
Tumor-Derived Exosomes (TEX) and Their Role in Immuno-Oncology



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. All sEV in Supernatants of Cultured Tumor Cells Are TEX
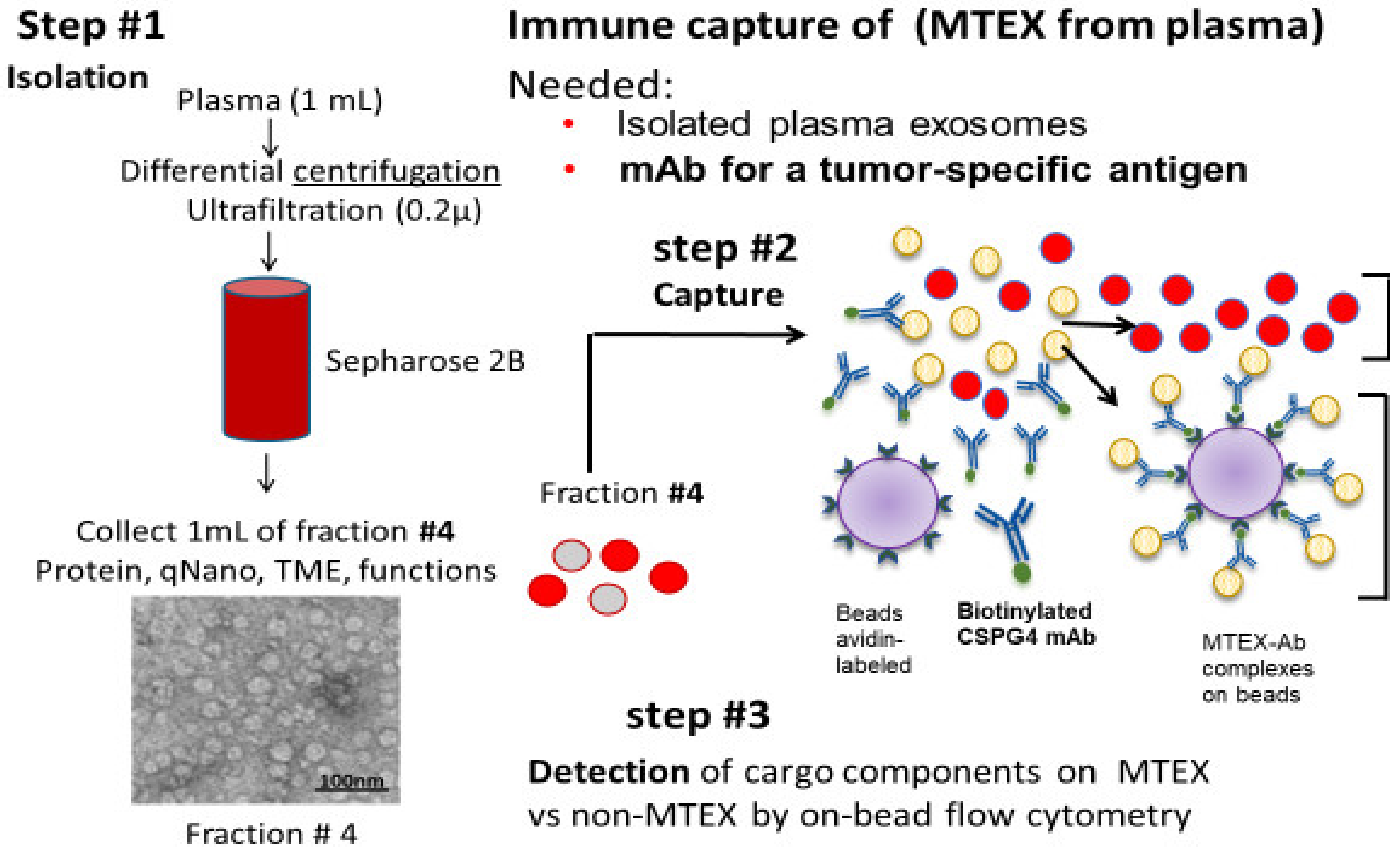
3. Cancer Patients’ Plasma Is a Complex Mix of Various sEV Types
4. TEX and Non-TEX Isolated from Cancer Patients’ Plasma Have Distinct Profiles
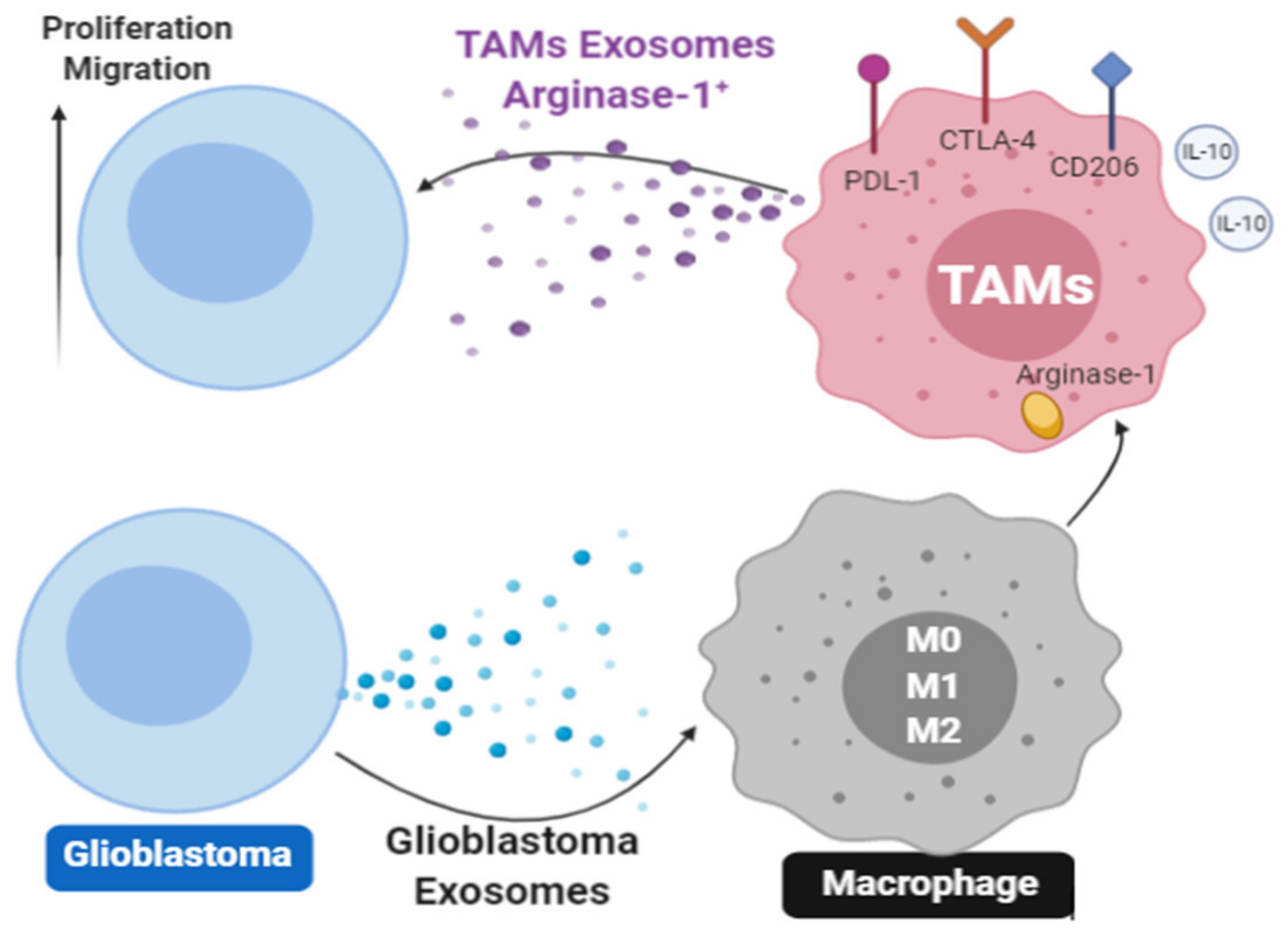
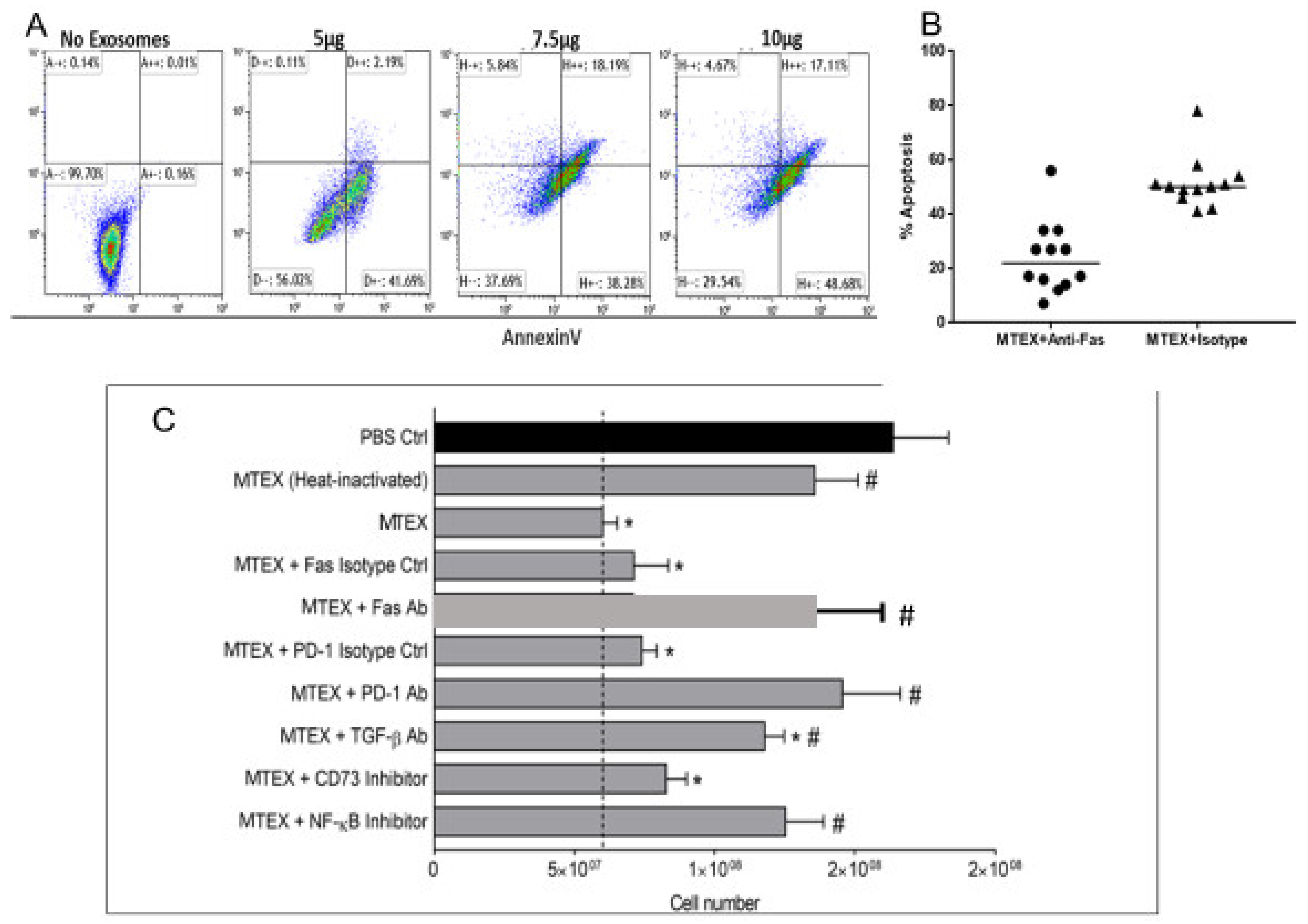
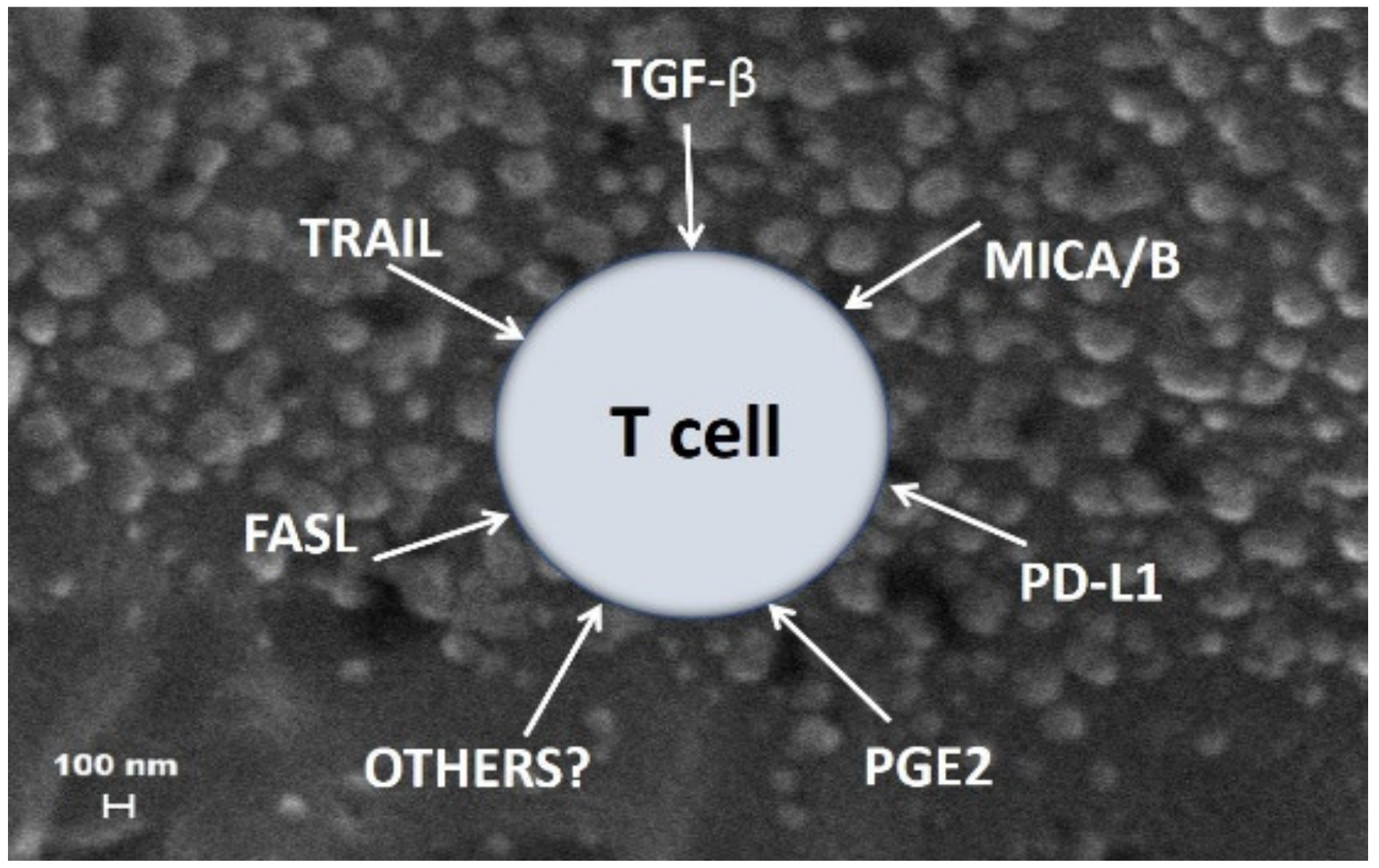
5. Mechanisms Involved in sEV-Mediated Immune Suppression in Cancer
6. Plasma sEV Derived from Non-Malignant Cells Are Reprogrammed to Mediate Immune Suppression in Cancer
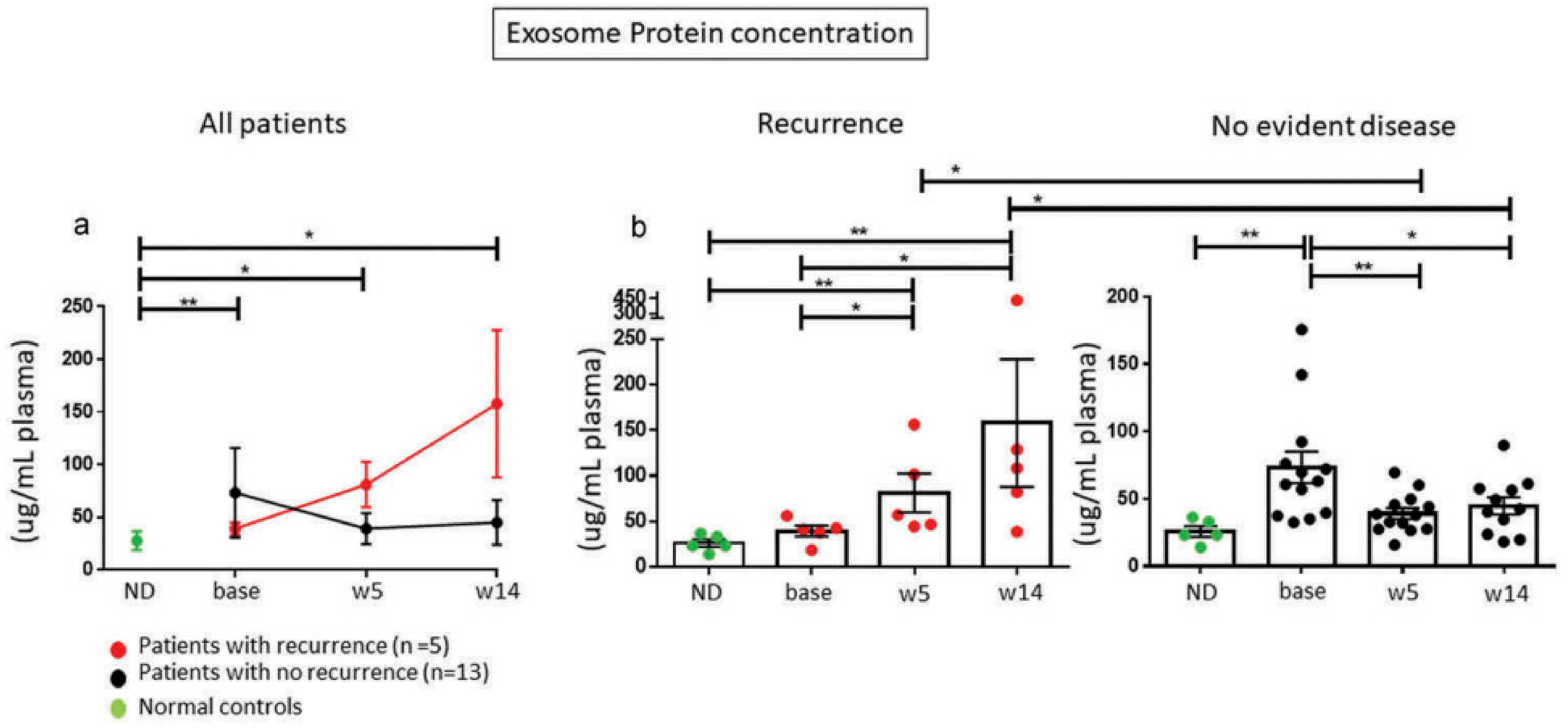
7. Plasma sEV as Monitors of Cancer Patients’ Responses to Oncological Therapy
8. Future Directions
Author Contributions
Funding
Conflicts of Interest
References
- Console, L.; Scalise, M.; Indiveri, C. Exosomes in inflammation and role as biomarkers. Clin. Chim. Acta 2019, 488, 165–171. [Google Scholar] [CrossRef]
- Huang, T.; Deng, C.X. Current Progresses of Exosomes as Cancer Diagnostic and Prognostic Biomarkers. Int. J. Biol. Sci. 2019, 15, 1–11. [Google Scholar] [CrossRef]
- Cui, S.; Cheng, Z.; Qin, W.; Jiang, L. Exosomes as a liquid biopsy for lung cancer. Lung Cancer 2018, 116, 46–54. [Google Scholar] [CrossRef]
- LeBleu, V.S.; Kalluri, R. Exosomes as a Multicomponent Biomarker Platform in Cancer. Trends Cancer 2020, 6, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. The potential of tumor-derived exosomes for noninvasive cancer monitoring: An update. Expert Rev. Mol. Diagn. 2018, 18, 1029–1040. [Google Scholar] [CrossRef]
- Mashouri, L.; Yousefi, H.; Aref, A.R.; Ahadi, A.M.; Molaei, F.; Alahari, S.K. Exosomes: Composition, biogenesis, and mechanisms in cancer metastasis and drug resistance. Mol. Cancer 2019, 18, 75. [Google Scholar] [CrossRef]
- Olejarz, W.; Dominiak, A.; Zolnierzak, A.; Kubiak-Tomaszewska, G.; Lorenc, T. Tumor-Derived Exosomes in Immunosuppression and Immunotherapy. J. Immunol. Res. 2020, 2020, 6272498. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. Tumor-Derived Exosomes and Their Role in Tumor-Induced Immune Suppression. Vaccines 2016, 4, 35. [Google Scholar] [CrossRef] [PubMed]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K.; et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zijlstra, A.; Di Vizio, D. Size matters in nanoscale communication. Nat. Cell Biol. 2018, 20, 228–230. [Google Scholar] [CrossRef] [PubMed]
