V.; Meegan, J.E.; Beard, R.S., Jr.; Yuan, S.Y. Role of Neutrophil Extracellular Traps and Vesicles in Regulating Vascular Endothelial Permeability. Front. Immunol. 2019, 10, 1037. [Google Scholar] [CrossRef] [Green Version]
- Amulic, B.; Cazalet, C.; Hayes, G.L.; Metzler, K.D.; Zychlinsky, A. Neutrophil function: From mechanisms to disease. Annu. Rev. Immunol. 2012, 30, 459–489. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, P.A.; Kilpatrick, L.E. Neutrophil Inflammation in COPD. In Smoking and Lung Inflammation: Basic, Pre-Clinical and Clinical Research Advances; Rogers, T.J., Criner, G.J., Cornwell, W.D., Eds.; Springer: New York, NY, USA, 2013; pp. 59–79. [Google Scholar]
- Orfanos, S.E.; Mavrommati, I.; Korovesi, I.; Roussos, C. Pulmonary endothelium in acute lung injury: From basic science to the critically ill. Intensive Care Med. 2004, 30, 1702–1714. [Google Scholar] [CrossRef]
- Reutershan, J.; Ley, K. Bench-to-bedside review: Acute respiratory distress syndrome—How neutrophils migrate into the lung. Crit. Care 2004, 8, 453–461. [Google Scholar] [CrossRef] [Green Version]
- Phillipson, M.; Kubes, P. The neutrophil in vascular inflammation. Nat. Med. 2011, 17, 1381–1390. [Google Scholar] [CrossRef]
- Kolaczkowska, E.; Kubes, P. Neutrophil recruitment and function in health and inflammation. Nat. Rev. Immunol. 2013, 13, 159–175. [Google Scholar] [CrossRef]
- Molteni, R.; Fabbri, M.; Bender, J.R.; Pardi, R. Pathophysiology of leukocyte-tissue interactions. Curr. Opin. Cell Biol. 2006, 18, 491–498. [Google Scholar] [CrossRef]
- Guo, R.-F.; Riedemann, N.C.; Laudes, I.J.; Sarma, V.J.; Kunkel, R.G.; Dilley, K.A.; Paulauskis, J.D.; Ward, P.A. Altered Neutrophil Trafficking During Sepsis. J. Immunol. 2002, 169, 307–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engelhardt, B.; Wolburg, H. Mini-review: Transendothelial migration of leukocytes: Through the front door or around the side of the house? Eur. J. Immunol. 2004, 34, 2955–2963. [Google Scholar] [CrossRef] [PubMed]
- Mickael, M.-E.; Kubick, N.; Klimovich, P.; Flournoy, P.H.; Bieńkowska, I.; Sacharczuk, M. Paracellular and Transcellular Leukocytes Diapedesis Are Divergent but Interconnected Evolutionary Events. Genes 2021, 12, 254. [Google Scholar] [CrossRef]
- Maas, S.L.; Soehnlein, O.; Viola, J.R. Organ-specific mechanisms of transendothelial neutrophil migration in the lung, liver, kidney, and aorta. Front. Immunol. 2018, 9, 2739. [Google Scholar] [CrossRef] [Green Version]
- Mizgerd, J.P.; Meek, B.B.; Kutkoski, G.J.; Bullard, D.C.; Beaudet, A.L.; Doerschuk, C.M. Selectins and neutrophil traffic: Margination and Streptococcus pneumoniae-induced emigration in murine lungs. J. Exp. Med. 1996, 184, 639–645. [Google Scholar] [CrossRef] [Green Version]
- Kornerup, K.N.; Salmon, G.P.; Pitchford, S.C.; Liu, W.L.; Page, C.P. Circulating platelet-neutrophil complexes are important for subsequent neutrophil activation and migration. J. Appl. Physiol. 2010, 109, 758–767. [Google Scholar] [CrossRef]
- Doerschuk, C.; Beyers, N.; Coxson, H.; Wiggs, B.; Hogg, J. Comparison of neutrophil and capillary diameters and their relation to neutrophil sequestration in the lung. J. Appl. Physiol. 1993, 74, 3040–3045. [Google Scholar] [CrossRef]
