
mTOR Signaling in the Inner Ear as Potential Target to Treat Hearing Loss


Abstract
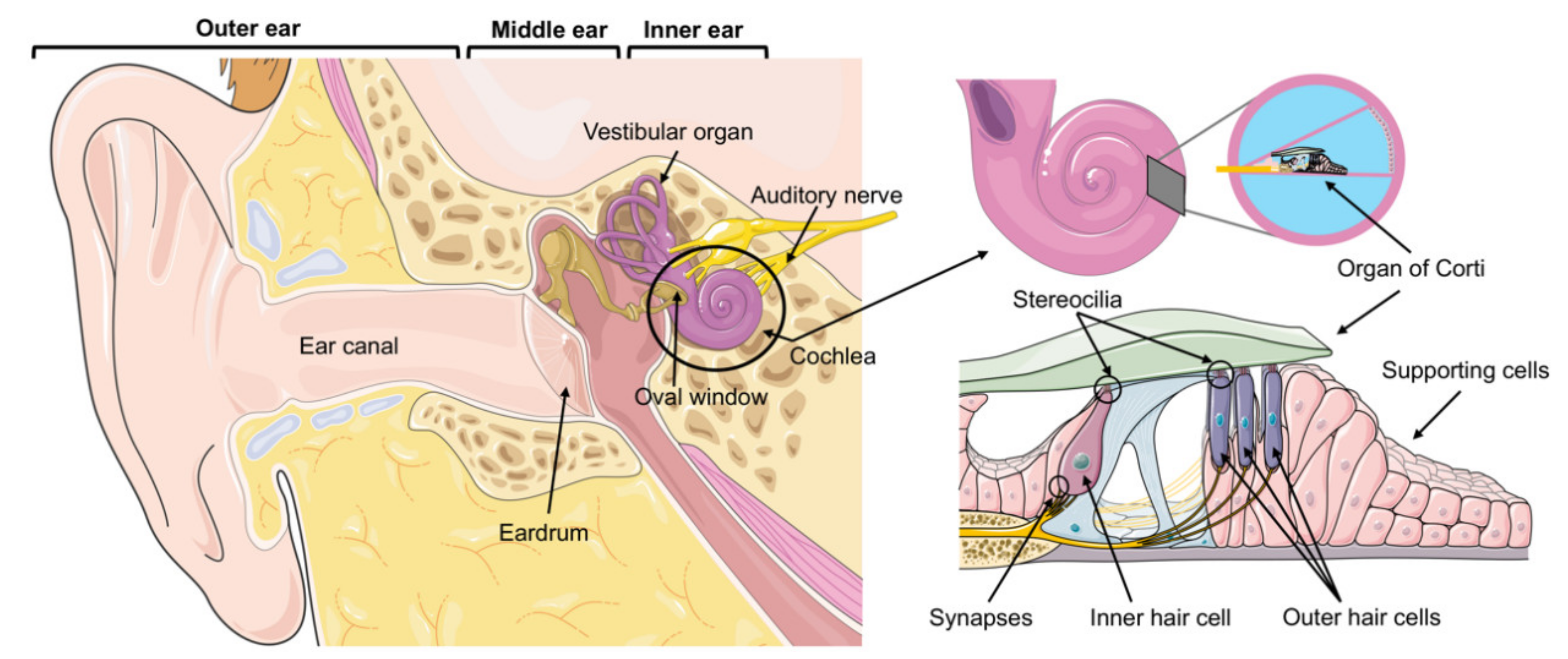
:1. Introduction
2. mTOR Signaling
2.1. mTORC1
2.2. mTORC2
2.3. Feedback Mechanisms
3. mTOR Signaling in Auditory Sensory Hair Cell Regeneration
4. mTOR Signaling in Auditory Sensory Hair Cell Survival and Death
5. Open Questions
6. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- WHO. Global Estimates on Prevalence of Hearing Loss. Available online: https://www.who.int/pbd/deafness/estimates/en/ (accessed on 16 February 2021).
- Geleoc, G.S.; Holt, J.R. Sound strategies for hearing restoration. Science 2014, 344, 1241062. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Puel, J.L. Toward Cochlear Therapies. Physiol. Rev. 2018, 98, 2477–2522. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Puel, J.L. Presbycusis: An Update on Cochlear Mechanisms and Therapies. J. Clin. Med. 2020, 9, 218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saxton, R.A.; Sabatini, D.M. mTOR Signaling in Growth, Metabolism, and Disease. Cell 2017, 169, 361–371. [Google Scholar] [CrossRef] [PubMed]
- Dazert, E.; Hall, M.N. mTOR signaling in disease. Curr. Opin. Cell Biol. 2011, 23, 744–755. [Google Scholar] [CrossRef] [PubMed]
- Heitman, J.; Movva, N.R.; Hall, M.N. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 1991, 253, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.E.; Strong, R.; Sharp, Z.D.; Nelson, J.F.; Astle, C.M.; Flurkey, K.; Nadon, N.L.; Wilkinson, J.E.; Frenkel, K.; Carter, C.S.; et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009, 460, 392–395. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, S.C.; Rabinovitch, P.S.; Kaeberlein, M. mTOR is a key modulator of ageing and age-related disease. Nature 2013, 493, 338–345. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.Y.; Sabatini, D.M. mTOR at the nexus of nutrition, growth, ageing and disease. Nat. Rev. Mol. Cell Biol. 2020, 21, 183–203. [Google Scholar] [CrossRef]
- Fu, W.; Hall, M.N. Regulation of mTORC2 Signaling. Genes 2020, 11, 1045. [Google Scholar] [CrossRef]
- Gonzalez, A.; Hall, M.N.; Lin, S.C.; Hardie, D.G. AMPK and TOR: The Yin and Yang of Cellular Nutrient Sensing and Growth Control. Cell Metab. 2020, 31, 472–492. [Google Scholar] [CrossRef]
- Aylett, C.H.; Sauer, E.; Imseng, S.; Boehringer, D.; Hall, M.N.; Ban, N.; Maier, T. Architecture of human mTOR complex 1. Science 2016, 351, 48–52. [Google Scholar] [CrossRef]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; Latek, R.R.; Guntur, K.V.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. GbetaL, a positive regulator of the rapamycin-sensitive pathway required for the nutrient-sensitive interaction between raptor and mTOR. Mol. Cell 2003, 11, 895–904. [Google Scholar] [CrossRef]
- Loewith, R.; Jacinto, E.; Wullschleger, S.; Lorberg, A.; Crespo, J.L.; Bonenfant, D.; Oppliger, W.; Jenoe, P.; Hall, M.N. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol. Cell 2002, 10, 457–468. [Google Scholar] [CrossRef]
- Kim, D.H.; Sarbassov, D.D.; Ali, S.M.; King, J.E.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 2002, 110, 163–175. [Google Scholar] [CrossRef] [Green Version]
- Bierer, B.E.; Mattila, P.S.; Standaert, R.F.; Herzenberg, L.A.; Burakoff, S.J.; Crabtree, G.; Schreiber, S.L. Two distinct signal transmission pathways in T lymphocytes are inhibited by complexes formed between an immunophilin and either FK506 or rapamycin. Proc. Natl. Acad. Sci. USA 1990, 87, 9231–9235. [Google Scholar] [CrossRef] [Green Version]
- Benjamin, D.; Colombi, M.; Moroni, C.; Hall, M.N. Rapamycin passes the torch: A new generation of mTOR inhibitors. Nat. Rev. Drug Discov. 2011, 10, 868–880. [Google Scholar] [CrossRef]
- Schalm, S.S.; Blenis, J. Identification of a conserved motif required for mTOR signaling. Curr. Biol. 2002, 12, 632–639. [Google Scholar] [CrossRef] [Green Version]
- Schalm, S.S.; Fingar, D.C.; Sabatini, D.M.; Blenis, J. TOS motif-mediated raptor binding regulates 4E-BP1 multisite phosphorylation and function. Curr. Biol. 2003, 13, 797–806. [Google Scholar] [CrossRef] [Green Version]
- Dibble, C.C.; Cantley, L.C. Regulation of mTORC1 by PI3K signaling. Trends Cell Biol. 2015, 25, 545–555. [Google Scholar] [CrossRef] [Green Version]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inoki, K.; Li, Y.; Zhu, T.; Wu, J.; Guan, K.L. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat. Cell Biol. 2002, 4, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Manning, B.D.; Tee, A.R.; Logsdon, M.N.; Blenis, J.; Cantley, L.C. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol. Cell 2002, 10, 151–162. [Google Scholar] [CrossRef]
- Potter, C.J.; Pedraza, L.G.; Xu, T. Akt regulates growth by directly phosphorylating Tsc2. Nat. Cell Biol. 2002, 4, 658–665. [Google Scholar] [CrossRef]
- Dibble, C.C.; Elis, W.; Menon, S.; Qin, W.; Klekota, J.; Asara, J.M.; Finan, P.M.; Kwiatkowski, D.J.; Murphy, L.O.; Manning, B.D. TBC1D7 is a third subunit of the TSC1-TSC2 complex upstream of mTORC1. Mol. Cell 2012, 47, 535–546. [Google Scholar] [CrossRef] [Green Version]
