
LRP5 Regulates HIF-1α Stability via Interaction with PHD2 in Ischemic Myocardium

Abstract
:1. Introduction
2. Results
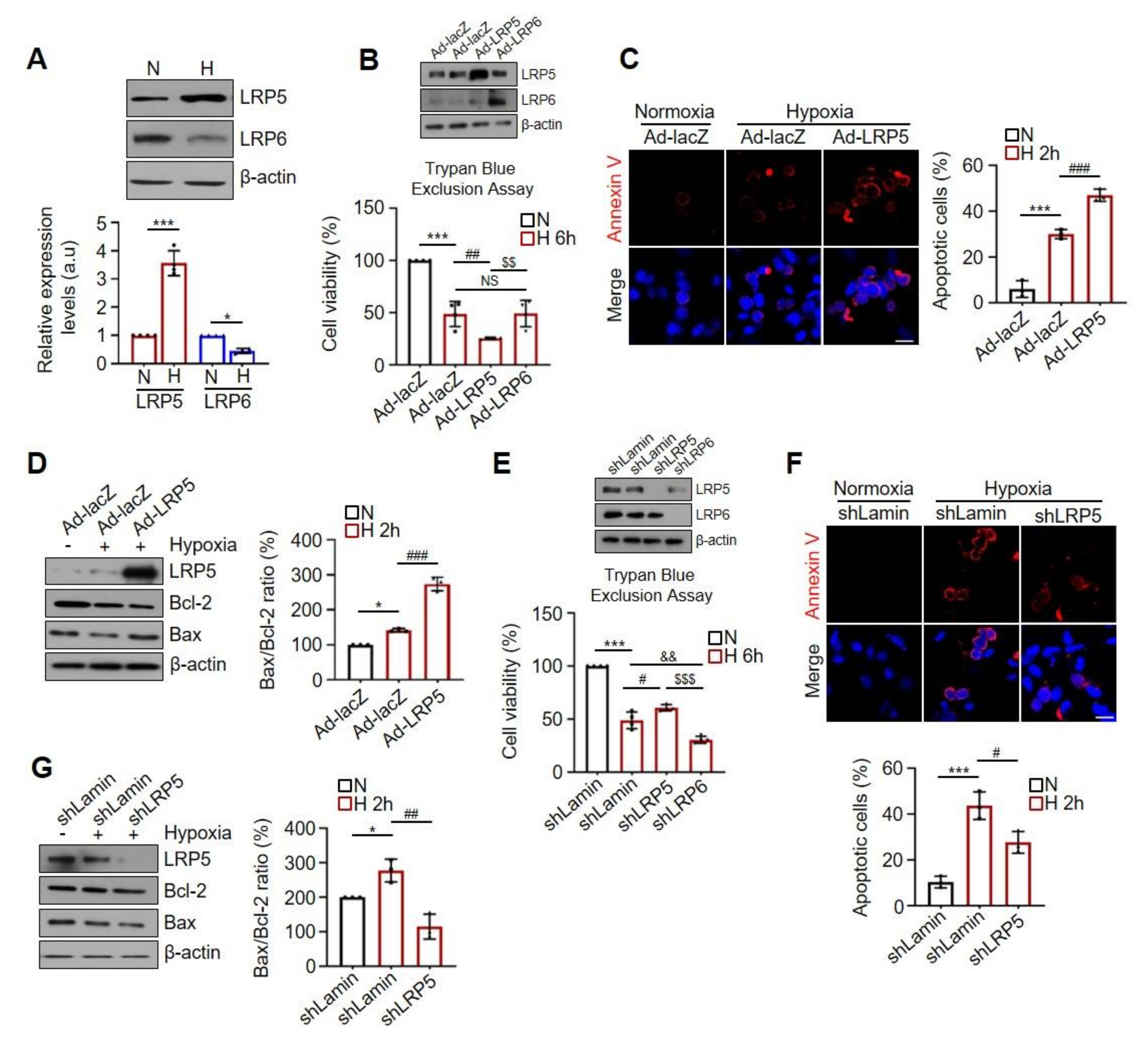
2.1. Differential Effects of LRP5 or LRP6 on Hypoxia-Induced Cardiomyocyte Death
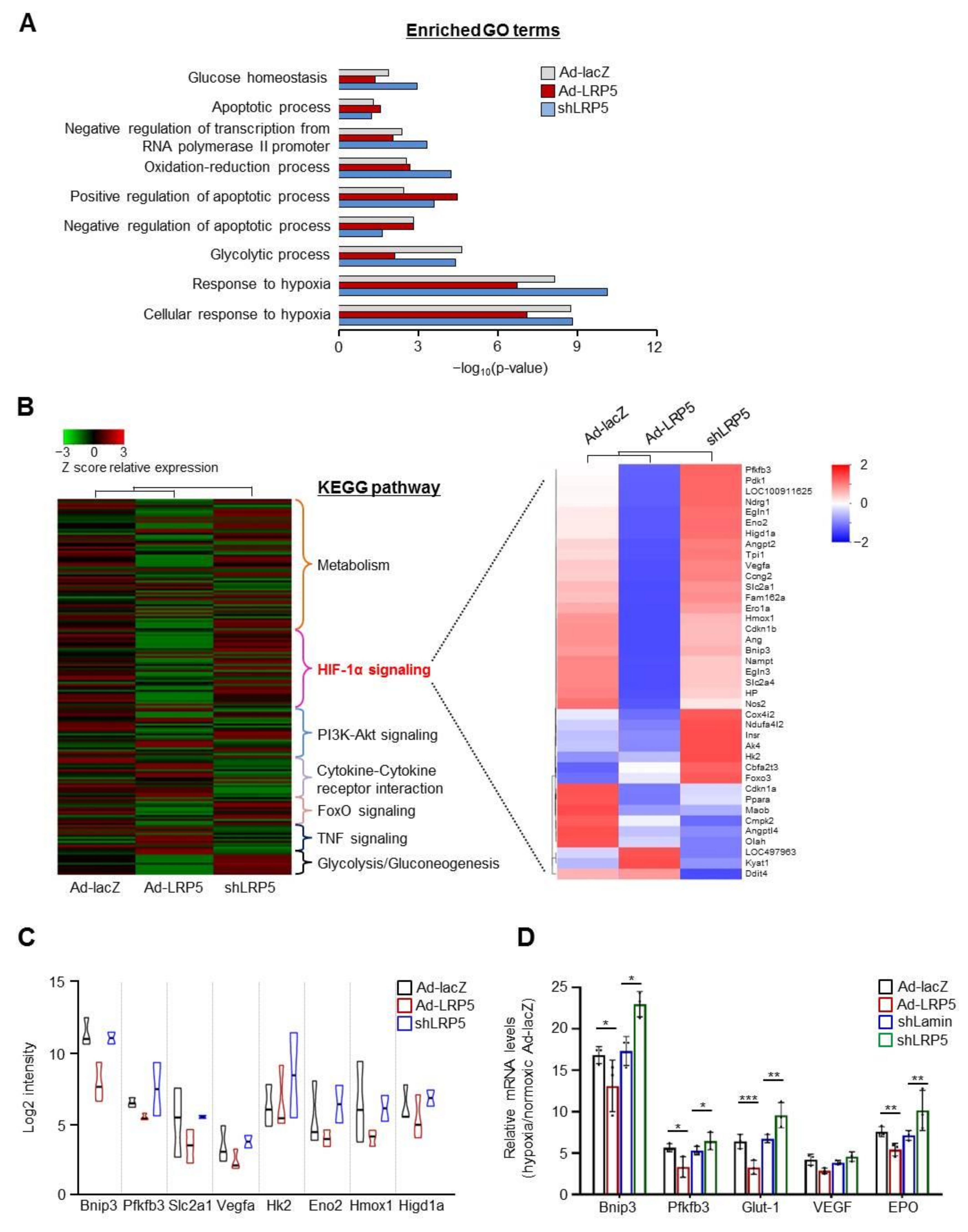
2.2. LRP5 Regulates the Expression of HIF1-α Transcriptional Target Genes
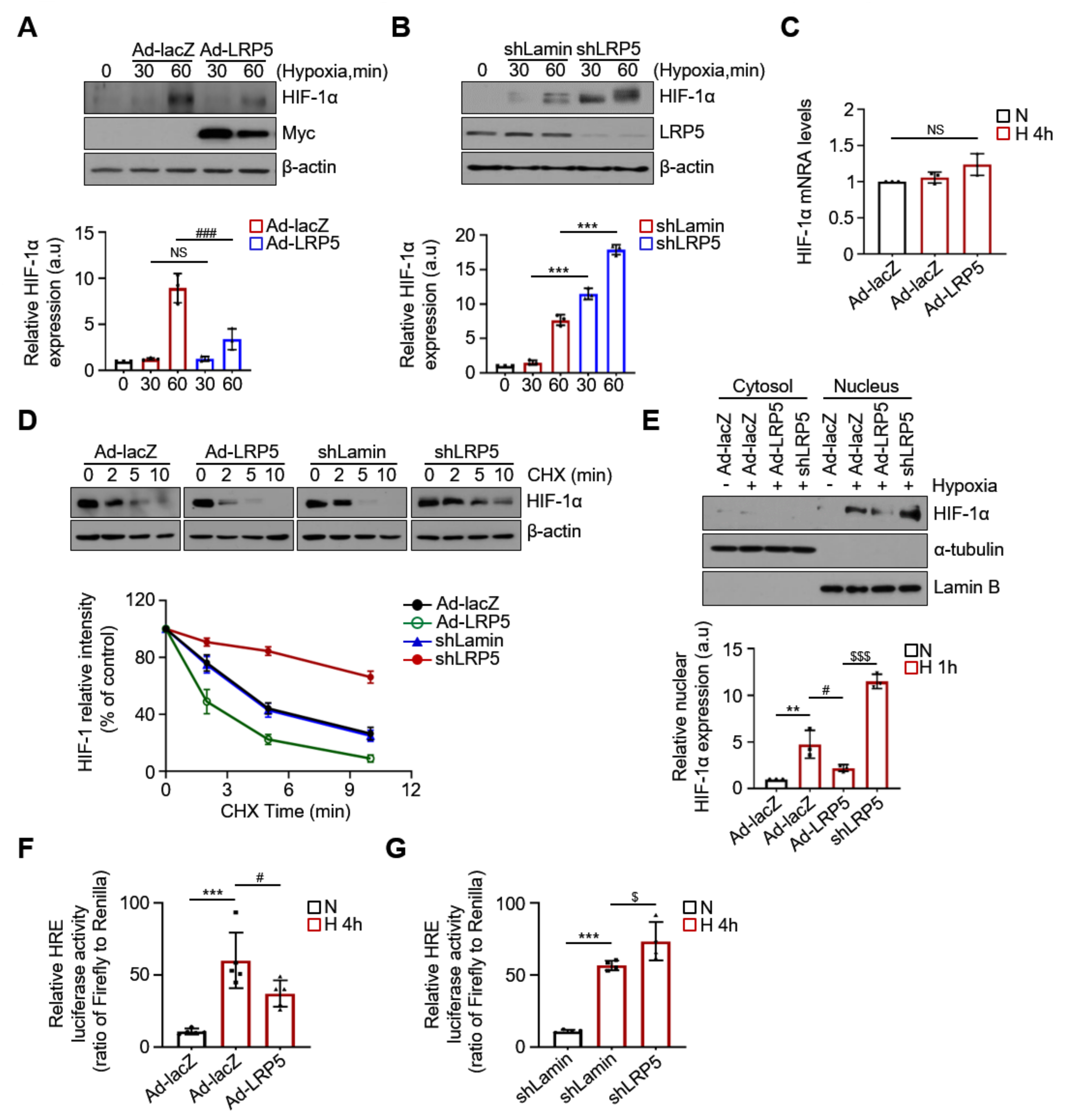
2.3. LRP5 Aggravates HIF-1α Stability in Hypoxic Cardiomyocytes
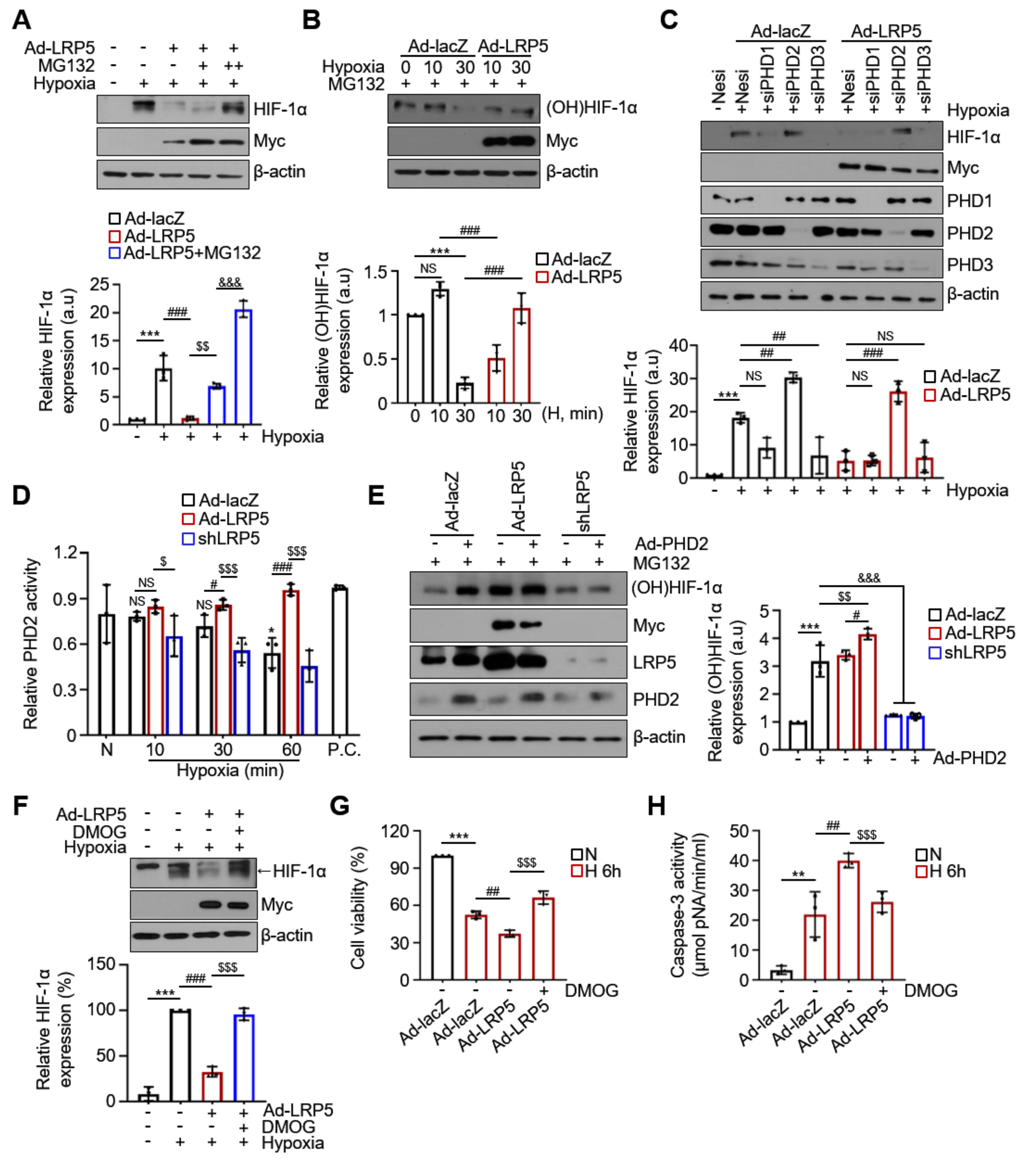
2.4. LRP5 Enhances Hydroxylation of HIF-1α
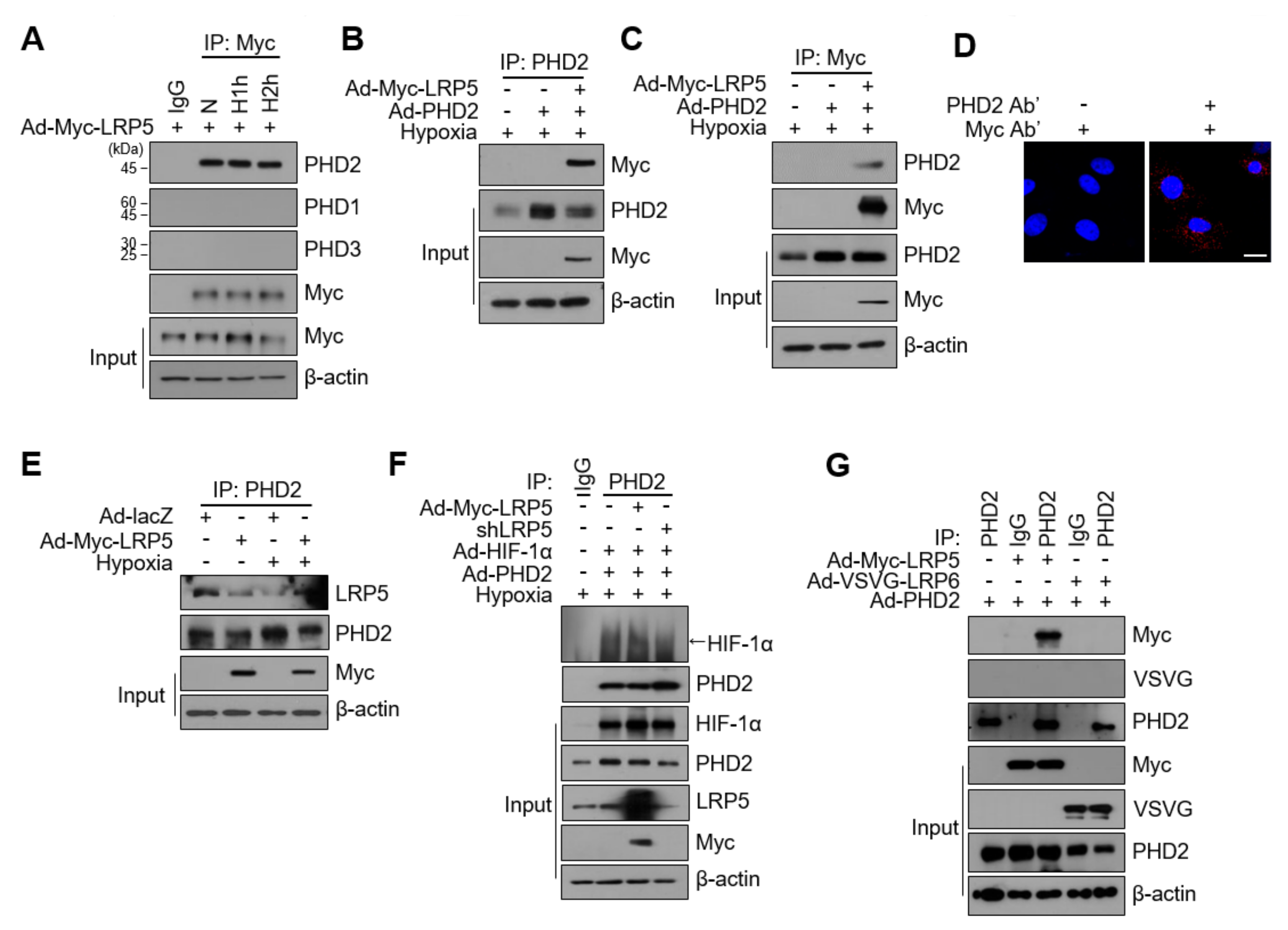
2.5. LRP5 Interacts with PHD2 in Cardiomyocytes
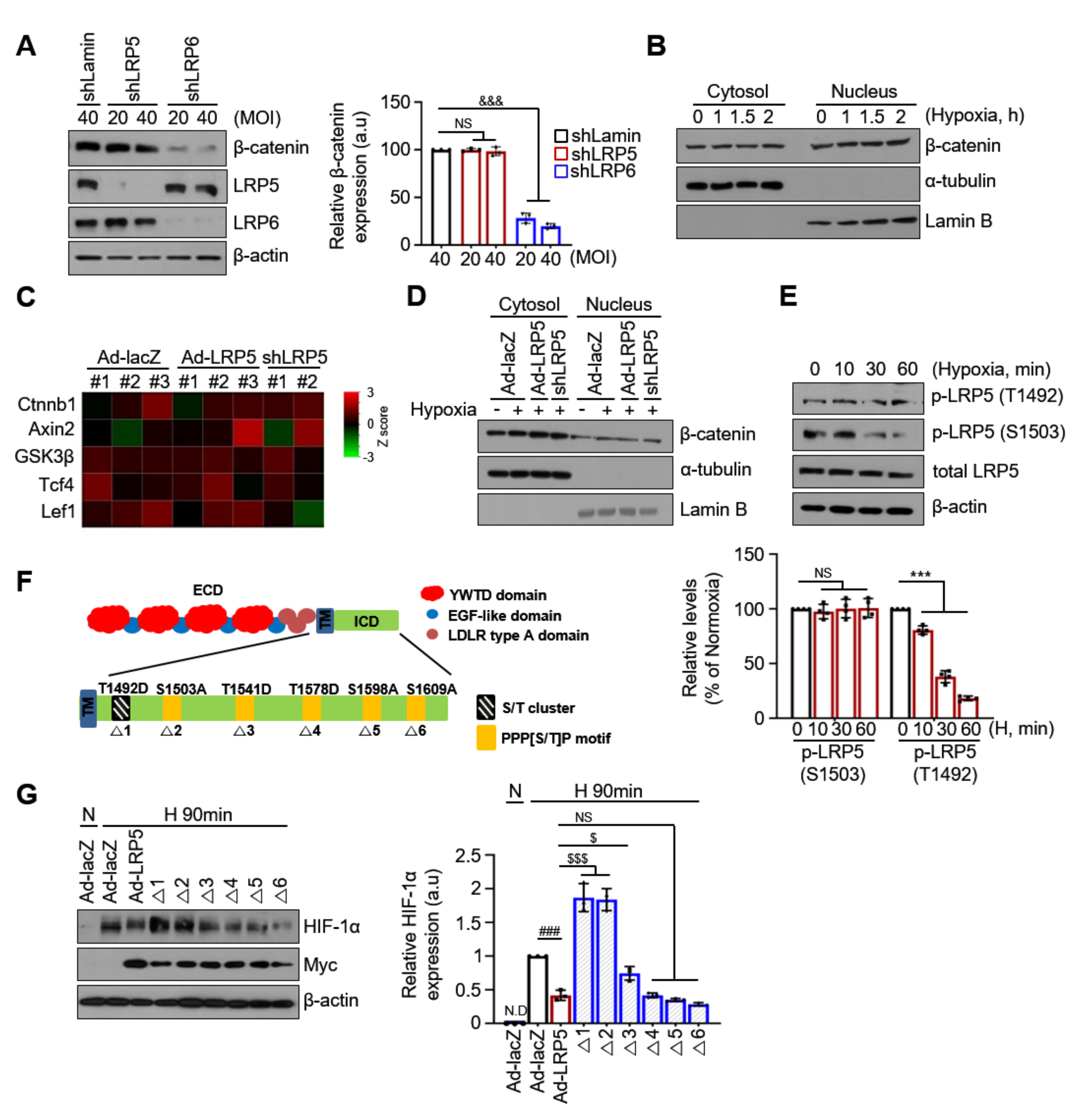
2.6. The Regulation of HIF-1α Stability by LRP5 Is Independent of Wnt/β-catenin Signaling
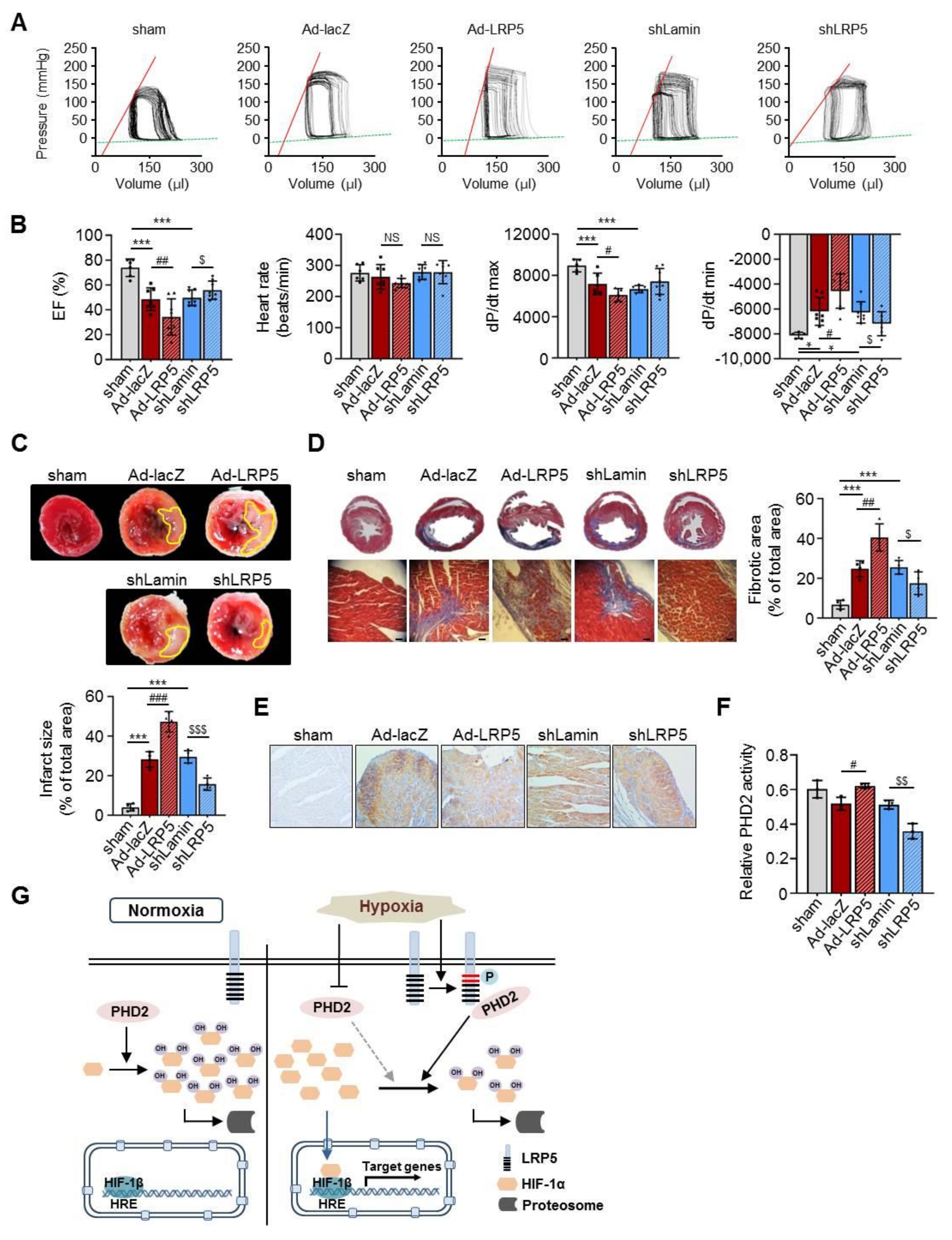
2.7. LRP5 Silencing Protects Ischemia/Reperfusion Injury In Vivo
3. Discussion
4. Materials and Methods
4.1. Primary Culture of Rat Cardiomyocytes
4.2. Production, Purification, and Administration of the Adenoviruses
4.3. siRNA Transfection
4.4. Hypoxia Treatment
4.5. Cell Viability
4.6. Luciferase Reporter Assay
4.7. Nuclear Extract Preparation
4.8. Apoptosis Assay
4.9. PHD2 Activity Assay
4.10. Proximity Ligation Assay
4.11. Immunoprecipitation Assay and Western Blotting
4.12. Gene Expression Analysis and Heat Map
4.13. Quantitative Real-Time PCR (RT-qPCR) Analysis
4.14. Immunofluorescence Staining
4.15. Animal Models of Ischemia/Reperfusion Injury
4.16. X-gal Staining of Tissues
4.17. Histological Analysis and Immunohistochemistry
4.18. Infarct Size Determination
4.19. Hemodynamic Analysis
4.20. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| Bnip3l | BCL2/adenovirus E1B interacting protein 3-like |
| BSA | Bovine serum albumin |
| CAD | C-terminal transactivation domain |
| CHX | Cycloheximide |
| Cx43 | Connexin 43 |
| DMOG | Dimethyloxalylglycine |
| EPO | Erythropoietin |
| FBS | Fetal bovine serum |
| FIH | Factor inhibiting HIF |
| Glut-1 | Glucose transporter-1 |
| GSK3β | Glycogen synthase kinase 3β |
| HIF-1α | Hypoxia-inducible factor-1α |
| HREs | Hypoxia-response elements |
| ICD | Intracellular domain |
| I/R | Ischemia/reperfusion |
| LDLR | Low-density lipoprotein receptor |
| LRP5 | Low-density lipoprotein receptor-related protein 5 |
| MI | Myocardial infarction |
| PHD2 | Prolyl hydroxylase 2 |
| PMSF | Phenylmethylsulfonyl fluoride |
| TCF | T-cell factor |
| α-MEM | Alpha-minimum essential medium |
References
- Semenza, G.L. Hypoxia-inducible factor 1: Master regulator of O2 homeostasis. Curr. Opin. Genet. Dev. 1998, 8, 588–594. [Google Scholar] [CrossRef]