- Doerschuk, C.M. Mechanisms of leukocyte sequestration in inflamed lungs. Microcirculation 2001, 8, 71–88. [Google Scholar] [CrossRef]
- Basit, A.; Reutershan, J.; Morris, M.A.; Solga, M.; Rose, C.E., Jr.; Ley, K. ICAM-1 and LFA-1 play critical roles in LPS-induced neutrophil recruitment into the alveolar space. Am. J. Physiol. Lung Cell. Mol. Physiol. 2006, 291, L200–L207. [Google Scholar] [CrossRef] [Green Version]
- Wong, J.; Johnston, B.; Lee, S.S.; Bullard, D.C.; Smith, C.W.; Beaudet, A.L.; Kubes, P. A minimal role for selectins in the recruitment of leukocytes into the inflamed liver microvasculature. J. Clin. Investig. 1997, 99, 2782–2790. [Google Scholar] [CrossRef] [Green Version]
- Devi, S.; Li, A.; Westhorpe, C.L.; Lo, C.Y.; Abeynaike, L.D.; Snelgrove, S.L.; Hall, P.; Ooi, J.D.; Sobey, C.G.; Kitching, A.R. Multiphoton imaging reveals a new leukocyte recruitment paradigm in the glomerulus. Nat. Med. 2013, 19, 107. [Google Scholar] [CrossRef]
- Kuligowski, M.P.; Kitching, A.R.; Hickey, M.J. Leukocyte recruitment to the inflamed glomerulus: A critical role for platelet-derived P-selectin in the absence of rolling. J. Immunol. 2006, 176, 6991–6999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, X. COVID-19: Immunopathology and its implications for therapy. Nat. Rev. Immunol. 2020, 20, 269–270. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vabret, N.; Britton, G.J.; Gruber, C.; Hegde, S.; Kim, J.; Kuksin, M.; Levantovsky, R.; Malle, L.; Moreira, A.; Park, M.D. Immunology of COVID-19: Current state of the science. Immunity 2020. [Google Scholar] [CrossRef] [PubMed]
- Varga, Z.; Flammer, A.J.; Steiger, P.; Haberecker, M.; Andermatt, R.; Zinkernagel, A.S.; Mehra, M.R.; Schuepbach, R.A.; Ruschitzka, F.; Moch, H. Endothelial cell infection and endotheliitis in COVID-19. Lancet 2020, 395, 1417–1418. [Google Scholar] [CrossRef]
- Gheblawi, M.; Wang, K.; Viveiros, A.; Nguyen, Q.; Zhong, J.-C.; Turner, A.J.; Raizada, M.K.; Grant, M.B.; Oudit, G.Y. Angiotensin-Converting Enzyme 2: SARS-CoV-2 Receptor and Regulator of the Renin-Angiotensin System: Celebrating the 20th Anniversary of the Discovery of ACE2. Circ. Res. 2020, 126, 1456–1474. [Google Scholar] [CrossRef] [PubMed]
- Sardu, C.; Gambardella, J.; Morelli, M.B.; Wang, X.; Marfella, R.; Santulli, G. Hypertension, thrombosis, kidney failure, and diabetes: Is COVID-19 an endothelial disease? A comprehensive evaluation of clinical and basic evidence. J. Clin. Med. 2020, 9, 1417. [Google Scholar] [CrossRef]
- Java, A.; Apicelli, A.J.; Liszewski, M.K.; Coler-Reilly, A.; Atkinson, J.P.; Kim, A.H.; Kulkarni, H.S. The complement system in COVID-19: Friend and foe? JCI Insight 2020, 5. [Google Scholar] [CrossRef]
- Bryce, C.; Grimes, Z.; Pujadas, E.; Ahuja, S.; Beasley, M.B.; Albrecht, R.; Hernandez, T.; Stock, A.; Zhao, Z.; Al Rasheed, M. Pathophysiology of SARS-CoV-2: Targeting of endothelial cells renders a complex disease with thrombotic microangiopathy and aberrant immune response. The Mount Sinai COVID-19 autopsy experience. MedRxiv 2020. [Google Scholar] [CrossRef]