- Hoshino, A.; Kim, H.S.; Bojmar, L.; Gyan, K.E.; Cioffi, M.; Hernandez, J.; Zambirinis, C.P.; Rodrigues, G.; Molina, H.; Heissel, S.; et al. Extracellular Vesicle and Particle Biomarkers Define Multiple Human Cancers. Cell 2020, 182, 1044–1061.e18. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Lyden, D. Asymmetric-flow field-flow fractionation technology for exomere and small extracellular vesicle separation and characterization. Nat. Protoc. 2019, 14, 1027–1053. [Google Scholar] [CrossRef] [PubMed]
- Mathieu, M.; Martin-Jaular, L.; Lavieu, G.; Thery, C. Specificities of secretion and uptake of exosomes and other extracellular vesicles for cell-to-cell communication. Nat. Cell Biol. 2019, 21, 9–17. [Google Scholar] [CrossRef] [PubMed]
- McAndrews, K.M.; Kalluri, R. Mechanisms associated with biogenesis of exosomes in cancer. Mol. Cancer 2019, 18, 52. [Google Scholar] [CrossRef]
- Hessvik, N.P.; Llorente, A. Current knowledge on exosome biogenesis and release. Cell Mol. Life Sci. 2018, 75, 193–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czystowska-Kuzmicz, M.; Whiteside, T.L. The potential role of tumor-derived exosomes in diagnosis, prognosis, and response to therapy in cancer. Expert Opin. Biol. 2020, 1–18. [Google Scholar]
- Whiteside, T.L. Exosome and mesenchymal stem cell cross-talk in the tumor microenvironment. Semin. Immunol. 2018, 35, 69–79. [Google Scholar] [CrossRef]
- Beccard, I.J.; Hofmann, L.; Schroeder, J.C.; Ludwig, S.; Laban, S.; Brunner, C.; Lotfi, R.; Hoffmann, T.K.; Jackson, E.K.; Schuler, P.J.; et al. Immune Suppressive Effects of Plasma-Derived Exosome Populations in Head and Neck Cancer. Cancers 2020, 12, 1997. [Google Scholar] [CrossRef] [PubMed]
- Theodoraki, M.N.; Hoffmann, T.K.; Whiteside, T.L. Separation of plasma-derived exosomes into CD3((+)) and CD3((−)) fractions allows for association of immune cell and tumour cell markers with disease activity in HNSCC patients. Clin. Exp. Immunol. 2018, 192, 271–283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atay, S.; Godwin, A.K. Tumor-derived exosomes: A message delivery system for tumor progression. Commun. Integr. Biol. 2014, 7, e28231. [Google Scholar] [CrossRef]
- Graner, M.W.; Schnell, S.; Olin, M.R. Tumor-derived exosomes, microRNAs, and cancer immune suppression. Semin. Immunopathol. 2018, 40, 505–515. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, A.; Tousif, S.; Wang, Y.; Hough, K.; Khan, S.; Strenkowski, J.; Chacko, B.K.; Darley-Usmar, V.M.; Deshane, J.S. Lung Tumor Cell-Derived Exosomes Promote M2 Macrophage Polarization. Cells 2020, 9, 1303. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. Tumor-Derived Exosomes and Their Role in Cancer Progression. Adv. Clin. Chem. 2016, 74, 103–141. [Google Scholar] [PubMed] [Green Version]