- Harris, A.L. Hypoxia—A key regulatory factor in tumour growth. Nat. Rev. Cancer 2002, 2, 38–47. [Google Scholar] [CrossRef] [PubMed]
- Kumar, H.; Choi, D.K. Hypoxia Inducible Factor Pathway and Physiological Adaptation: A Cell Survival Pathway? Mediat. Inflamm. 2015, 2015, 584758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Semenza, G.L. HIF-1 and mechanisms of hypoxia sensing. Curr. Opin. Cell Biol. 2001, 13, 167–171. [Google Scholar] [CrossRef]
- Epstein, A.C.; Gleadle, J.M.; McNeill, L.A.; Hewitson, K.S.; O’Rourke, J.; Mole, D.R.; Mukherji, M.; Metzen, E.; Wilson, M.I.; Dhanda, A.; et al. C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 2001, 107, 43–54. [Google Scholar] [CrossRef] [Green Version]
- Maxwell, P.H.; Wiesener, M.S.; Chang, G.W.; Clifford, S.C.; Vaux, E.C.; Cockman, M.E.; Wykoff, C.C.; Pugh, C.W.; Maher, E.R.; Ratcliffe, P.J. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999, 399, 271–275. [Google Scholar] [CrossRef]
- Lando, D.; Peet, D.J.; Whelan, D.A.; Gorman, J.J.; Whitelaw, M.L. Asparagine hydroxylation of the HIF transactivation domain a hypoxic switch. Science 2002, 295, 858–861. [Google Scholar] [CrossRef]
- Hewitson, K.S.; McNeill, L.A.; Riordan, M.V.; Tian, Y.M.; Bullock, A.N.; Welford, R.W.; Elkins, J.M.; Oldham, N.J.; Bhattacharya, S.; Gleadle, J.M.; et al. Hypoxia-inducible factor (HIF) asparagine hydroxylase is identical to factor inhibiting HIF (FIH) and is related to the cupin structural family. J. Biol. Chem. 2002, 277, 26351–26355. [Google Scholar] [CrossRef] [Green Version]
- Mahon, P.C.; Hirota, K.; Semenza, G.L. FIH-1: A novel protein that interacts with HIF-1alpha and VHL to mediate repression of HIF-1 transcriptional activity. Genes Dev. 2001, 15, 2675–2686. [Google Scholar] [CrossRef] [Green Version]
- Schofield, C.J.; Ratcliffe, P.J. Oxygen sensing by HIF hydroxylases. Nat. Rev. Mol. Cell Biol. 2004, 5, 343–354. [Google Scholar] [CrossRef]
- Wenger, R.H. Cellular adaptation to hypoxia: O2-sensing protein hydroxylases, hypoxia-inducible transcription factors, and O2-regulated gene expression. FASEB J. 2002, 16, 1151–1162. [Google Scholar] [CrossRef] [Green Version]
- Eckle, T.; Kohler, D.; Lehmann, R.; El Kasmi, K.; Eltzschig, H.K. Hypoxia-inducible factor-1 is central to cardioprotection: A new paradigm for ischemic preconditioning. Circulation 2008, 118, 166–175. [Google Scholar] [CrossRef] [Green Version]