- Jin, Y.; Ji, W.; Yang, H.; Chen, S.; Zhang, W.; Duan, G. Endothelial activation and dysfunction in COVID-19: From basic mechanisms to potential therapeutic approaches. Signal Transduct. Target. Ther. 2020, 5, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Teuwen, L.-A.; Geldhof, V.; Pasut, A.; Carmeliet, P. COVID-19: The vasculature unleashed. Nat. Rev. Immunol. 2020, 20, 389–391. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Tripathi, D.M.; Yadav, A. The enigma of endothelium in COVID-19. Front. Physiol. 2020, 11, 989. [Google Scholar] [CrossRef]
- Zuo, Y.; Yalavarthi, S.; Shi, H.; Gockman, K.; Zuo, M.; Madison, J.A.; Blair, C.; Weber, A.; Barnes, B.J.; Egeblad, M.; et al. Neutrophil extracellular traps in COVID-19. JCI Insight 2020, 5, e138999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Libby, P.; Lüscher, T. COVID-19 is, in the end, an endothelial disease. Eur. Heart J. 2020, 41, 3038–3044. [Google Scholar] [CrossRef]
- Garg, M. RNA sequencing: A revolutionary tool for transcriptomics. In Advances in Animal Genomics; Elsevier: Amsterdam, The Netherlands, 2021; pp. 61–73. [Google Scholar]
- Stark, R.; Grzelak, M.; Hadfield, J. RNA sequencing: The teenage years. Nat. Rev. Genet. 2019, 20, 631–656. [Google Scholar] [CrossRef]
- Vanlandewijck, M.; He, L.; Mäe, M.A.; Andrae, J.; Ando, K.; Del Gaudio, F.; Nahar, K.; Lebouvier, T.; Laviña, B.; Gouveia, L. A molecular atlas of cell types and zonation in the brain vasculature. Nature 2018, 554, 475–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jambusaria, A.; Hong, Z.; Zhang, L.; Srivastava, S.; Jana, A.; Toth, P.T.; Dai, Y.; Malik, A.B.; Rehman, J. Endothelial heterogeneity across distinct vascular beds during homeostasis and inflammation. eLife 2020, 9, e51413. [Google Scholar] [CrossRef]
- Kalucka, J.; de Rooij, L.P.; Goveia, J.; Rohlenova, K.; Dumas, S.J.; Meta, E.; Conchinha, N.V.; Taverna, F.; Teuwen, L.-A.; Veys, K. Single-cell transcriptome atlas of murine endothelial cells. Cell 2020, 180, 764–779.e20. [Google Scholar] [CrossRef]
- Goveia, J.; Rohlenova, K.; Taverna, F.; Treps, L.; Conradi, L.-C.; Pircher, A.; Geldhof, V.; de Rooij, L.P.; Kalucka, J.; Sokol, L. An Integrated Gene Expression Landscape Profiling Approach to Identify Lung Tumor Endothelial Cell Heterogeneity and Angiogenic Candidates. Cancer Cell 2020, 37, 21–36.e13. [Google Scholar] [CrossRef]
- Raju, S.M.; Jahnavi, V.; Kamaraju, R.S.; Sritharan, V.; Rajkumar, K.; Natarajan, S.; Kumar, A.D.; Burgula, S. Continuous evaluation of changes in the serum proteome from early to late stages of sepsis caused by Klebsiella pneumoniae. Mol. Med. Rep. 2016, 13, 4835–4844. [Google Scholar] [CrossRef]
- Faria, S.S.; Morris, C.F.M.; Silva, A.R.; Fonseca, M.P.; Forget, P.; Castro, M.S.; Fontes, W. A Timely Shift from Shotgun to Targeted Proteomics and How It Can Be Groundbreaking for Cancer Research. Front. Oncol. 2017, 7. [Google Scholar] [CrossRef] [Green Version]
- Arora, A.; Somasundaram, K. Targeted Proteomics Comes to the Benchside and the Bedside: Is it Ready for Us? Bioessays 2019, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borras, E.; Sabido, E. What is targeted proteomics? A concise revision of targeted acquisition and targeted data analysis in mass spectrometry. Proteomics 2017, 17. [Google Scholar] [CrossRef]