- Kim, J.W.; Wieckowski, E.; Taylor, D.D.; Reichert, T.E.; Watkins, S.; Whiteside, T.L. Fas ligand-positive membranous vesicles isolated from sera of patients with oral cancer induce apoptosis of activated T lymphocytes. Clin. Cancer Res. 2005, 11, 1010–1020. [Google Scholar]
- Taylor, D.D.; Gercel-Taylor, C.; Lyons, K.S.; Stanson, J.; Whiteside, T.L. T-cell apoptosis and suppression of T-cell receptor/CD3-zeta by Fas ligand-containing membrane vesicles shed from ovarian tumors. Clin. Cancer Res. 2003, 9, 5113–5119. [Google Scholar]
- Wieckowski, E.U.; Visus, C.; Szajnik, M.; Szczepanski, M.J.; Storkus, W.J.; Whiteside, T.L. Tumor-derived microvesicles promote regulatory T cell expansion and induce apoptosis in tumor-reactive activated CD8+ T lymphocytes. J. Immunol. 2009, 183, 3720–3730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boniface, J.D.; Poschke, I.; Mao, Y.; Kiessling, R. Tumor-dependent down-regulation of the zeta-chain in T-cells is detectable in early breast cancer and correlates with immune cell function. Int. J. Cancer 2012, 131, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. Down-regulation of zeta-chain expression in T cells: A biomarker of prognosis in cancer? Cancer Immunol. Immunother. 2004, 53, 865–878. [Google Scholar] [CrossRef] [PubMed]
- Clayton, A.; Mitchell, J.P.; Court, J.; Mason, M.D.; Tabi, Z. Human tumor-derived exosomes selectively impair lymphocyte responses to interleukin-2. Cancer Res. 2007, 67, 7458–7466. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Czystowska, M.; Han, J.; Szczepanski, M.J.; Szajnik, M.; Quadrini, K.; Brandwein, H.; Hadden, J.W.; Signorelli, K.; Whiteside, T.L. IRX-2, a novel immunotherapeutic, protects human T cells from tumor-induced cell death. Cell Death Differ. 2009, 16, 708–718. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludwig, S.; Sharma, P.; Theodoraki, M.N.; Pietrowska, M.; Yerneni, S.S.; Lang, S.; Ferrone, S.; Whiteside, T.L. Molecular and Functional Profiles of Exosomes From HPV(+) and HPV(-) Head and Neck Cancer Cell Lines. Front. Oncol. 2018, 8, 445. [Google Scholar] [CrossRef]
- Szajnik, M.; Czystowska, M.; Szczepanski, M.J.; Mandapathil, M.; Whiteside, T.L. Tumor-derived microvesicles induce, expand and up-regulate biological activities of human regulatory T cells (Treg). PLoS ONE 2010, 5, e11469. [Google Scholar] [CrossRef] [Green Version]