- Natarajan, R.; Salloum, F.N.; Fisher, B.J.; Kukreja, R.C.; Fowler, A.A., 3rd. Hypoxia inducible factor-1 activation by prolyl 4-hydroxylase-2 gene silencing attenuates myocardial ischemia reperfusion injury. Circ. Res. 2006, 98, 133–140. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.E.; Yang, D.; Li, L.; Wang, W.; Peng, Y.; Chen, C.; Chen, P.; Xia, X.; Wang, H.; Jiang, J.; et al. Prolyl hydroxylase domain protein 2 silencing enhances the survival and paracrine function of transplanted adipose-derived stem cells in infarcted myocardium. Circ. Res. 2013, 113, 288–300. [Google Scholar] [CrossRef]
- Pinson, K.I.; Brennan, J.; Monkley, S.; Avery, B.J.; Skarnes, W.C. An LDL-receptor-related protein mediates Wnt signalling in mice. Nature 2000, 407, 535–538. [Google Scholar] [CrossRef]
- Tamai, K.; Semenov, M.; Kato, Y.; Spokony, R.; Liu, C.; Katsuyama, Y.; Hess, F.; Saint-Jeannet, J.P.; He, X. LDL-receptor-related proteins in Wnt signal transduction. Nature 2000, 407, 530–535. [Google Scholar] [CrossRef]
- Laine, C.M.; Chung, B.D.; Susic, M.; Prescott, T.; Semler, O.; Fiskerstrand, T.; D’Eufemia, P.; Castori, M.; Pekkinen, M.; Sochett, E.; et al. Novel mutations affecting LRP5 splicing in patients with osteoporosis-pseudoglioma syndrome (OPPG). Eur. J. Hum. Genet. 2011, 19, 875–881. [Google Scholar] [CrossRef] [Green Version]
- Bjorklund, P.; Svedlund, J.; Olsson, A.K.; Akerstrom, G.; Westin, G. The internally truncated LRP5 receptor presents a therapeutic target in breast cancer. PLoS ONE 2009, 4, e4243. [Google Scholar] [CrossRef]
- Li, Y.; Lu, W.; He, X.; Schwartz, A.L.; Bu, G. LRP6 expression promotes cancer cell proliferation and tumorigenesis by altering beta-catenin subcellular distribution. Oncogene 2004, 23, 9129–9135. [Google Scholar] [CrossRef] [Green Version]
- Borrell-Pages, M.; Romero, J.C.; Badimon, L. LRP5 deficiency down-regulates Wnt signalling and promotes aortic lipid infiltration in hypercholesterolaemic mice. J. Cell. Mol. Med. 2015, 19, 770–777. [Google Scholar] [CrossRef]
- Alfaro, M.P.; Pagni, M.; Vincent, A.; Atkinson, J.; Hill, M.F.; Cates, J.; Davidson, J.M.; Rottman, J.; Lee, E.; Young, P.P. The Wnt modulator sFRP2 enhances mesenchymal stem cell engraftment, granulation tissue formation and myocardial repair. Proc. Natl. Acad. Sci. USA 2008, 105, 18366–18371. [Google Scholar] [CrossRef] [Green Version]
- Barandon, L.; Casassus, F.; Leroux, L.; Moreau, C.; Allieres, C.; Lamaziere, J.M.; Dufourcq, P.; Couffinhal, T.; Duplaa, C. Secreted frizzled-related protein-1 improves postinfarction scar formation through a modulation of inflammatory response. Arterioscler. Thromb. Vasc. Biol. 2011, 31, e80–e87. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; Deb, A.; Zhang, Z.; Pachori, A.; He, W.; Guo, J.; Pratt, R.; Dzau, V.J. Secreted frizzled related protein 2 protects cells from apoptosis by blocking the effect of canonical Wnt3a. J. Mol. Cell. Cardiol. 2009, 46, 370–377. [Google Scholar] [CrossRef] [Green Version]