- Kavallaris, M.; Marshall, G.M. Proteomics and disease: Opportunities and challenges. Med. J. Aust. 2005, 182, 575–579. [Google Scholar] [CrossRef]
- Gautier, V.; Cayrol, C.; Farache, D.; Roga, S.; Monsarrat, B.; Burlet-Schiltz, O.; Gonzalez de Peredo, A.; Girard, J.P. Extracellular IL-33 cytokine, but not endogenous nuclear IL-33, regulates protein expression in endothelial cells. Sci. Rep. 2016, 6, 34255. [Google Scholar] [CrossRef]
- Mohr, T.; Haudek-Prinz, V.; Slany, A.; Grillari, J.; Micksche, M.; Gerner, C. Proteome profiling in IL-1beta and VEGF-activated human umbilical vein endothelial cells delineates the interlink between inflammation and angiogenesis. PLoS ONE 2017, 12, e0179065. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paik, D.T.; Tian, L.; Williams, I.M.; Rhee, S.; Zhang, H.; Liu, C.; Mishra, R.; Wu, S.M.; Red-Horse, K.; Wu, J.C. Single-Cell RNA Sequencing Unveils Unique Transcriptomic Signatures of Organ-Specific Endothelial Cells. Circulation 2020, 142, 1848–1862. [Google Scholar] [CrossRef] [PubMed]
- Cleuren, A.C.A.; van der Ent, M.A.; Jiang, H.; Hunker, K.L.; Yee, A.; Siemieniak, D.R.; Molema, G.; Aird, W.C.; Ganesh, S.K.; Ginsburg, D. The in vivo endothelial cell translatome is highly heterogeneous across vascular beds. Proc. Natl. Acad. Sci. USA 2019, 116, 23618–23624. [Google Scholar] [CrossRef] [PubMed]
- Toledo, A.G.; Golden, G.; Campos, A.R.; Cuello, H.; Sorrentino, J.; Lewis, N.; Varki, N.; Nizet, V.; Smith, J.W.; Esko, J.D. Proteomic atlas of organ vasculopathies triggered by Staphylococcus aureus sepsis. Nat. Commun. 2019, 10, 13. [Google Scholar] [CrossRef] [Green Version]
- Wikswo, J.P. The relevance and potential roles of microphysiological systems in biology and medicine. Exp. Biol. Med. 2014, 239, 1061–1072. [Google Scholar] [CrossRef] [PubMed]
- Huh, D.; Matthews, B.D.; Mammoto, A.; Montoya-Zavala, M.; Hsin, H.Y.; Ingber, D.E. Reconstituting organ-level lung functions on a chip. Science 2010, 328, 1662–1668. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Mackay, S.; Gordon, D.M.; Anderson, J.D.; Haithcock, D.W.; Garson, C.J.; Tearney, G.J.; Solomon, G.M.; Pant, K.; Prabhakarpandian, B. Co-cultured microfluidic model of the airway optimized for microscopy and micro-optical coherence tomography imaging. Biomed. Opt. Express 2019, 10, 5414–5430. [Google Scholar] [CrossRef]
- Jalili-Firoozinezhad, S.; Prantil-Baun, R.; Jiang, A.; Potla, R.; Mammoto, T.; Weaver, J.C.; Ferrante, T.C.; Kim, H.J.; Cabral, J.M.; Levy, O. Modeling radiation injury-induced cell death and countermeasure drug responses in a human Gut-on-a-Chip. Cell Death Dis. 2018, 9, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Benam, K.H.; Villenave, R.; Lucchesi, C.; Varone, A.; Hubeau, C.; Lee, H.-H.; Alves, S.E.; Salmon, M.; Ferrante, T.C.; Weaver, J.C. Small airway-on-a-chip enables analysis of human lung inflammation and drug responses in vitro. Nat. Methods 2016, 13, 151–157. [Google Scholar] [CrossRef]
- Jang, K.-J.; Mehr, A.P.; Hamilton, G.A.; McPartlin, L.A.; Chung, S.; Suh, K.-Y.; Ingber, D.E. Human kidney proximal tubule-on-a-chip for drug transport and nephrotoxicity assessment. Integr. Biol. 2013, 5, 1119–1129. [Google Scholar] [CrossRef]