- Whiteside, T.L. Immune modulation of T-cell and NK (natural killer) cell activities by TEXs (tumour-derived exosomes). Biochem. Soc. Trans. 2013, 41, 245–251. [Google Scholar] [CrossRef]
- Muller, L.; Simms, P.; Hong, C.S.; Nishimura, M.I.; Jackson, E.K.; Watkins, S.C.; Whiteside, T.L. Human tumor-derived exosomes (TEX) regulate Treg functions via cell surface signaling rather than uptake mechanisms. Oncoimmunology 2017, 6, e1261243. [Google Scholar] [CrossRef] [PubMed]
- Muller, L.; Mitsuhashi, M.; Simms, P.; Gooding, W.E.; Whiteside, T.L. Tumor-derived exosomes regulate expression of immune function-related genes in human T cell subsets. Sci. Rep. 2016, 6, 20254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Szczepanski, M.J.; Szajnik, M.; Welsh, A.; Whiteside, T.L.; Boyiadzis, M. Blast-derived microvesicles in sera from patients with acute myeloid leukemia suppress natural killer cell function via membrane-associated transforming growth factor-beta1. Haematologica 2011, 96, 1302–1309. [Google Scholar] [CrossRef]
- Syn, N.; Wang, L.; Sethi, G.; Thiery, J.P.; Goh, B.C. Exosome-Mediated Metastasis: From Epithelial-Mesenchymal Transition to Escape from Immunosurveillance. Trends Pharm. Sci. 2016, 37, 606–617. [Google Scholar] [CrossRef] [PubMed]
- Schuler, P.J.; Saze, Z.; Hong, C.S.; Muller, L.; Gillespie, D.G.; Cheng, D.; Harasymczuk, M.; Mandapathil, M.; Lang, S.; Jackson, E.K.; et al. Human CD4(+) CD39(+) regulatory T cells produce adenosine upon co-expression of surface CD73 or contact with CD73(+) exosomes or CD73(+) cells. Clin. Exp. Immunol. 2014, 177, 531–543. [Google Scholar] [CrossRef] [PubMed]
- Figueiro, F.; Muller, L.; Funk, S.; Jackson, E.K.; Battastini, A.M.; Whiteside, T.L. Phenotypic and functional characteristics of CD39high human regulatory B cells (Breg). Oncoimmunology 2016, 5, e1082703. [Google Scholar] [CrossRef] [Green Version]
- Bretz, N.P.; Ridinger, J.; Rupp, A.K.; Rimbach, K.; Keller, S.; Rupp, C.; Marme, F.; Umansky, L.; Umansky, V.; Eigenbrod, T.; et al. Body fluid exosomes promote secretion of inflammatory cytokines in monocytic cells via Toll-like receptor signaling. J. Biol. Chem. 2013, 288, 36691–36702. [Google Scholar] [CrossRef] [Green Version]
- Xiang, X.; Poliakov, A.; Liu, C.; Liu, Y.; Deng, Z.B.; Wang, J.; Cheng, Z.; Shah, S.V.; Wang, G.J.; Zhang, L.; et al. Induction of myeloid-derived suppressor cells by tumor exosomes. Int. J. Cancer 2009, 124, 2621–2633. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Xiang, X.; Zhuang, X.; Zhang, S.; Liu, C.; Cheng, Z.; Michalek, S.; Grizzle, W.; Zhang, H.G. Contribution of MyD88 to the tumor exosome-mediated induction of myeloid derived suppressor cells. Am. J. Pathol. 2010, 176, 2490–2499. [Google Scholar] [CrossRef] [PubMed]