- Saraswati, S.; Alfaro, M.P.; Thorne, C.A.; Atkinson, J.; Lee, E.; Young, P.P. Pyrvinium, a potent small molecule Wnt inhibitor, promotes wound repair and post-MI cardiac remodeling. PLoS ONE 2010, 5, e15521. [Google Scholar] [CrossRef] [Green Version]
- Wo, D.; Peng, J.; Ren, D.N.; Qiu, L.; Chen, J.; Zhu, Y.; Yan, Y.; Yan, H.; Wu, J.; Ma, E.; et al. Opposing Roles of Wnt Inhibitors IGFBP-4 and Dkk1 in Cardiac Ischemia by Differential Targeting of LRP5/6 and beta-catenin. Circulation 2016, 134, 1991–2007. [Google Scholar] [CrossRef]
- Abe, T.; Zhou, P.; Jackman, K.; Capone, C.; Casolla, B.; Hochrainer, K.; Kahles, T.; Ross, M.E.; Anrather, J.; Iadecola, C. Lipoprotein receptor-related protein-6 protects the brain from ischemic injury. Stroke 2013, 44, 2284–2291. [Google Scholar] [CrossRef] [Green Version]
- Borrell-Pages, M.; Vilahur, G.; Romero, J.C.; Casani, L.; Bejar, M.T.; Badimon, L. LRP5/canonical Wnt signalling and healing of ischemic myocardium. Basic Res. Cardiol. 2016, 111, 67. [Google Scholar] [CrossRef]
- MacDonald, B.T.; Semenov, M.V.; Huang, H.; He, X. Dissecting molecular differences between Wnt coreceptors LRP5 and LRP6. PLoS ONE 2011, 6, e23537. [Google Scholar] [CrossRef]
- MacDonald, B.T.; Tamai, K.; He, X. Wnt/beta-catenin signaling: Components, mechanisms, and diseases. Dev. Cell 2009, 17, 9–26. [Google Scholar] [CrossRef] [Green Version]
- Chin, E.N.; Martin, J.A.; Kim, S.; Fakhraldeen, S.A.; Alexander, C.M. Lrp5 Has a Wnt-Independent Role in Glucose Uptake and Growth for Mammary Epithelial Cells. Mol. Cell. Biol. 2015, 36, 871–885. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Li, C.; Liang, D.; Lv, F.; Yuan, T.; The, E.; Ma, X.; Wu, Y.; Zhen, L.; Xie, D.; et al. LRP6 acts as a scaffold protein in cardiac gap junction assembly. Nat. Commun. 2016, 7, 11775. [Google Scholar] [CrossRef] [PubMed]
- Fujino, T.; Asaba, H.; Kang, M.J.; Ikeda, Y.; Sone, H.; Takada, S.; Kim, D.H.; Ioka, R.X.; Ono, M.; Tomoyori, H.; et al. Low-density lipoprotein receptor-related protein 5 (LRP5) is essential for normal cholesterol metabolism and glucose-induced insulin secretion. Proc. Natl. Acad. Sci. USA 2003, 100, 229–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clevers, H.; Nusse, R. Wnt/beta-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Freeman, R.S.; Hasbani, D.M.; Lipscomb, E.A.; Straub, J.A.; Xie, L. SM-20, EGL-9, and the EGLN family of hypoxia-inducible factor prolyl hydroxylases. Mol. Cells 2003, 16, 1–12. [Google Scholar]