- Soroush, F.; Zhang, T.; King, D.J.; Tang, Y.; Deosarkar, S.; Prabhakarpandian, B.; Kilpatrick, L.E.; Kiani, M.F. A novel microfluidic assay reveals a key role for protein kinase C delta in regulating human neutrophil-endothelium interaction. J. Leukoc. Biol. 2016, 100, 1027–1035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roth, N.M.; Kiani, M.F. A “geographic information systems” based technique for the study of microvascular networks. Ann. Biomed. Eng. 1999, 27, 42–47. [Google Scholar] [CrossRef]
- Prabhakarpandian, B.; Shen, M.-C.; Pant, K.; Kiani, M.F. Microfluidic devices for modeling cell-cell and particle-cell interactions in the microvasculature. Microvasc. Res. 2011, 82, 210–220. [Google Scholar] [CrossRef] [Green Version]
- Prabhakarpandian, B.; Pant, K.; Scott, R.; Patillo, C.; Irimia, D.; Kiani, M.; Sundaram, S. Synthetic microvascular networks for quantitative analysis of particle adhesion. Biomed. Microdevices 2008, 10, 585–595. [Google Scholar] [CrossRef] [Green Version]
- Rosano, J.; Tousi, N.; Scott, R.; Krynska, B.; Rizzo, V.; Prabhakarpandian, B.; Pant, K.; Sundaram, S.; Kiani, M. A physiologically realistic in vitro model of microvascular networks. Biomed. Microdevices 2009, 11, 1051–1057. [Google Scholar] [CrossRef] [Green Version]
- Tousi, N.; Wang, B.; Pant, K.; Kiani, M.F.; Prabhakarpandian, B. Preferential adhesion of leukocytes near bifurcations is endothelium independent. Microvasc. Res. 2010, 80, 384–388. [Google Scholar] [CrossRef] [Green Version]
- Prabhakarpandian, B.; Wang, Y.I.; Rea-Ramsey, A.; Sundaram, S.; Kiani, M.F.; Pant, K. Bifurcations: Focal Points of Particle Adhesion in Microvascular Networks. Microcirculation 2011, 18, 380–389. [Google Scholar] [CrossRef] [Green Version]
- Lamberti, G.; Prabhakarpandian, B.; Garson, C.; Smith, A.; Pant, K.; Wang, B.; Kiani, M.F. Bioinspired Microfluidic Assay for In Vitro Modeling of Leukocyte-Endothelium Interactions. Anal. Chem. 2014, 86, 8344–8351. [Google Scholar] [CrossRef] [Green Version]
- Faber, S.C.; McCullough, S.D. Through the looking glass: In vitro models for inhalation toxicology and interindividual variability in the airway. Appl. In Vitro Toxicol. 2018, 4, 115–128. [Google Scholar] [CrossRef] [Green Version]
- Van Riet, S.; Ninaber, D.K.; Mikkers, H.M.; Tetley, T.D.; Jost, C.R.; Mulder, A.A.; Pasman, T.; Baptista, D.; Poot, A.A.; Truckenmüller, R. In vitro modelling of alveolar repair at the air-liquid interface using alveolar epithelial cells derived from human induced pluripotent stem cells. Sci. Rep. 2020, 10, 1–12. [Google Scholar] [CrossRef]
- Pasman, T.; Baptista, D.; van Riet, S.; Truckenmüller, R.K.; Hiemstra, P.S.; Rottier, R.J.; Hamelmann, N.M.; Paulusse, J.M.; Stamatialis, D.; Poot, A.A. Development of an In Vitro Airway Epithelial–Endothelial Cell Culture Model on a Flexible Porous Poly (Trimethylene Carbonate) Membrane Based on Calu-3 Airway Epithelial Cells and Lung Microvascular Endothelial Cells. Membranes 2021, 11, 197. [Google Scholar] [CrossRef]
- Caffrey, T.M.; Button, E.B.; Robert, J. Toward three-dimensional in vitro models to study neurovascular unit functions in health and disease. Neural Regen. Res. 2021, 16, 2132. [Google Scholar]