- Theodoraki, M.N.; Hoffmann, T.K.; Jackson, E.K.; Whiteside, T.L. Exosomes in HNSCC plasma as surrogate markers of tumour progression and immune competence. Clin. Exp. Immunol. 2018, 194, 67–78. [Google Scholar] [CrossRef] [Green Version]
- Azambuja, J.H.; Ludwig, N.; Yerneni, S.; Rao, A.; Braganhol, E.; Whiteside, T.L. Molecular profiles and immunomodulatory activities of glioblastoma-derived exosomes. Neurooncol. Adv. 2020, 2, vdaa056. [Google Scholar] [CrossRef] [PubMed]
- Gao, L.; Wang, L.; Dai, T.; Jin, K.; Zhang, Z.; Wang, S.; Xie, F.; Fang, P.; Yang, B.; Huang, H.; et al. Tumor-derived exosomes antagonize innate antiviral immunity. Nat. Immunol. 2018, 19, 233–245. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, N.; Yerneni, S.S.; Razzo, B.M.; Whiteside, T.L. Exosomes from HNSCC Promote Angiogenesis through Reprogramming of Endothelial Cells. Mol. Cancer Res. 2018, 16, 1798–1808. [Google Scholar] [CrossRef] [Green Version]
- Razzo, B.M.; Ludwig, N.; Hong, C.S.; Sharma, P.; Fabian, K.P.; Fecek, R.J.; Storkus, W.J.; Whiteside, T.L. Tumor-derived exosomes promote carcinogenesis of murine oral squamous cell carcinoma. Carcinogenesis 2019, 41, 625–633. [Google Scholar] [CrossRef]
- Brinton, L.T.; Sloane, H.S.; Kester, M.; Kelly, K.A. Formation and role of exosomes in cancer. Cell Mol. Life Sci. 2015, 72, 659–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludwig, N.; Whiteside, T.L.; Reichert, T.E. Challenges in Exosome Isolation and Analysis in Health and Disease. Int. J. Mol. Sci. 2019, 20, 4684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Yuan, X.; Shi, H.; Wu, L.; Qian, H.; Xu, W. Exosomes in cancer: Small particle, big player. J. Hematol. Oncol. 2015, 8, 83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gurunathan, S.; Kang, M.H.; Jeyaraj, M.; Qasim, M.; Kim, J.H. Review of the Isolation, Characterization, Biological Function, and Multifarious Therapeutic Approaches of Exosomes. Cells 2019, 8, 307. [Google Scholar] [CrossRef] [Green Version]
- Yang, D.; Zhang, W.; Zhang, H.; Zhang, F.; Chen, L.; Ma, L.; Larcher, L.M.; Chen, S.; Liu, N.; Zhao, Q.; et al. Progress, opportunity, and perspective on exosome isolation—Efforts for efficient exosome-based theranostics. Theranostics 2020, 10, 3684–3707. [Google Scholar] [CrossRef]
- Zhu, L.; Sun, H.T.; Wang, S.; Huang, S.L.; Zheng, Y.; Wang, C.Q.; Hu, B.Y.; Qin, W.; Zou, T.T.; Fu, Y.; et al. Isolation and characterization of exosomes for cancer research. J. Hematol. Oncol. 2020, 13, 152. [Google Scholar] [CrossRef] [PubMed]