- Menon, S.; Dibble, C.C.; Talbott, G.; Hoxhaj, G.; Valvezan, A.J.; Takahashi, H.; Cantley, L.C.; Manning, B.D. Spatial control of the TSC complex integrates insulin and nutrient regulation of mTORC1 at the lysosome. Cell 2014, 156, 771–785. [Google Scholar] [CrossRef] [Green Version]
- Inoki, K.; Li, Y.; Xu, T.; Guan, K.L. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003, 17, 1829–1834. [Google Scholar] [CrossRef] [Green Version]
- Long, X.; Lin, Y.; Ortiz-Vega, S.; Yonezawa, K.; Avruch, J. Rheb binds and regulates the mTOR kinase. Curr. Biol. 2005, 15, 702–713. [Google Scholar] [CrossRef] [Green Version]
- Saito, K.; Araki, Y.; Kontani, K.; Nishina, H.; Katada, T. Novel role of the small GTPase Rheb: Its implication in endocytic pathway independent of the activation of mammalian target of rapamycin. J. Biochem. 2005, 137, 423–430. [Google Scholar] [CrossRef]
- Sancak, Y.; Peterson, T.R.; Shaul, Y.D.; Lindquist, R.A.; Thoreen, C.C.; Bar-Peled, L.; Sabatini, D.M. The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 2008, 320, 1496–1501. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.; Goraksha-Hicks, P.; Li, L.; Neufeld, T.P.; Guan, K.L. Regulation of TORC1 by Rag GTPases in nutrient response. Nat. Cell Biol. 2008, 10, 935–945. [Google Scholar] [CrossRef] [Green Version]
- Sancak, Y.; Bar-Peled, L.; Zoncu, R.; Markhard, A.L.; Nada, S.; Sabatini, D.M. Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 2010, 141, 290–303. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, A.; Hall, M.N. Nutrient sensing and TOR signaling in yeast and mammals. EMBO J. 2017, 36, 397–408. [Google Scholar] [CrossRef] [Green Version]
- Rogala, K.B.; Gu, X.; Kedir, J.F.; Abu-Remaileh, M.; Bianchi, L.F.; Bottino, A.M.S.; Dueholm, R.; Niehaus, A.; Overwijn, D.; Fils, A.P.; et al. Structural basis for the docking of mTORC1 on the lysosomal surface. Science 2019, 366, 468–475. [Google Scholar] [CrossRef]
- Bar-Peled, L.; Chantranupong, L.; Cherniack, A.D.; Chen, W.W.; Ottina, K.A.; Grabiner, B.C.; Spear, E.D.; Carter, S.L.; Meyerson, M.; Sabatini, D.M. A Tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science 2013, 340, 1100–1106. [Google Scholar] [CrossRef] [Green Version]
- Shen, K.; Huang, R.K.; Brignole, E.J.; Condon, K.J.; Valenstein, M.L.; Chantranupong, L.; Bomaliyamu, A.; Choe, A.; Hong, C.; Yu, Z.; et al. Architecture of the human GATOR1 and GATOR1-Rag GTPases complexes. Nature 2018, 556, 64–69. [Google Scholar] [CrossRef]
- Peng, M.; Yin, N.; Li, M.O. SZT2 dictates GATOR control of mTORC1 signalling. Nature 2017, 543, 433–437. [Google Scholar] [CrossRef] [Green Version]
- Wolfson, R.L.; Chantranupong, L.; Wyant, G.A.; Gu, X.; Orozco, J.M.; Shen, K.; Condon, K.J.; Petri, S.; Kedir, J.; Scaria, S.M.; et al. KICSTOR recruits GATOR1 to the lysosome and is necessary for nutrients to regulate mTORC1. Nature 2017, 543, 438–442. [Google Scholar] [CrossRef] [Green Version]
- Wolfson, R.L.; Chantranupong, L.; Saxton, R.A.; Shen, K.; Scaria, S.M.; Cantor, J.R.; Sabatini, D.M. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science 2016, 351, 43–48. [Google Scholar] [CrossRef] [Green Version]
- Chantranupong, L.; Scaria, S.M.; Saxton, R.A.; Gygi, M.P.; Shen, K.; Wyant, G.A.; Wang, T.; Harper, J.W.; Gygi, S.P.; Sabatini, D.M. The CASTOR Proteins Are Arginine Sensors for the mTORC1 Pathway. Cell 2016, 165, 153–164. [Google Scholar] [CrossRef] [Green Version]
- Petit, C.S.; Roczniak-Ferguson, A.; Ferguson, S.M. Recruitment of folliculin to lysosomes supports the amino acid-dependent activation of Rag GTPases. J. Cell Biol. 2013, 202, 1107–1122. [Google Scholar] [CrossRef] [Green Version]
- Tsun, Z.Y.; Bar-Peled, L.; Chantranupong, L.; Zoncu, R.; Wang, T.; Kim, C.; Spooner, E.; Sabatini, D.M. The folliculin tumor suppressor is a GAP for the RagC/D GTPases that signal amino acid levels to mTORC1. Mol. Cell 2013, 52, 495–505. [Google Scholar] [CrossRef] [Green Version]
- Gu, X.; Orozco, J.M.; Saxton, R.A.; Condon, K.J.; Liu, G.Y.; Krawczyk, P.A.; Scaria, S.M.; Harper, J.W.; Gygi, S.P.; Sabatini, D.M. SAMTOR is an S-adenosylmethionine sensor for the mTORC1 pathway. Science 2017, 358, 813–818. [Google Scholar] [CrossRef] [Green Version]
- Zoncu, R.; Bar-Peled, L.; Efeyan, A.; Wang, S.; Sancak, Y.; Sabatini, D.M. mTORC1 senses lysosomal amino acids through an inside-out mechanism that requires the vacuolar H(+)-ATPase. Science 2011, 334, 678–683. [Google Scholar] [CrossRef] [Green Version]
- Rebsamen, M.; Pochini, L.; Stasyk, T.; de Araujo, M.E.; Galluccio, M.; Kandasamy, R.K.; Snijder, B.; Fauster, A.; Rudashevskaya, E.L.; Bruckner, M.; et al. SLC38A9 is a component of the lysosomal amino acid sensing machinery that controls mTORC1. Nature 2015, 519, 477–481. [Google Scholar] [CrossRef]
- Reiling, J.H.; Sabatini, D.M. Stress and mTORture signaling. Oncogene 2006, 25, 6373–6383. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Zhao, L.; Liu, J.; Liu, A.; Jia, C.; Ma, D.; Jiang, Y.; Bai, X. Multi-mechanisms are involved in reactive oxygen species regulation of mTORC1 signaling. Cell. Signal. 2010, 22, 1469–1476. [Google Scholar] [CrossRef] [PubMed]