- Halldorsson, S.; Lucumi, E.; Gómez-Sjöberg, R.; Fleming, R.M. Advantages and challenges of microfluidic cell culture in polydimethylsiloxane devices. Biosens. Bioelectron. 2015, 63, 218–231. [Google Scholar] [CrossRef] [Green Version]
- Chistiakov, D.A.; Orekhov, A.N.; Bobryshev, Y.V. Effects of shear stress on endothelial cells: Go with the flow. Acta Physiol. 2017, 219, 382–408. [Google Scholar] [CrossRef]
- Kinstlinger, I.S.; Calderon, G.A.; Royse, M.K.; Means, A.K.; Grigoryan, B.; Miller, J.S. Perfusion and endothelialization of engineered tissues with patterned vascular networks. Nat. Protoc. 2021, 16, 3089–3113. [Google Scholar] [CrossRef] [PubMed]
- Grigoryan, B.; Paulsen, S.J.; Corbett, D.C.; Sazer, D.W.; Fortin, C.L.; Zaita, A.J.; Greenfield, P.T.; Calafat, N.J.; Gounley, J.P.; Ta, A.H. Multivascular networks and functional intravascular topologies within biocompatible hydrogels. Science 2019, 364, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Wimmer, R.A.; Leopoldi, A.; Aichinger, M.; Wick, N.; Hantusch, B.; Novatchkova, M.; Taubenschmid, J.; Hämmerle, M.; Esk, C.; Bagley, J.A. Human blood vessel organoids as a model of diabetic vasculopathy. Nature 2019, 565, 505–510. [Google Scholar] [CrossRef] [PubMed]
- Novo, P.; Janasek, D. Current advances and challenges in microfluidic free-flow electrophoresis—A critical review. Anal. Chim. Acta 2017, 991, 9–29. [Google Scholar] [CrossRef]
- Michael, I.J.; Kim, T.-H.; Sunkara, V.; Cho, Y.-K. Challenges and opportunities of centrifugal microfluidics for extreme point-of-care testing. Micromachines 2016, 7, 32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Needs, S.; Donmez, S.I.; Bull, S.; McQuaid, C.; Osborn, H.M.; Edwards, A. Challenges in microfluidic and point-of-care phenotypic antimicrobial resistance tests. Front. Mech. Eng. 2020, 6. [Google Scholar] [CrossRef]
- Soroush, F.; Tang, Y.; Guglielmo, K.; Engelmann, A.; Liverani, E.; Langston, J.; Sun, S.; Kunapuli, S.; Kiani, M.F.; Kilpatrick, L.E. Protein Kinase C-Delta (PKCdelta) Tyrosine Phosphorylation is a Critical Regulator of Neutrophil-Endothelial Cell Interaction in Inflammation. Shock 2018, 51, 538–547. [Google Scholar] [CrossRef]
- Efron, P.A.; Mohr, A.M.; Moore, F.A.; Moldawer, L.L. The future of murine sepsis and trauma research models. J. Leukoc. Biol. 2015, 98, 945–952. [Google Scholar] [CrossRef]
- Drake, A.C. Of mice and men: What rodent models don’t tell us. Cell. Mol. Immunol. 2013, 10, 284–285. [Google Scholar] [CrossRef]
- Fink, M.P.; Warren, H.S. Strategies to improve drug development for sepsis. Nat. Rev. Drug Discov. 2014, 13, 741–758. [Google Scholar] [CrossRef]
- Hillyer, P.; Mordelet, E.; Flynn, G.; Male, D. Chemokines, chemokine receptors and adhesion molecules on different human endothelia: Discriminating the tissue-specific functions that affect leucocyte migration. Clin. Exp. Immunol. 2003, 134, 431–441. [Google Scholar] [CrossRef]
- NAGMSC Working Group on Sepsis. 2019. Available online: https://www.nigms.nih.gov/News/reports/Documents/nagmsc-working-group-on-sepsis-final-report.pdf (accessed on 25 May 2021).