- Villar, D.; Vara-Vega, A.; Landazuri, M.O.; Del Peso, L. Identification of a region on hypoxia-inducible-factor prolyl 4-hydroxylases that determines their specificity for the oxygen degradation domains. Biochem. J. 2007, 408, 231–240. [Google Scholar] [CrossRef] [Green Version]
- Berra, E.; Benizri, E.; Ginouves, A.; Volmat, V.; Roux, D.; Pouyssegur, J. HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1alpha in normoxia. EMBO J. 2003, 22, 4082–4090. [Google Scholar] [CrossRef] [Green Version]
- Daskalopoulos, E.P.; Hermans, K.C.; Janssen, B.J.; Blankesteijn, W.M. Targeting the Wnt/frizzled signaling pathway after myocardial infarction: A new tool in the therapeutic toolbox? Trends Cardiovasc. Med. 2013, 23, 121–127. [Google Scholar] [CrossRef]
- Aisagbonhi, O.; Rai, M.; Ryzhov, S.; Atria, N.; Feoktistov, I.; Hatzopoulos, A.K. Experimental myocardial infarction triggers canonical Wnt signaling and endothelial-to-mesenchymal transition. Dis. Models Mech. 2011, 4, 469–483. [Google Scholar] [CrossRef] [Green Version]
- Paik, D.T.; Rai, M.; Ryzhov, S.; Sanders, L.N.; Aisagbonhi, O.; Funke, M.J.; Feoktistov, I.; Hatzopoulos, A.K. Wnt10b Gain-of-Function Improves Cardiac Repair by Arteriole Formation and Attenuation of Fibrosis. Circ. Res. 2015, 117, 804–816. [Google Scholar] [CrossRef] [Green Version]
- Zelarayan, L.C.; Noack, C.; Sekkali, B.; Kmecova, J.; Gehrke, C.; Renger, A.; Zafiriou, M.P.; van der Nagel, R.; Dietz, R.; de Windt, L.J.; et al. Beta-Catenin downregulation attenuates ischemic cardiac remodeling through enhanced resident precursor cell differentiation. Proc. Natl. Acad. Sci. USA 2008, 105, 19762–19767. [Google Scholar] [CrossRef] [Green Version]
- van de Schans, V.A.; van den Borne, S.W.; Strzelecka, A.E.; Janssen, B.J.; van der Velden, J.L.; Langen, R.C.; Wynshaw-Boris, A.; Smits, J.F.; Blankesteijn, W.M. Interruption of Wnt signaling attenuates the onset of pressure overload-induced cardiac hypertrophy. Hypertension 2007, 49, 473–480. [Google Scholar] [CrossRef] [Green Version]
- Woulfe, K.C.; Gao, E.; Lal, H.; Harris, D.; Fan, Q.; Vagnozzi, R.; DeCaul, M.; Shang, X.; Patel, S.; Woodgett, J.R.; et al. Glycogen synthase kinase-3beta regulates post-myocardial infarction remodeling and stress-induced cardiomyocyte proliferation in vivo. Circ. Res. 2010, 106, 1635–1645. [Google Scholar] [CrossRef]
- Min, J.K.; Park, H.; Choi, H.J.; Kim, Y.; Pyun, B.J.; Agrawal, V.; Song, B.W.; Jeon, J.; Maeng, Y.S.; Rho, S.S.; et al. The WNT antagonist Dickkopf2 promotes angiogenesis in rodent and human endothelial cells. J. Clin. Investig. 2011, 121, 1882–1893. [Google Scholar] [CrossRef] [Green Version]