- Thedieck, K.; Holzwarth, B.; Prentzell, M.T.; Boehlke, C.; Klasener, K.; Ruf, S.; Sonntag, A.G.; Maerz, L.; Grellscheid, S.N.; Kremmer, E.; et al. Inhibition of mTORC1 by astrin and stress granules prevents apoptosis in cancer cells. Cell 2013, 154, 859–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dames, S.A.; Mulet, J.M.; Rathgeb-Szabo, K.; Hall, M.N.; Grzesiek, S. The solution structure of the FATC domain of the protein kinase target of rapamycin suggests a role for redox-dependent structural and cellular stability. J. Biol. Chem. 2005, 280, 20558–20564. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarbassov, D.D.; Sabatini, D.M. Redox regulation of the nutrient-sensitive raptor-mTOR pathway and complex. J. Biol. Chem. 2005, 280, 39505–39509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshida, S.; Hong, S.; Suzuki, T.; Nada, S.; Mannan, A.M.; Wang, J.; Okada, M.; Guan, K.L.; Inoki, K. Redox regulates mammalian target of rapamycin complex 1 (mTORC1) activity by modulating the TSC1/TSC2-Rheb GTPase pathway. J. Biol. Chem. 2011, 286, 32651–32660. [Google Scholar] [CrossRef] [Green Version]
- Shimobayashi, M.; Hall, M.N. Making new contacts: The mTOR network in metabolism and signalling crosstalk. Nat. Rev. Mol. Cell Biol. 2014, 15, 155–162. [Google Scholar] [CrossRef]
- Settembre, C.; Zoncu, R.; Medina, D.L.; Vetrini, F.; Erdin, S.; Erdin, S.; Huynh, T.; Ferron, M.; Karsenty, G.; Vellard, M.C.; et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 2012, 31, 1095–1108. [Google Scholar] [CrossRef] [Green Version]
- Roczniak-Ferguson, A.; Petit, C.S.; Froehlich, F.; Qian, S.; Ky, J.; Angarola, B.; Walther, T.C.; Ferguson, S.M. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal. 2012, 5, ra42. [Google Scholar] [CrossRef] [Green Version]
- Martina, J.A.; Chen, Y.; Gucek, M.; Puertollano, R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 2012, 8, 903–914. [Google Scholar] [CrossRef] [Green Version]
- Ben-Sahra, I.; Hoxhaj, G.; Ricoult, S.J.H.; Asara, J.M.; Manning, B.D. mTORC1 induces purine synthesis through control of the mitochondrial tetrahydrofolate cycle. Science 2016, 351, 728–733. [Google Scholar] [CrossRef] [Green Version]
- Duvel, K.; Yecies, J.L.; Menon, S.; Raman, P.; Lipovsky, A.I.; Souza, A.L.; Triantafellow, E.; Ma, Q.; Gorski, R.; Cleaver, S.; et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 2010, 39, 171–183. [Google Scholar] [CrossRef] [Green Version]
- Burnett, P.E.; Barrow, R.K.; Cohen, N.A.; Snyder, S.H.; Sabatini, D.M. RAFT1 phosphorylation of the translational regulators p70 S6 kinase and 4E-BP1. Proc. Natl. Acad. Sci. USA 1998, 95, 1432–1437. [Google Scholar] [CrossRef] [Green Version]
- Hara, K.; Yonezawa, K.; Kozlowski, M.T.; Sugimoto, T.; Andrabi, K.; Weng, Q.P.; Kasuga, M.; Nishimoto, I.; Avruch, J. Regulation of eIF-4E BP1 phosphorylation by mTOR. J. Biol. Chem. 1997, 272, 26457–26463. [Google Scholar] [CrossRef] [Green Version]
- Peterson, T.R.; Sengupta, S.S.; Harris, T.E.; Carmack, A.E.; Kang, S.A.; Balderas, E.; Guertin, D.A.; Madden, K.L.; Carpenter, A.E.; Finck, B.N.; et al. mTOR complex 1 regulates lipin 1 localization to control the SREBP pathway. Cell 2011, 146, 408–420. [Google Scholar] [CrossRef] [Green Version]
- Ben-Sahra, I.; Howell, J.J.; Asara, J.M.; Manning, B.D. Stimulation of de novo pyrimidine synthesis by growth signaling through mTOR and S6K1. Science 2013, 339, 1323–1328. [Google Scholar] [CrossRef] [Green Version]
- Robitaille, A.M.; Christen, S.; Shimobayashi, M.; Cornu, M.; Fava, L.L.; Moes, S.; Prescianotto-Baschong, C.; Sauer, U.; Jenoe, P.; Hall, M.N. Quantitative phosphoproteomics reveal mTORC1 activates de novo pyrimidine synthesis. Science 2013, 339, 1320–1323. [Google Scholar] [CrossRef]
- Zid, B.M.; Rogers, A.N.; Katewa, S.D.; Vargas, M.A.; Kolipinski, M.C.; Lu, T.A.; Benzer, S.; Kapahi, P. 4E-BP extends lifespan upon dietary restriction by enhancing mitochondrial activity in Drosophila. Cell 2009, 139, 149–160. [Google Scholar] [CrossRef] [Green Version]
- Morita, M.; Gravel, S.P.; Chenard, V.; Sikstrom, K.; Zheng, L.; Alain, T.; Gandin, V.; Avizonis, D.; Arguello, M.; Zakaria, C.; et al. mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell Metab. 2013, 18, 698–711. [Google Scholar] [CrossRef] [Green Version]
- Cunningham, J.T.; Rodgers, J.T.; Arlow, D.H.; Vazquez, F.; Mootha, V.K.; Puigserver, P. mTOR controls mitochondrial oxidative function through a YY1-PGC-1alpha transcriptional complex. Nature 2007, 450, 736–740. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, N.; Hara, T.; Kaizuka, T.; Kishi, C.; Takamura, A.; Miura, Y.; Iemura, S.; Natsume, T.; Takehana, K.; Yamada, N.; et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol. Biol. Cell 2009, 20, 1981–1991. [Google Scholar] [CrossRef] [Green Version]
- Ganley, I.G.; Lam du, H.; Wang, J.; Ding, X.; Chen, S.; Jiang, X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J. Biol. Chem. 2009, 284, 12297–12305. [Google Scholar] [CrossRef] [Green Version]