- Low, L.A.; Mummery, C.; Berridge, B.R.; Austin, C.P.; Tagle, D.A. Organs-on-chips: Into the next decade. Nat. Rev. Drug Discov. 2020, 345–361. [Google Scholar] [CrossRef]
- Cong, Y.; Han, X.; Wang, Y.; Chen, Z.; Lu, Y.; Liu, T.; Wu, Z.; Jin, Y.; Luo, Y.; Zhang, X. Drug Toxicity Evaluation Based on Organ-on-a-Chip Technology: A Review. Micromachines 2020, 11, 381. [Google Scholar] [CrossRef] [Green Version]
- Kimura, H.; Sakai, Y.; Fujii, T. Organ/body-on-a-chip based on microfluidic technology for drug discovery. Drug Metab. Pharmacokinet. 2018, 33, 43–48. [Google Scholar] [CrossRef]
- Mondrinos, M.J.; Zhang, T.; Sun, S.; Kennedy, P.A.; King, D.J.; Wolfson, M.R.; Knight, L.C.; Scalia, R.; Kilpatrick, L.E. Pulmonary Endothelial Protein Kinase C-Delta (PKCd) Regulates Neutrophil Migration in Acute Lung Inflammation. Am. J. Pathol. 2014, 184, 200–213. [Google Scholar] [CrossRef] [Green Version]
- Liverani, E.; Tursi, S.A.; Cornwell, W.D.; Mondrinos, M.J.; Sun, S.; Buttaro, B.A.; Wolfson, M.R.; Rogers, T.J.; Tükel, Ç.; Kilpatrick, L.E. Protein kinase C-delta inhibition is organ-protective, enhances pathogen clearance, and improves survival in sepsis. FASEB J. 2020, 34, 2497–2510. [Google Scholar] [CrossRef] [Green Version]
- Kilpatrick, L.E.; Standage, S.W.; Li, H.; Raj, N.R.; Korchak, H.M.; Wolfson, M.R.; Deutschman, C.S. Protection against sepsis-induced lung injury by selective inhibition of protein kinase C-d (d-PKC). J. Leukoc. Biol. 2011, 89, 3–10. [Google Scholar] [CrossRef]
- Mondrinos, M.J.; Knight, L.C.; Kennedy, P.A.; Wu, J.; Kauffman, M.; Baker, S.T.; Wolfson, M.R.; Kilpatrick, L.E. Biodistribution and Efficacy of Targeted Pulmonary Delivery of a Protein Kinase C-d Inhibitory Peptide: Impact on Indirect Lung Injury. J. Pharmacol. Exp. Ther. 2015, 355, 86–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kilpatrick, L.E.; Kiani, M.F. Experimental Approaches to Evaluate Leukocyte-Endothelial Cell Interactions in Sepsis and Inflammation. Shock 2020, 53, 585–595. [Google Scholar] [CrossRef]
- Jiang, L.; Li, S.; Zheng, J.; Li, Y.; Huang, H. Recent progress in microfluidic models of the blood-brain barrier. Micromachines 2019, 10, 375. [Google Scholar] [CrossRef] [Green Version]
- Mondrinos, M.J.; Kennedy, P.A.; Lyons, M.; Deutschman, C.S.; Kilpatrick, L.E. Protein kinase C and acute respiratory distress syndrome. Shock 2013, 39, 467. [Google Scholar] [CrossRef] [Green Version]
- Liverani, E.; Mondrinos, M.J.; Sun, S.; Kunapuli, S.P.; Kilpatrick, L.E. Role of Protein Kinase C-delta in regulating platelet activation and platelet-leukocyte interaction during sepsis. PLoS ONE 2018, 13, e0195379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrile, R.; van der Meer, A.D.; Park, H.; Fraser, J.P.; Simic, D.; Teng, F.; Conegliano, D.; Nguyen, J.; Jain, A.; Zhou, M. Organ-on-chip recapitulates thrombosis induced by an anti-CD154 monoclonal antibody: Translational potential of advanced microengineered systems. Clin. Pharmacol. Ther. 2018, 104, 1240–1248. [Google Scholar] [CrossRef]