- Zhong, Z.; Baker, J.J.; Zylstra-Diegel, C.R.; Williams, B.O. Lrp5 and Lrp6 play compensatory roles in mouse intestinal development. J. Cell. Biochem. 2012, 113, 31–38. [Google Scholar] [CrossRef] [Green Version]
- Joeng, K.S.; Schumacher, C.A.; Zylstra-Diegel, C.R.; Long, F.; Williams, B.O. Lrp5 and Lrp6 redundantly control skeletal development in the mouse embryo. Dev. Biol. 2011, 359, 222–229. [Google Scholar] [CrossRef] [Green Version]
- Mi, K.; Johnson, G.V. Role of the intracellular domains of LRP5 and LRP6 in activating the Wnt canonical pathway. J. Cell. Biochem. 2005, 95, 328–338. [Google Scholar] [CrossRef]
- Holmen, S.L.; Giambernardi, T.A.; Zylstra, C.R.; Buckner-Berghuis, B.D.; Resau, J.H.; Hess, J.F.; Glatt, V.; Bouxsein, M.L.; Ai, M.; Warman, M.L.; et al. Decreased BMD and limb deformities in mice carrying mutations in both Lrp5 and Lrp6. J. Am. Soc. Bone Miner. Res. 2004, 19, 2033–2040. [Google Scholar] [CrossRef]
- Joiner, D.M.; Ke, J.; Zhong, Z.; Xu, H.E.; Williams, B.O. LRP5 and LRP6 in development and disease. Trends Endocrinol. Metab. TEM 2013, 24, 31–39. [Google Scholar] [CrossRef] [Green Version]
- Kelly, O.G.; Pinson, K.I.; Skarnes, W.C. The Wnt co-receptors Lrp5 and Lrp6 are essential for gastrulation in mice. Development 2004, 131, 2803–2815. [Google Scholar] [CrossRef] [Green Version]
- Appelhoff, R.J.; Tian, Y.M.; Raval, R.R.; Turley, H.; Harris, A.L.; Pugh, C.W.; Ratcliffe, P.J.; Gleadle, J.M. Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J. Biol. Chem. 2004, 279, 38458–38465. [Google Scholar] [CrossRef] [Green Version]
- Serocki, M.; Bartoszewska, S.; Janaszak-Jasiecka, A.; Ochocka, R.J.; Collawn, J.F.; Bartoszewski, R. miRNAs regulate the HIF switch during hypoxia: A novel therapeutic target. Angiogenesis 2018, 21, 183–202. [Google Scholar] [CrossRef] [Green Version]
- Holmquist, L.; Jögi, A.; Påhlman, S. Phenotypic persistence after reoxygenation of hypoxic neuroblastoma cells. Int. J. Cancer 2005, 116, 218–225. [Google Scholar] [CrossRef]
- Holmquist-Mengelbier, L.; Fredlund, E.; Löfstedt, T.; Noguera, R.; Navarro, S.; Nilsson, H.; Pietras, A.; Vallon-Christersson, J.; Borg, A.; Gradin, K.; et al. Recruitment of HIF-1alpha and HIF-2alpha to common target genes is differentially regulated in neuroblastoma: HIF-2alpha promotes an aggressive phenotype. Cancer Cell 2006, 10, 413–423. [Google Scholar] [CrossRef] [Green Version]
- Bartoszewski, R.; Moszyńska, A.; Serocki, M.; Cabaj, A.; Polten, A.; Ochocka, R.; Dell’Italia, L.; Bartoszewska, S.; Króliczewski, J.; Dąbrowski, M.; et al. Primary endothelial cell-specific regulation of hypoxia-inducible factor (HIF)-1 and HIF-2 and their target gene expression profiles during hypoxia. FASEB J. 2019, 33, 7929–7941. [Google Scholar] [CrossRef]