- Thery, C.; Amigorena, S.; Raposo, G.; Clayton, A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 2006, 30, 3–22. [Google Scholar] [CrossRef]
- Hong, C.S.; Funk, S.; Muller, L.; Boyiadzis, M.; Whiteside, T.L. Isolation of biologically active and morphologically intact exosomes from plasma of patients with cancer. J. Extracell. Vesicles 2016, 5, 29289. [Google Scholar] [CrossRef]
- Hong, C.S.; Sharma, P.; Yerneni, S.S.; Simms, P.; Jackson, E.K.; Whiteside, T.L.; Boyiadzis, M. Circulating exosomes carrying an immunosuppressive cargo interfere with cellular immunotherapy in acute myeloid leukemia. Sci. Rep. 2017, 7, 14684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ludwig, S.; Floros, T.; Theodoraki, M.N.; Hong, C.S.; Jackson, E.K.; Lang, S.; Whiteside, T.L. Suppression of Lymphocyte Functions by Plasma Exosomes Correlates with Disease Activity in Patients with Head and Neck Cancer. Clin. Cancer Res. 2017, 23, 4843–4854. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, P.; Ludwig, S.; Muller, L.; Hong, C.S.; Kirkwood, J.M.; Ferrone, S.; Whiteside, T.L. Immunoaffinity-based isolation of melanoma cell-derived exosomes from plasma of patients with melanoma. J. Extracell. Vesicles 2018, 7, 1435138. [Google Scholar] [CrossRef] [PubMed]
- Campoli, M.; Ferrone, S.; Wang, X. Functional and clinical relevance of chondroitin sulfate proteoglycan 4. Adv. Cancer Res. 2010, 109, 73–121. [Google Scholar]
- Wang, X.; Katayama, A.; Wang, Y.; Yu, L.; Favoino, E.; Sakakura, K.; Favole, A.; Tsuchikawa, T.; Silver, S.; Watkins, S.C.; et al. Functional characterization of an scFv-Fc antibody that immunotherapeutically targets the common cancer cell surface proteoglycan CSPG4. Cancer Res. 2011, 71, 7410–7422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Sabbatino, F.; Wang, X.; Ferrone, S. Detection of chondroitin sulfate proteoglycan 4 (CSPG4) in melanoma. Methods Mol. Biol. 2014, 1102, 523–535. [Google Scholar]
- Chang, C.C.; Campoli, M.; Luo, W.; Zhao, W.; Zaenker, K.S.; Ferrone, S. Immunotherapy of melanoma targeting human high molecular weight melanoma-associated antigen: Potential role of nonimmunological mechanisms. Ann. N. Y. Acad. Sci. 2004, 1028, 340–350. [Google Scholar] [CrossRef] [PubMed]
- Whiteside, T.L. Exosomes carrying immunoinhibitory proteins and their role in cancer. Clin. Exp. Immunol. 2017, 189, 259–267. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Diergaarde, B.; Ferrone, S.; Kirkwood, J.M.; Whiteside, T.L. Melanoma cell-derived exosomes in plasma of melanoma patients suppress functions of immune effector cells. Sci. Rep. 2020, 10, 92. [Google Scholar] [CrossRef] [PubMed]