- Jung, C.H.; Jun, C.B.; Ro, S.H.; Kim, Y.M.; Otto, N.M.; Cao, J.; Kundu, M.; Kim, D.H. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol. Biol. Cell 2009, 20, 1992–2003. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.; Kundu, M.; Viollet, B.; Guan, K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011, 13, 132–141. [Google Scholar] [CrossRef] [Green Version]
- Brown, E.J.; Albers, M.W.; Shin, T.B.; Ichikawa, K.; Keith, C.T.; Lane, W.S.; Schreiber, S.L. A mammalian protein targeted by G1-arresting rapamycin-receptor complex. Nature 1994, 369, 756–758. [Google Scholar] [CrossRef]
- Sabatini, D.M.; Erdjument-Bromage, H.; Lui, M.; Tempst, P.; Snyder, S.H. RAFT1: A mammalian protein that binds to FKBP12 in a rapamycin-dependent fashion and is homologous to yeast TORs. Cell 1994, 78, 35–43. [Google Scholar] [CrossRef]
- Sabers, C.J.; Martin, M.M.; Brunn, G.J.; Williams, J.M.; Dumont, F.J.; Wiederrecht, G.; Abraham, R.T. Isolation of a protein target of the FKBP12-rapamycin complex in mammalian cells. J. Biol. Chem. 1995, 270, 815–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacinto, E.; Loewith, R.; Schmidt, A.; Lin, S.; Ruegg, M.A.; Hall, A.; Hall, M.N. Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat. Cell Biol. 2004, 6, 1122–1128. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Ali, S.M.; Kim, D.H.; Guertin, D.A.; Latek, R.R.; Erdjument-Bromage, H.; Tempst, P.; Sabatini, D.M. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr. Biol. 2004, 14, 1296–1302. [Google Scholar] [CrossRef] [Green Version]
- Jacinto, E.; Facchinetti, V.; Liu, D.; Soto, N.; Wei, S.; Jung, S.Y.; Huang, Q.; Qin, J.; Su, B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 2006, 127, 125–137. [Google Scholar] [CrossRef] [Green Version]
- Frias, M.A.; Thoreen, C.C.; Jaffe, J.D.; Schroder, W.; Sculley, T.; Carr, S.A.; Sabatini, D.M. mSin1 is necessary for Akt/PKB phosphorylation, and its isoforms define three distinct mTORC2s. Curr. Biol. 2006, 16, 1865–1870. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.; Inoki, K.; Ikenoue, T.; Guan, K.L. Identification of Sin1 as an essential TORC2 component required for complex formation and kinase activity. Genes Dev. 2006, 20, 2820–2832. [Google Scholar] [CrossRef] [Green Version]
- Stuttfeld, E.; Aylett, C.H.; Imseng, S.; Boehringer, D.; Scaiola, A.; Sauer, E.; Hall, M.N.; Maier, T.; Ban, N. Architecture of the human mTORC2 core complex. eLife 2018, 7, e33101. [Google Scholar] [CrossRef]
- Chen, X.; Liu, M.; Tian, Y.; Li, J.; Qi, Y.; Zhao, D.; Wu, Z.; Huang, M.; Wong, C.C.L.; Wang, H.W.; et al. Cryo-EM structure of human mTOR complex 2. Cell Res. 2018, 28, 518–528. [Google Scholar] [CrossRef] [Green Version]
- Scaiola, A.; Mangia, F.; Imseng, S.; Boehringer, D.; Berneiser, K.; Shimobayashi, M.; Stuttfeld, E.; Hall, M.N.; Ban, N.; Maier, T. The 3.2-A resolution structure of human mTORC2. Sci. Adv. 2020, 6, eabc1251. [Google Scholar] [CrossRef] [PubMed]
- Liu, P.; Gan, W.; Chin, Y.R.; Ogura, K.; Guo, J.; Zhang, J.; Wang, B.; Blenis, J.; Cantley, L.C.; Toker, A.; et al. PtdIns(3,4,5)P3-Dependent Activation of the mTORC2 Kinase Complex. Cancer Discov. 2015, 5, 1194–1209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gan, X.; Wang, J.; Su, B.; Wu, D. Evidence for direct activation of mTORC2 kinase activity by phosphatidylinositol 3,4,5-trisphosphate. J. Biol. Chem. 2011, 286, 10998–11002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovalski, J.R.; Bhaduri, A.; Zehnder, A.M.; Neela, P.H.; Che, Y.; Wozniak, G.G.; Khavari, P.A. The Functional Proximal Proteome of Oncogenic Ras Includes mTORC2. Mol. Cell 2019, 73, 830–844.e812. [Google Scholar] [CrossRef] [Green Version]
- Tumaneng, K.; Schlegelmilch, K.; Russell, R.C.; Yimlamai, D.; Basnet, H.; Mahadevan, N.; Fitamant, J.; Bardeesy, N.; Camargo, F.D.; Guan, K.L. YAP mediates crosstalk between the Hippo and PI(3)K-TOR pathways by suppressing PTEN via miR-29. Nat. Cell Biol. 2012, 14, 1322–1329. [Google Scholar] [CrossRef]
- Esen, E.; Chen, J.; Karner, C.M.; Okunade, A.L.; Patterson, B.W.; Long, F. WNT-LRP5 signaling induces Warburg effect through mTORC2 activation during osteoblast differentiation. Cell Metab. 2013, 17, 745–755. [Google Scholar] [CrossRef] [Green Version]
- Daulat, A.M.; Bertucci, F.; Audebert, S.; Serge, A.; Finetti, P.; Josselin, E.; Castellano, R.; Birnbaum, D.; Angers, S.; Borg, J.P. PRICKLE1 Contributes to Cancer Cell Dissemination through Its Interaction with mTORC2. Dev. Cell 2016, 37, 311–325. [Google Scholar] [CrossRef]
- Kazyken, D.; Magnuson, B.; Bodur, C.; Acosta-Jaquez, H.A.; Zhang, D.; Tong, X.; Barnes, T.M.; Steinl, G.K.; Patterson, N.E.; Altheim, C.H.; et al. AMPK directly activates mTORC2 to promote cell survival during acute energetic stress. Sci. Signal. 2019, 12, eaav3249. [Google Scholar] [CrossRef]
- Betz, C.; Hall, M.N. Where is mTOR and what is it doing there? J. Cell Biol. 2013, 203, 563–574. [Google Scholar] [CrossRef] [Green Version]
- Pearce, L.R.; Komander, D.; Alessi, D.R. The nuts and bolts of AGC protein kinases. Nat. Rev. Mol. Cell Biol. 2010, 11, 9–22. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Guertin, D.A.; Ali, S.M.; Sabatini, D.M. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 2005, 307, 1098–1101. [Google Scholar] [CrossRef] [Green Version]