- Koh, M.Y.; Lemos, R., Jr.; Liu, X.; Powis, G. The hypoxia-associated factor switches cells from HIF-1α- to HIF-2α-dependent signaling promoting stem cell characteristics, aggressive tumor growth and invasion. Cancer Res. 2011, 71, 4015–4027. [Google Scholar] [CrossRef] [Green Version]
- Lin, Q.; Cong, X.; Yun, Z. Differential hypoxic regulation of hypoxia-inducible factors 1alpha and 2alpha. Mol. Cancer Res. 2011, 9, 757–765. [Google Scholar] [CrossRef] [Green Version]
- Uchida, T.; Rossignol, F.; Matthay, M.A.; Mounier, R.; Couette, S.; Clottes, E.; Clerici, C. Prolonged hypoxia differentially regulates hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha expression in lung epithelial cells: Implication of natural antisense HIF-1alpha. J. Biol. Chem. 2004, 279, 14871–14878. [Google Scholar] [CrossRef] [Green Version]
- Oerlemans, M.I.; Goumans, M.J.; van Middelaar, B.; Clevers, H.; Doevendans, P.A.; Sluijter, J.P. Active Wnt signaling in response to cardiac injury. Basic Res. Cardiol. 2010, 105, 631–641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Staal, F.J.; Luis, T.C.; Tiemessen, M.M. WNT signalling in the immune system: WNT is spreading its wings. Nat. Rev. Immunol. 2008, 8, 581–593. [Google Scholar] [CrossRef] [PubMed]
- Holdstock, L.; Meadowcroft, A.M.; Maier, R.; Johnson, B.M.; Jones, D.; Rastogi, A.; Zeig, S.; Lepore, J.J.; Cobitz, A.R. Four-Week Studies of Oral Hypoxia-Inducible Factor-Prolyl Hydroxylase Inhibitor GSK1278863 for Treatment of Anemia. J. Am. Soc. Nephrol. 2016, 27, 1234–1244. [Google Scholar] [CrossRef] [PubMed]
- Ju, S.; Park, S.; Lim, L.; Choi, D.H.; Song, H. Low density lipoprotein receptor-related protein 1 regulates cardiac hypertrophy induced by pressure overload. Int. J. Cardiol. 2020, 299, 235–242. [Google Scholar] [CrossRef]
- Chang, W.; Song, B.W.; Lim, S.; Song, H.; Shim, C.Y.; Cha, M.J.; Ahn, D.H.; Jung, Y.G.; Lee, D.H.; Chung, J.H.; et al. Mesenchymal stem cells pretreated with delivered Hph-1-Hsp70 protein are protected from hypoxia-mediated cell death and rescue heart functions from myocardial injury. Stem Cells 2009, 27, 2283–2292. [Google Scholar] [CrossRef]
- Song, H.; Song, B.W.; Cha, M.J.; Choi, I.G.; Hwang, K.C. Modification of mesenchymal stem cells for cardiac regeneration. Expert Opin. Biol. Ther. 2010, 10, 309–319. [Google Scholar] [CrossRef]
- Pfaffl, M.W. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001, 29, e45. [Google Scholar] [CrossRef]