- Theodoraki, M.-N.; Hong, C.-S.; Donnenberg, V.S.; Donnenberg, A.D.; Whiteside, T.L. Evaluation of Exosome Proteins by on-Bead Flow Cytometry. Cytometry Part A 2020. [Google Scholar] [CrossRef]
- Rivoltini, L.; Chiodoni, C.; Squarcina, P.; Tortoreto, M.; Villa, A.; Vergani, B.; Burdek, M.; Botti, L.; Arioli, I.; Cova, A.; et al. TNF-Related Apoptosis-Inducing Ligand (TRAIL)-Armed Exosomes Deliver Proapoptotic Signals to Tumor Site. Clin. Cancer Res. 2016, 22, 3499–3512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulcahy, L.A.; Pink, R.C.; Carter, D.R. Routes and mechanisms of extracellular vesicle uptake. J. Extracell Vesicles 2014, 3, 24641. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peinado, H.; Aleckovic, M.; Lavotshkin, S.; Matei, I.; Costa-Silva, B.; Moreno-Bueno, G.; Hergueta-Redondo, M.; Williams, C.; Garcia-Santos, G.; Ghajar, C.; et al. Melanoma exosomes educate bone marrow progenitor cells toward a pro-metastatic phenotype through MET. Nat. Med. 2012, 18, 883–891. [Google Scholar] [CrossRef] [Green Version]
- Azambuja, J.H.; Ludwig, N.; Yerneni, S.S.; Braganhol, E.; Whiteside, T.L. Arginase-1+ Exosomes from Reprogrammed Macrophages Promote Glioblastoma Progression. Int. J. Mol. Sci. 2020, 21, 3990. [Google Scholar] [CrossRef] [PubMed]
- Barbato, S.; Solaini, G.; Fabbri, M. MicroRNAs in Oncogenesis and Tumor Suppression. Int. Rev. Cell Mol. Biol. 2017, 333, 229–268. [Google Scholar]
- Whiteside, T.L. Exosomes and tumor-mediated immune suppression. J. Clin. Investig. 2016, 126, 1216–1223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theodoraki, M.N.; Yerneni, S.S.; Brunnner, C.; Theodorakis, J.; Hoffmann, T.K.; Whiteside, T.L. Plasma-derived exosomes reverse epithelial-to-mesenchymal transition after photodynamic therapy of patients with head and neck cancer. Oncoscience 2018, in press. [Google Scholar] [CrossRef] [Green Version]
- Theodoraki, M.-N.; Matsumoto, A.; Beccard, I.; Hoffmann, T.K.; Whiteside, T.L. CD44v3 protein-carrying tumor-derived exosomes in HNSCC patients’ plasma as potential noninvasive biomarkers of disease activity. Oncoimmunology 2020, 9, 1747732. [Google Scholar] [CrossRef] [Green Version]