- Hresko, R.C.; Mueckler, M. mTOR.RICTOR is the Ser473 kinase for Akt/protein kinase B in 3T3-L1 adipocytes. J. Biol. Chem. 2005, 280, 40406–40416. [Google Scholar] [CrossRef] [Green Version]
- Oh, W.J.; Wu, C.C.; Kim, S.J.; Facchinetti, V.; Julien, L.A.; Finlan, M.; Roux, P.P.; Su, B.; Jacinto, E. mTORC2 can associate with ribosomes to promote cotranslational phosphorylation and stability of nascent Akt polypeptide. EMBO J. 2010, 29, 3939–3951. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Martinez, J.M.; Alessi, D.R. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum- and glucocorticoid-induced protein kinase 1 (SGK1). Biochem. J. 2008, 416, 375–385. [Google Scholar] [CrossRef] [Green Version]
- Gu, Y.; Albuquerque, C.P.; Braas, D.; Zhang, W.; Villa, G.R.; Bi, J.; Ikegami, S.; Masui, K.; Gini, B.; Yang, H.; et al. mTORC2 Regulates Amino Acid Metabolism in Cancer by Phosphorylation of the Cystine-Glutamate Antiporter xCT. Mol. Cell 2017, 67, 128–138. [Google Scholar] [CrossRef] [Green Version]
- Shah, O.J.; Wang, Z.; Hunter, T. Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr. Biol. 2004, 14, 1650–1656. [Google Scholar] [CrossRef] [Green Version]
- Harrington, L.S.; Findlay, G.M.; Gray, A.; Tolkacheva, T.; Wigfield, S.; Rebholz, H.; Barnett, J.; Leslie, N.R.; Cheng, S.; Shepherd, P.R.; et al. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J. Cell Biol. 2004, 166, 213–223. [Google Scholar] [CrossRef] [Green Version]
- Um, S.H.; Frigerio, F.; Watanabe, M.; Picard, F.; Joaquin, M.; Sticker, M.; Fumagalli, S.; Allegrini, P.R.; Kozma, S.C.; Auwerx, J.; et al. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature 2004, 431, 200–205. [Google Scholar] [CrossRef]
- Yu, Y.; Yoon, S.O.; Poulogiannis, G.; Yang, Q.; Ma, X.M.; Villen, J.; Kubica, N.; Hoffman, G.R.; Cantley, L.C.; Gygi, S.P.; et al. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science 2011, 332, 1322–1326. [Google Scholar] [CrossRef] [Green Version]
- Hsu, P.P.; Kang, S.A.; Rameseder, J.; Zhang, Y.; Ottina, K.A.; Lim, D.; Peterson, T.R.; Choi, Y.; Gray, N.S.; Yaffe, M.B.; et al. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science 2011, 332, 1317–1322. [Google Scholar] [CrossRef] [Green Version]
- Lim, M.A.; Riedel, H.; Liu, F. Grb10: More than a simple adaptor protein. Front. Biosci. 2004, 9, 387–403. [Google Scholar] [CrossRef] [PubMed]
- Liberman, M.C.; Kujawa, S.G. Cochlear synaptopathy in acquired sensorineural hearing loss: Manifestations and mechanisms. Hear. Res. 2017, 349, 138–147. [Google Scholar] [CrossRef] [PubMed]
- Carlson, M.L. Cochlear Implantation in Adults. N. Engl. J. Med. 2020, 382, 1531–1542. [Google Scholar] [CrossRef] [PubMed]
- Kujawa, S.G.; Liberman, M.C. Adding insult to injury: Cochlear nerve degeneration after “temporary” noise-induced hearing loss. J. Neurosci. 2009, 29, 14077–14085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sergeyenko, Y.; Lall, K.; Liberman, M.C.; Kujawa, S.G. Age-related cochlear synaptopathy: An early-onset contributor to auditory functional decline. J. Neurosci. 2013, 33, 13686–13694. [Google Scholar] [CrossRef]
- Rubel, E.W.; Furrer, S.A.; Stone, J.S. A brief history of hair cell regeneration research and speculations on the future. Hear. Res. 2013, 297, 42–51. [Google Scholar] [CrossRef] [Green Version]
- Cotanche, D.A. Regeneration of hair cell stereociliary bundles in the chick cochlea following severe acoustic trauma. Hear. Res. 1987, 30, 181–195. [Google Scholar] [CrossRef]
- Cruz, R.M.; Lambert, P.R.; Rubel, E.W. Light microscopic evidence of hair cell regeneration after gentamicin toxicity in chick cochlea. Arch. Otolaryngol. Head Neck Surg. 1987, 113, 1058–1062. [Google Scholar] [CrossRef]
- Corwin, J.T.; Cotanche, D.A. Regeneration of sensory hair cells after acoustic trauma. Science 1988, 240, 1772–1774. [Google Scholar] [CrossRef]
- Ryals, B.M.; Rubel, E.W. Hair cell regeneration after acoustic trauma in adult Coturnix quail. Science 1988, 240, 1774–1776. [Google Scholar] [CrossRef]
- Atkinson, P.J.; Huarcaya Najarro, E.; Sayyid, Z.N.; Cheng, A.G. Sensory hair cell development and regeneration: Similarities and differences. Development 2015, 142, 1561–1571. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- White, P.M.; Doetzlhofer, A.; Lee, Y.S.; Groves, A.K.; Segil, N. Mammalian cochlear supporting cells can divide and trans-differentiate into hair cells. Nature 2006, 441, 984–987. [Google Scholar] [CrossRef] [PubMed]
- Warchol, M.E.; Lambert, P.R.; Goldstein, B.J.; Forge, A.; Corwin, J.T. Regenerative proliferation in inner ear sensory epithelia from adult guinea pigs and humans. Science 1993, 259, 1619–1622. [Google Scholar] [CrossRef]
- Forge, A.; Li, L.; Corwin, J.T.; Nevill, G. Ultrastructural evidence for hair cell regeneration in the mammalian inner ear. Science 1993, 259, 1616–1619. [Google Scholar] [CrossRef]
- Safety, Tolerability and Efficacy for CGF166 in Patients With Unilateral or Bilateral Severe-to-Profound Hearing Loss. Available online: https://ClinicalTrials.gov/show/NCT02132130 (accessed on 15 April 2021).