- Song, H.; Hwang, H.J.; Chang, W.; Song, B.W.; Cha, M.J.; Kim, I.K.; Lim, S.; Choi, E.J.; Ham, O.; Lee, C.Y.; et al. Cardiomyocytes from phorbol myristate acetate-activated mesenchymal stem cells restore electromechanical function in infarcted rat hearts. Proc. Natl. Acad. Sci. USA 2011, 108, 296–301. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| qPCR Primers | ||
|---|---|---|
| Gene | Forward (5′–3′) | Reverse (5′-3′) |
| LRP5 | AGGCCCTACATCATTCGAGG | GGGGTCTGAGTCCGAATTCA |
| BNIP3 | CTGCACTTCAGCAATGGG | CTCTTGGAGCTGCTTCGT |
| PFKFB3 | CAGTCCTGAAACTGACGC | GACAGCCTCTGACCTCTC |
| GLUT-1 | GCCTGAGACCAGTTGAAAGCAC | CTGCTTAGGTAAAGTTACAGGAG |
| VEGF | TTACTGCTGTACCTCCACC | ACAGGACGGCTTGAAGATG |
| EPO | AGGGTCACGAAGCCATGAAG | GATTTCGGCTGTTGCCAGTG |
| HIF-1α | GGTGGATATGTCTGGGTTGAG | TTCAACTGGTTTGAGGACAGA |
| β-CATENIN | AGGGGTCCTCTGTGAACTTG | CAGCAGTCTCATTCCAAGCC |
| WNT3a | ATTTGGAGGAATGGTCTCTCG | GCAGGTCTTCACTTCGCAAC |
| AXIN2 | CGCTAGGCGGAATGAAGA | GTATGCACCATCTTGGTC |
| GAPDH | CAGTGCCAGCCTCGTCTCAT | TGGTAACCAGGCGTCCGATG |
| β-ACTIN | AGGGAAATCGTGCGTGAC | CGCTCATTGCCGATAGTG |
| α-TUBULIN | GGTTGAGCCCTACAATTCC | CAATGTCGAGGTTTCTACG |
| Site-Directed Mutagenesis | ||
| Mutants | Forward (5′–3′) | Reverse (5′-3′) |
| T1492D | TCCAGCACGAAGGCCgacCTGTACCCGCCGATC | GATCGGCGGGTACAGgtcGGCCTTCGTGCTGGA |
| S1503A | ATCCTGAACCCGCCGCCCgCCCCGGCCACGGACCCCT | AGGGGTCCGTGGCCGGGGcGGGCGGCGGGTTCAGGAT |
| T1541D | AATGGCGCCCCCGACGgacCCCTGCAGCACCGACG | CGTCGGTGCTGCAGGGgtcCGTCGGGGGCGCCATT |
| T1578D | TATCCACCCCCACCCgacCCCCACAGCCAGTAC | GTACTGGCTGTGGGGgtcGGGTGGGGGTGGATA |
| S1598A | GGACAGCTGCCCGCCCgCGCCCGCCACCGAGAG | CTCTCGGTGGCGGGCGcGGGCGGGCAGCTGTCC |
| S1609A | CTTCCCGCCCCCTCCGgCCCCCTGCACGGACTC | GAGTCCGTGCAGGGGGcCGGAGGGGGCGGGAAG |
| Oligos for siRNA | ||
| Gene | Sequence (5′–3′) | |
| PHD1 | GCUAGCAUCGGGACAGAAAGG | |
| PHD2 | GCGGAGGUAUUCUUCGAAUUU | |
| PHD3 | GGCUGGAUCUGGAGAAGAUCG | |
| Negative control | UUCUCCGAACGUGUCACGU | |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ju, S.; Lim, L.; Wi, K.; Park, C.; Ki, Y.-J.; Choi, D.-H.; Song, H. LRP5 Regulates HIF-1α Stability via Interaction with PHD2 in Ischemic Myocardium. Int. J. Mol. Sci. 2021, 22, 6581. https://doi.org/10.3390/ijms22126581
Ju S, Lim L, Wi K, Park C, Ki Y-J, Choi D-H, Song H. LRP5 Regulates HIF-1α Stability via Interaction with PHD2 in Ischemic Myocardium. International Journal of Molecular Sciences. 2021; 22(12):6581. https://doi.org/10.3390/ijms22126581
Chicago/Turabian StyleJu, Sujin, Leejin Lim, Kwanhwan Wi, Changwon Park, Young-Jae Ki, Dong-Hyun Choi, and Heesang Song. 2021. "LRP5 Regulates HIF-1α Stability via Interaction with PHD2 in Ischemic Myocardium" International Journal of Molecular Sciences 22, no. 12: 6581. https://doi.org/10.3390/ijms22126581