- Orian-Rousseau, V.; Sleeman, J. CD44 is a multidomain signaling platform that integrates extracellular matrix cues with growth factor and cytokine signals. Adv. Cancer Res. 2014, 123, 231–254. [Google Scholar] [PubMed]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H.; et al. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018, 560, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Zhang, P.; Wang, Y.; Wang, J.; Su, M.; Wang, Y.; Zhou, L.; Zhou, J.; Xiong, W.; Zeng, Z.; et al. The Biogenesis, Biology, and Clinical Significance of Exosomal PD-L1 in Cancer. Front. Immunol. 2020, 11, 604. [Google Scholar] [CrossRef] [PubMed]
- Theodoraki, M.N.; Yerneni, S.S.; Hoffmann, T.K.; Gooding, W.E.; Whiteside, T.L. Clinical Significance of PD-L1(+) Exosomes in Plasma of Head and Neck Cancer Patients. Clin. Cancer Res. 2018, 24, 896–905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, B.; Hu, X.; Ding, Y.; Liu, M. Tumor-derived exosomes in the PD-1/PD-L1 axis: Significant regulators as well as promising clinical targets. J. Cell. Physiol. 2020. [Google Scholar] [CrossRef]
- Xie, F.; Xu, M.; Lu, J.; Mao, L.; Wang, S. The role of exosomal PD-L1 in tumor progression and immunotherapy. Mol. Cancer 2019, 18, 146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Theodoraki, M.N.; Yerneni, S.; Gooding, W.E.; Ohr, J.; Clump, D.A.; Bauman, J.E.; Ferris, R.L.; Whiteside, T.L. Circulating exosomes measure responses to therapy in head and neck cancer patients treated with cetuximab, ipilimumab, and IMRT. Oncoimmunology 2019, 8, 1593805. [Google Scholar] [CrossRef] [PubMed]
- Cordonnier, M.; Nardin, C.; Chanteloup, G.; Derangere, V.; Algros, M.P.; Arnould, L.; Garrido, C.; Aubin, F.; Gobbo, J. Tracking the evolution of circulating exosomal-PD-L1 to monitor melanoma patients. J. Extracell. Vesicles 2020, 9, 1710899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zebrowska, A.; Widlak, P.; Whiteside, T.; Pietrowska, M. Signaling of Tumor-Derived sEV Impacts Melanoma Progression. Int. J. Mol. Sci. 2020, 21, 5066. [Google Scholar] [CrossRef] [PubMed]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Whiteside, T.L.; Diergaarde, B.; Hong, C.-S. Tumor-Derived Exosomes (TEX) and Their Role in Immuno-Oncology. Int. J. Mol. Sci. 2021, 22, 6234. https://doi.org/10.3390/ijms22126234
Whiteside TL, Diergaarde B, Hong C-S. Tumor-Derived Exosomes (TEX) and Their Role in Immuno-Oncology. International Journal of Molecular Sciences. 2021; 22(12):6234. https://doi.org/10.3390/ijms22126234
Chicago/Turabian StyleWhiteside, Theresa L., Brenda Diergaarde, and Chang-Sook Hong. 2021. "Tumor-Derived Exosomes (TEX) and Their Role in Immuno-Oncology" International Journal of Molecular Sciences 22, no. 12: 6234. https://doi.org/10.3390/ijms22126234