- Mulvaney, J.; Dabdoub, A. Atoh1, an essential transcription factor in neurogenesis and intestinal and inner ear development: Function, regulation, and context dependency. J. Assoc. Res. Otolaryngol. 2012, 13, 281–293. [Google Scholar] [CrossRef] [Green Version]
- Zheng, J.L.; Gao, W.Q. Overexpression of Math1 induces robust production of extra hair cells in postnatal rat inner ears. Nat. Neurosci. 2000, 3, 580–586. [Google Scholar] [CrossRef]
- Rutten, R.J. A First-in-Human Study of the Safety and Efficacy of a New Drug, a Gamma Secretase Inhibitor, to Treat People with Sensorineural Hearing Loss; ISRCTN: London, UK, 2017. [Google Scholar] [CrossRef]
- McLean, W.J.; Hinton, A.S.; Herby, J.T.J.; Salt, A.N.; Hartsock, J.J.; Wilson, S.; Lucchino, D.L.; Lenarz, T.; Warnecke, A.; Prenzler, N.; et al. Improved Speech Intelligibility in Subjects With Stable Sensorineural Hearing Loss Following Intratympanic Dosing of FX-322 in a Phase 1b Study. Otol. Neurotol. 2021. [Google Scholar] [CrossRef]
- Montcouquiol, M.; Corwin, J.T. Intracellular signals that control cell proliferation in mammalian balance epithelia: Key roles for phosphatidylinositol-3 kinase, mammalian target of rapamycin, and S6 kinases in preference to calcium, protein kinase C, and mitogen-activated protein kinase. J. Neurosci. 2001, 21, 570–580. [Google Scholar]
- Witte, M.C.; Montcouquiol, M.; Corwin, J.T. Regeneration in avian hair cell epithelia: Identification of intracellular signals required for S-phase entry. Eur. J. Neurosci. 2001, 14, 829–838. [Google Scholar] [CrossRef]
- Shu, Y.; Li, W.; Huang, M.; Quan, Y.Z.; Scheffer, D.; Tian, C.; Tao, Y.; Liu, X.; Hochedlinger, K.; Indzhykulian, A.A.; et al. Renewed proliferation in adult mouse cochlea and regeneration of hair cells. Nat. Commun. 2019, 10, 5530. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.J.; Park, Y.J.; Park, J.Y.; Jeong, H.O.; Kim, D.H.; Ha, Y.M.; Kim, J.M.; Song, Y.M.; Heo, H.S.; Yu, B.P.; et al. Inhibitory effect of mTOR activator MHY1485 on autophagy: Suppression of lysosomal fusion. PLoS ONE 2012, 7, e43418. [Google Scholar] [CrossRef] [PubMed]
- Li, X.J.; Doetzlhofer, A. LIN28B/let-7 control the ability of neonatal murine auditory supporting cells to generate hair cells through mTOR signaling. Proc. Natl. Acad. Sci. USA 2020, 117, 22225–22236. [Google Scholar] [CrossRef] [PubMed]
- Xiong, W.; Wei, W.; Qi, Y.; Du, Z.; Qu, T.; Liu, K.; Gong, S. Autophagy is Required for Remodeling in Postnatal Developing Ribbon Synapses of Cochlear Inner Hair Cells. Neuroscience 2020, 431, 1–16. [Google Scholar] [CrossRef]
- Kim, H.J.; Woo, H.M.; Ryu, J.; Bok, J.; Kim, J.W.; Choi, S.B.; Park, M.H.; Park, H.Y.; Koo, S.K. Conditional deletion of pten leads to defects in nerve innervation and neuronal survival in inner ear development. PLoS ONE 2013, 8, e55609. [Google Scholar] [CrossRef] [Green Version]
- Bodmer, D. An update on drug design strategies to prevent acquired sensorineural hearing loss. Expert Opin. Drug Discov. 2017, 12, 1161–1167. [Google Scholar] [CrossRef]
- Fang, B.; Xiao, H. Rapamycin alleviates cisplatin-induced ototoxicity in vivo. Biochem. Biophys. Res. Commun. 2014, 448, 443–447. [Google Scholar] [CrossRef]
- Kim, Y.J.; Tian, C.; Kim, J.; Shin, B.; Choo, O.S.; Kim, Y.S.; Choung, Y.H. Autophagic flux, a possible mechanism for delayed gentamicin-induced ototoxicity. Sci. Rep. 2017, 7, 41356. [Google Scholar] [CrossRef] [Green Version]
- Yuan, H.; Wang, X.; Hill, K.; Chen, J.; Lemasters, J.; Yang, S.M.; Sha, S.H. Autophagy attenuates noise-induced hearing loss by reducing oxidative stress. Antioxid. Redox Signal. 2015, 22, 1308–1324. [Google Scholar] [CrossRef] [Green Version]
- Fu, X.; Sun, X.; Zhang, L.; Jin, Y.; Chai, R.; Yang, L.; Zhang, A.; Liu, X.; Bai, X.; Li, J.; et al. Tuberous sclerosis complex-mediated mTORC1 overactivation promotes age-related hearing loss. J. Clin. Investig. 2018, 128, 4938–4955. [Google Scholar] [CrossRef]
- Altschuler, R.A.; Kabara, L.; Martin, C.; Kanicki, A.; Stewart, C.E.; Kohrman, D.C.; Dolan, D.F. Rapamycin Added to Diet in Late Mid-Life Delays Age-Related Hearing Loss in UMHET4 Mice. Front. Cell. Neurosci. 2021, 15, 658972. [Google Scholar] [CrossRef]
- Altschuler, R.A.; Kanicki, A.; Martin, C.; Kohrman, D.C.; Miller, R.A. Rapamycin but not acarbose decreases age-related loss of outer hair cells in the mouse Cochlea. Hear. Res. 2018, 370, 11–15. [Google Scholar] [CrossRef] [PubMed]
- He, Z.; Guo, L.; Shu, Y.; Fang, Q.; Zhou, H.; Liu, Y.; Liu, D.; Lu, L.; Zhang, X.; Ding, X.; et al. Autophagy protects auditory hair cells against neomycin-induced damage. Autophagy 2017, 13, 1884–1904. [Google Scholar] [CrossRef] [PubMed]
- Guo, S.; Xu, N.; Chen, P.; Liu, Y.; Qi, X.; Liu, S.; Li, C.; Tang, J. Rapamycin Protects Spiral Ganglion Neurons from Gentamicin-Induced Degeneration In Vitro. J. Assoc. Res. Otolaryngol. 2019, 20, 475–487. [Google Scholar] [CrossRef]
- Ye, B.; Wang, Q.; Hu, H.; Shen, Y.; Fan, C.; Chen, P.; Ma, Y.; Wu, H.; Xiang, M. Restoring autophagic flux attenuates cochlear spiral ganglion neuron degeneration by promoting TFEB nuclear translocation via inhibiting MTOR. Autophagy 2019, 15, 998–1016. [Google Scholar] [CrossRef]
- Ebnoether, E.; Ramseier, A.; Cortada, M.; Bodmer, D.; Levano-Huaman, S. Sesn2 gene ablation enhances susceptibility to gentamicin-induced hair cell death via modulation of AMPK/mTOR signaling. Cell Death Discov. 2017, 3, 17024. [Google Scholar] [CrossRef] [Green Version]
- Francis, S.P.; Katz, J.; Fanning, K.D.; Harris, K.A.; Nicholas, B.D.; Lacy, M.; Pagana, J.; Agris, P.F.; Shin, J.B. A novel role of cytosolic protein synthesis inhibition in aminoglycoside ototoxicity. J. Neurosci. 2013, 33, 3079–3093. [Google Scholar] [CrossRef] [Green Version]
- Nicholas, B.D.; Francis, S.; Wagner, E.L.; Zhang, S.; Shin, J.B. Protein Synthesis Inhibition and Activation of the c-Jun N-Terminal Kinase Are Potential Contributors to Cisplatin Ototoxicity. Front. Cell. Neurosci. 2017, 11, 303. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Xie, X.; Deng, M.; Chen, X.; Gan, L. Generation and characterization of Atoh1-Cre knock-in mouse line. Genesis 2010, 48, 407–413. [Google Scholar] [CrossRef] [Green Version]
- Ye, B.; Fan, C.; Shen, Y.; Wang, Q.; Hu, H.; Xiang, M. The Antioxidative Role of Autophagy in Hearing Loss. Front. Neurosci. 2018, 12, 1010. [Google Scholar] [CrossRef] [Green Version]