- Nguyen, D.-H.T.; Stapleton, S.C.; Yang, M.T.; Cha, S.S.; Choi, C.K.; Galie, P.A.; Chen, C.S. Biomimetic model to reconstitute angiogenic sprouting morphogenesis in vitro. Proc. Natl. Acad. Sci. USA 2013, 110, 6712–6717. [Google Scholar] [CrossRef] [Green Version]
- Yu, F.; Kumar, N.D.O.S.; Foo, L.C.; Ng, S.H.; Hunziker, W.; Choudhury, D. A pump-free tricellular blood–brain barrier on-a-chip model to understand barrier property and evaluate drug response. Biotechnol. Bioeng. 2020, 117, 1127–1136. [Google Scholar] [CrossRef]
- Huh, D.; Leslie, D.C.; Matthews, B.D.; Fraser, J.P.; Jurek, S.; Hamilton, G.A.; Thorneloe, K.S.; McAlexander, M.A.; Ingber, D.E. A human disease model of drug toxicity–induced pulmonary edema in a lung-on-a-chip microdevice. Sci. Transl. Med. 2012, 4, ra147–ra159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, X.; Li, K.; Zhang, X.; Liu, C.; Guo, B.; Wen, W.; Gao, X. Nanofiber membrane supported lung-on-a-chip microdevice for anti-cancer drug testing. Lab Chip 2018, 18, 486–495. [Google Scholar] [CrossRef]
- Zhang, M.; Xu, C.; Jiang, L.; Qin, J. A 3D human lung-on-a-chip model for nanotoxicity testing. Toxicol. Res. 2018, 7, 1048–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, Y.; Soroush, F.; Sheffield, J.B.; Wang, B.; Prabhakarpandian, B.; Kiani, M.F. A biomimetic microfluidic tumor microenvironment platform mimicking the EPR effect for rapid screening of drug delivery systems. Sci. Rep. 2017, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EC Phenotypes | Location | Characteristics | Specialized Function | Diagram |
|---|---|---|---|---|
| Continuous |
|
|
|  |
| Fenestrated |
|
|
|  |
| Discontinuous |
|
|
|  |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, Q.; Wijerathne, H.; Langston, J.C.; Kiani, M.F.; Kilpatrick, L.E. Emerging Approaches to Understanding Microvascular Endothelial Heterogeneity: A Roadmap for Developing Anti-Inflammatory Therapeutics. Int. J. Mol. Sci. 2021, 22, 7770. https://doi.org/10.3390/ijms22157770
Yang Q, Wijerathne H, Langston JC, Kiani MF, Kilpatrick LE. Emerging Approaches to Understanding Microvascular Endothelial Heterogeneity: A Roadmap for Developing Anti-Inflammatory Therapeutics. International Journal of Molecular Sciences. 2021; 22(15):7770. https://doi.org/10.3390/ijms22157770
Chicago/Turabian StyleYang, Qingliang, Harshani Wijerathne, Jordan C. Langston, Mohammad F. Kiani, and Laurie E. Kilpatrick. 2021. "Emerging Approaches to Understanding Microvascular Endothelial Heterogeneity: A Roadmap for Developing Anti-Inflammatory Therapeutics" International Journal of Molecular Sciences 22, no. 15: 7770. https://doi.org/10.3390/ijms22157770