- Fujimoto, C.; Iwasaki, S.; Urata, S.; Morishita, H.; Sakamaki, Y.; Fujioka, M.; Kondo, K.; Mizushima, N.; Yamasoba, T. Autophagy is essential for hearing in mice. Cell Death Dis. 2017, 8, e2780. [Google Scholar] [CrossRef]
- Zhou, H.; Qian, X.; Xu, N.; Zhang, S.; Zhu, G.; Zhang, Y.; Liu, D.; Cheng, C.; Zhu, X.; Liu, Y.; et al. Disruption of Atg7-dependent autophagy causes electromotility disturbances, outer hair cell loss, and deafness in mice. Cell Death Dis. 2020, 11, 913. [Google Scholar] [CrossRef] [PubMed]
- Nuttall, A.L.; Wilson, T.; Omelchenko, I.; Shi, X.R. AMPK activation by AICAR induces mitochondrial biogenesis in the inner ear. FASEB J. 2012, 26. [Google Scholar] [CrossRef]
- Foller, M.; Jaumann, M.; Dettling, J.; Saxena, A.; Pakladok, T.; Munoz, C.; Ruth, P.; Sopjani, M.; Seebohm, G.; Ruttiger, L.; et al. AMP-activated protein kinase in BK-channel regulation and protection against hearing loss following acoustic overstimulation. FASEB J. 2012, 26, 4243–4253. [Google Scholar] [CrossRef] [PubMed]
- Hill, K.; Yuan, H.; Wang, X.; Sha, S.H. Noise-Induced Loss of Hair Cells and Cochlear Synaptopathy Are Mediated by the Activation of AMPK. J. Neurosci. 2016, 36, 7497–7510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKay, S.E.; Yan, W.; Nouws, J.; Thormann, M.J.; Raimundo, N.; Khan, A.; Santos-Sacchi, J.; Song, L.; Shadel, G.S. Auditory Pathology in a Transgenic mtTFB1 Mouse Model of Mitochondrial Deafness. Am. J. Pathol. 2015, 185, 3132–3140. [Google Scholar] [CrossRef]
- Zhao, J.; Li, G.; Zhao, X.; Lin, X.; Gao, Y.; Raimundo, N.; Li, G.L.; Shang, W.; Wu, H.; Song, L. Down-regulation of AMPK signaling pathway rescues hearing loss in TFB1 transgenic mice and delays age-related hearing loss. Aging 2020, 12, 5590–5611. [Google Scholar] [CrossRef]
- Egan, D.F.; Shackelford, D.B.; Mihaylova, M.M.; Gelino, S.; Kohnz, R.A.; Mair, W.; Vasquez, D.S.; Joshi, A.; Gwinn, D.M.; Taylor, R.; et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 2011, 331, 456–461. [Google Scholar] [CrossRef] [Green Version]
- Bach, M.; Larance, M.; James, D.E.; Ramm, G. The serine/threonine kinase ULK1 is a target of multiple phosphorylation events. Biochem. J. 2011, 440, 283–291. [Google Scholar] [CrossRef] [Green Version]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef] [Green Version]
- Howell, J.J.; Hellberg, K.; Turner, M.; Talbott, G.; Kolar, M.J.; Ross, D.S.; Hoxhaj, G.; Saghatelian, A.; Shaw, R.J.; Manning, B.D. Metformin Inhibits Hepatic mTORC1 Signaling via Dose-Dependent Mechanisms Involving AMPK and the TSC Complex. Cell Metab. 2017, 25, 463–471. [Google Scholar] [CrossRef] [Green Version]
- Chang, J.; Jung, H.H.; Yang, J.Y.; Choi, J.; Im, G.J.; Chae, S.W. Protective role of antidiabetic drug metformin against gentamicin induced apoptosis in auditory cell line. Hear. Res. 2011, 282, 92–96. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.; Jung, H.H.; Yang, J.Y.; Lee, S.; Choi, J.; Im, G.J.; Chae, S.W. Protective effect of metformin against cisplatin-induced ototoxicity in an auditory cell line. J. Assoc. Res. Otolaryngol. 2014, 15, 149–158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, Z.; Zhang, T.; Zhan, T.; Cheng, G.; Zhang, W.; Jia, H.; Yang, H. Metformin alleviates cisplatin-induced ototoxicity by autophagy induction possibly via the AMPK/FOXO3a pathway. J. Neurophysiol. 2021, 125, 1202–1212. [Google Scholar] [CrossRef] [PubMed]
- Glutz, A.; Leitmeyer, K.; Setz, C.; Brand, Y.; Bodmer, D. Metformin Protects Auditory Hair Cells from Gentamicin-Induced Toxicity in vitro. Audiol. Neurootol. 2015, 20, 360–369. [Google Scholar] [CrossRef]
- Oishi, N.; Kendall, A.; Schacht, J. Metformin protects against gentamicin-induced hair cell death in vitro but not ototoxicity in vivo. Neurosci. Lett. 2014, 583, 65–69. [Google Scholar] [CrossRef] [Green Version]
- Gedik, O.; Dogan, R.; Babademez, M.A.; Karatas, E.; Aydin, M.S.; Kocyigit, A.; Esrefoglu, M.; Ozturan, O. Therapeutic effects of metformin for noise induced hearing loss. Am. J. Otolaryngol. 2020, 41, 102328. [Google Scholar] [CrossRef]
- Muri, L.; Le, N.D.; Zemp, J.; Grandgirard, D.; Leib, S.L. Metformin mediates neuroprotection and attenuates hearing loss in experimental pneumococcal meningitis. J. Neuroinflamm. 2019, 16, 156. [Google Scholar] [CrossRef] [Green Version]
- Hosoya, M.; Fujioka, M.; Sone, T.; Okamoto, S.; Akamatsu, W.; Ukai, H.; Ueda, H.R.; Ogawa, K.; Matsunaga, T.; Okano, H. Cochlear Cell Modeling Using Disease-Specific iPSCs Unveils a Degenerative Phenotype and Suggests Treatments for Congenital Progressive Hearing Loss. Cell Rep. 2017, 18, 68–81. [Google Scholar] [CrossRef]
- Hosoya, M.; Saeki, T.; Saegusa, C.; Matsunaga, T.; Okano, H.; Fujioka, M.; Ogawa, K. Estimating the concentration of therapeutic range using disease-specific iPS cells: Low-dose rapamycin therapy for Pendred syndrome. Regen Ther. 2019, 10, 54–63. [Google Scholar] [CrossRef]
- Fujioka, M.; Akiyama, T.; Hosoya, M.; Kikuchi, K.; Fujiki, Y.; Saito, Y.; Yoshihama, K.; Ozawa, H.; Tsukada, K.; Nishio, S.Y.; et al. A phase I/IIa double blind single institute trial of low dose sirolimus for Pendred syndrome/DFNB4. Medicine 2020, 99, e19763. [Google Scholar] [CrossRef]
- Parmigiani, A.; Budanov, A.V. Sensing the Environment Through Sestrins: Implications for Cellular Metabolism. Int. Rev. Cell Mol. Biol. 2016, 327, 1–42. [Google Scholar] [CrossRef]
- Zhang, C.; Sun, W.; Li, J.; Xiong, B.; Frye, M.D.; Ding, D.; Salvi, R.; Kim, M.J.; Someya, S.; Hu, B.H. Loss of sestrin 2 potentiates the early onset of age-related sensory cell degeneration in the cochlea. Neuroscience 2017, 361, 179–191. [Google Scholar] [CrossRef] [Green Version]
- Bodmer, D.; Levano-Huaman, S. Sesn2/AMPK/mTOR signaling mediates balance between survival and apoptosis in sensory hair cells under stress. Cell Death Dis. 2017, 8, e3068. [Google Scholar] [CrossRef]
- Jiang, H.; Sha, S.H.; Schacht, J. Kanamycin alters cytoplasmic and nuclear phosphoinositide signaling in the organ of Corti in vivo. J. Neurochem. 2006, 99, 269–276. [Google Scholar] [CrossRef]
- Chung, W.H.; Pak, K.; Lin, B.; Webster, N.; Ryan, A.F. A PI3K pathway mediates hair cell survival and opposes gentamicin toxicity in neonatal rat organ of Corti. J. Assoc. Res. Otolaryngol. 2006, 7, 373–382. [Google Scholar] [CrossRef] [Green Version]
- Sha, S.H.; Chen, F.Q.; Schacht, J. PTEN attenuates PIP3/Akt signaling in the cochlea of the aging CBA/J mouse. Hear. Res. 2010, 264, 86–92. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Yuan, H.; Talaska, A.E.; Hill, K.; Sha, S.H. Increased Sensitivity to Noise-Induced Hearing Loss by Blockade of Endogenous PI3K/Akt Signaling. J. Assoc. Res. Otolaryngol. 2015, 16, 347–356. [Google Scholar] [CrossRef] [Green Version]
- Jadali, A.; Kwan, K.Y. Activation of PI3K signaling prevents aminoglycoside-induced hair cell death in the murine cochlea. Biol. Open 2016, 5, 698–708. [Google Scholar] [CrossRef] [Green Version]
- Brand, Y.; Levano, S.; Radojevic, V.; Naldi, A.M.; Setz, C.; Ryan, A.F.; Pak, K.; Hemmings, B.A.; Bodmer, D. All Akt isoforms (Akt1, Akt2, Akt3) are involved in normal hearing, but only Akt2 and Akt3 are involved in auditory hair cell survival in the mammalian inner ear. PLoS ONE 2015, 10, e0121599. [Google Scholar] [CrossRef]
- Yamahara, K.; Yamamoto, N.; Nakagawa, T.; Ito, J. Insulin-like growth factor 1: A novel treatment for the protection or regeneration of cochlear hair cells. Hear. Res. 2015, 330, 2–9. [Google Scholar] [CrossRef]
- Rodriguez-de la Rosa, L.; Lassaletta, L.; Calvino, M.; Murillo-Cuesta, S.; Varela-Nieto, I. The Role of Insulin-Like Growth Factor 1 in the Progression of Age-Related Hearing Loss. Front. Aging Neurosci. 2017, 9, 411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iwai, K.; Nakagawa, T.; Endo, T.; Matsuoka, Y.; Kita, T.; Kim, T.S.; Tabata, Y.; Ito, J. Cochlear protection by local insulin-like growth factor-1 application using biodegradable hydrogel. Laryngoscope 2006, 116, 529–533. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, Y.; Yamamoto, N.; Nakagawa, T.; Ito, J. Insulin-like growth factor 1 inhibits hair cell apoptosis and promotes the cell cycle of supporting cells by activating different downstream cascades after pharmacological hair cell injury in neonatal mice. Mol. Cell. Neurosci. 2013, 56, 29–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, L.; Kita, T.; Katsuno, T.; Yamamoto, N.; Omori, K.; Nakagawa, T. Insulin-Like Growth Factor 1 on the Maintenance of Ribbon Synapses in Mouse Cochlear Explant Cultures. Front. Cell. Neurosci. 2020, 14, 571155. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, T.; Kumakawa, K.; Usami, S.; Hato, N.; Tabuchi, K.; Takahashi, M.; Fujiwara, K.; Sasaki, A.; Komune, S.; Sakamoto, T.; et al. A randomized controlled clinical trial of topical insulin-like growth factor-1 therapy for sudden deafness refractory to systemic corticosteroid treatment. BMC Med. 2014, 12, 219. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Protein(s)/Protein Complex | mTORC1 Signaling Related Function | References |
|---|---|---|
| mTOR | Serine/threonine kinase that forms the catalytic subunit of mTORC1 | [10,71,72,73] |
| mLST8 | mTORC1 subunit | [14,15] |
| RAPTOR | Defining and essential subunit of mTORC1 | [15,16] |
| FKBP12 | Forms a complex with rapamycin and inhibits mTORC1 | [17,18] |
| AKT | Inhibits the TSC upon growth factor signaling | [23,24,25] |
| TSC1 TSC2 TBC1D7 | Form the TSC which has GAP activity towards RHEB and therefore inhibits mTORC1 | [26,28] |
| RHEB | GTP-bound RHEB activates mTORC1 upon growth factor signaling | [28,29] |
| RAG proteins | Signal amino acid availability to mTORC1 | [31,32] |
| RAGULATOR complex | Binds the RAGs and is involved in amino acid signaling to mTORC1 | [33] |
| GATOR1 complex | Negatively regulates RAGs to inhibit mTORC1 | [36] |
| GATOR2 complex | Negatively regulates GATOR1 to activate mTORC1 | [36] |
| KICSTOR complex | Translocates GATOR1 to the lysosome to negatively regulate mTORC1 | [38,39] |
| SESTRIN-2 | Leucine sensor, inhibits mTORC1 by binding GATOR2 when leucine levels are low | [40] |
| CASTOR-1 CASTOR-2 | Arginine sensors, inhibit mTORC1 by binding GATOR2 when arginine levels are low | [41] |
| FLCN | Amino acid sensor positively regulating mTORC1 | [42,43] |
| SAMTOR | SAM (methionine) sensor, binds GATOR1 and KICSTOR to inhibit mTORC1 when SAM/methionine levels are low | [44] |
| v-ATPase | Lysosomal amino acid sensor | [45] |
| SLC38A9 | Lysosomal amino acid sensor | [46] |
| AMPK | Senses cellular energy status and inhibits mTORC1 by different mechanisms when cellular energy (ATP) is low | [12] |
| S6K | mTORC1 effector, is phosphorylated and activated by mTORC1 to promote anabolism | [10,59] |
| 4E-BP | mTORC1 substrate, is phosphorylated and inhibited by mTORC1 to promote anabolism | [10,59,60] |
| LIPIN1 | mTORC1 substrate, is phosphorylated and inhibited by mTORC1 to promote lipid synthesis | [53,61] |
| ATF4 | Is activated by mTORC1 to promote nucleotide synthesis | [57] |
| HIF1α | Is activated by mTORC1 to promote glycolysis | [58] |
| ULK1 | mTORC1 substrate, is phosphorylated and inhibited by mTORC1 to inhibit autophagy | [67,68,69,70] |
| ATG13 | mTORC1 substrate, is phosphorylated and inhibited by mTORC1 to inhibit autophagy | [67,68,69] |
| TFEB | mTORC1 substrate, is phosphorylated and inhibited by mTORC1 to inhibit autophagy and lysosomal biogenesis | [12,54,55,56] |
| Protein | mTORC2 Signaling Related Function | References |
|---|---|---|
| mTOR | Serine/threonine kinase that forms the catalytic subunit of mTORC2 | [10,71,72,73] |
| mLST8 | mTORC2 subunit | [14,15] |
| mSIN1 | mTORC2 subunit | [77,78,79] |
| RICTOR | Defining and essential subunit of mTORC2 | [74,76] |
| AKT | mTORC2 effector, phosphorylated and activated by mTORC2 | [92,93,94] |
| PKC | mTORC2 effector, phosphorylated and activated by mTORC2, modulates the actin cytoskeleton | [15,74,76] |
| SGK1 | mTORC2 effector, phosphorylated and activated by mTORC2 | [95] |
| SLC7A11 | mTORC2 substrate, phosphorylated and inhibited by mTORC2 | [96] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cortada, M.; Levano, S.; Bodmer, D. mTOR Signaling in the Inner Ear as Potential Target to Treat Hearing Loss. Int. J. Mol. Sci. 2021, 22, 6368. https://doi.org/10.3390/ijms22126368
Cortada M, Levano S, Bodmer D. mTOR Signaling in the Inner Ear as Potential Target to Treat Hearing Loss. International Journal of Molecular Sciences. 2021; 22(12):6368. https://doi.org/10.3390/ijms22126368
Chicago/Turabian StyleCortada, Maurizio, Soledad Levano, and Daniel Bodmer. 2021. "mTOR Signaling in the Inner Ear as Potential Target to Treat Hearing Loss" International Journal of Molecular Sciences 22, no. 12: 6368. https://doi.org/10.3390/ijms22126368