
[β-Glu2]TRH Is a Functional Antagonist of Thyrotropin-Releasing Hormone (TRH) in the Rodent Brain




Abstract
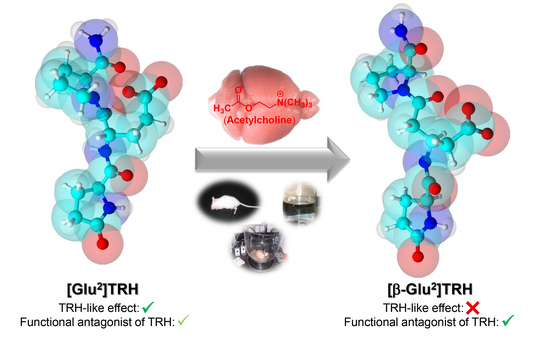
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
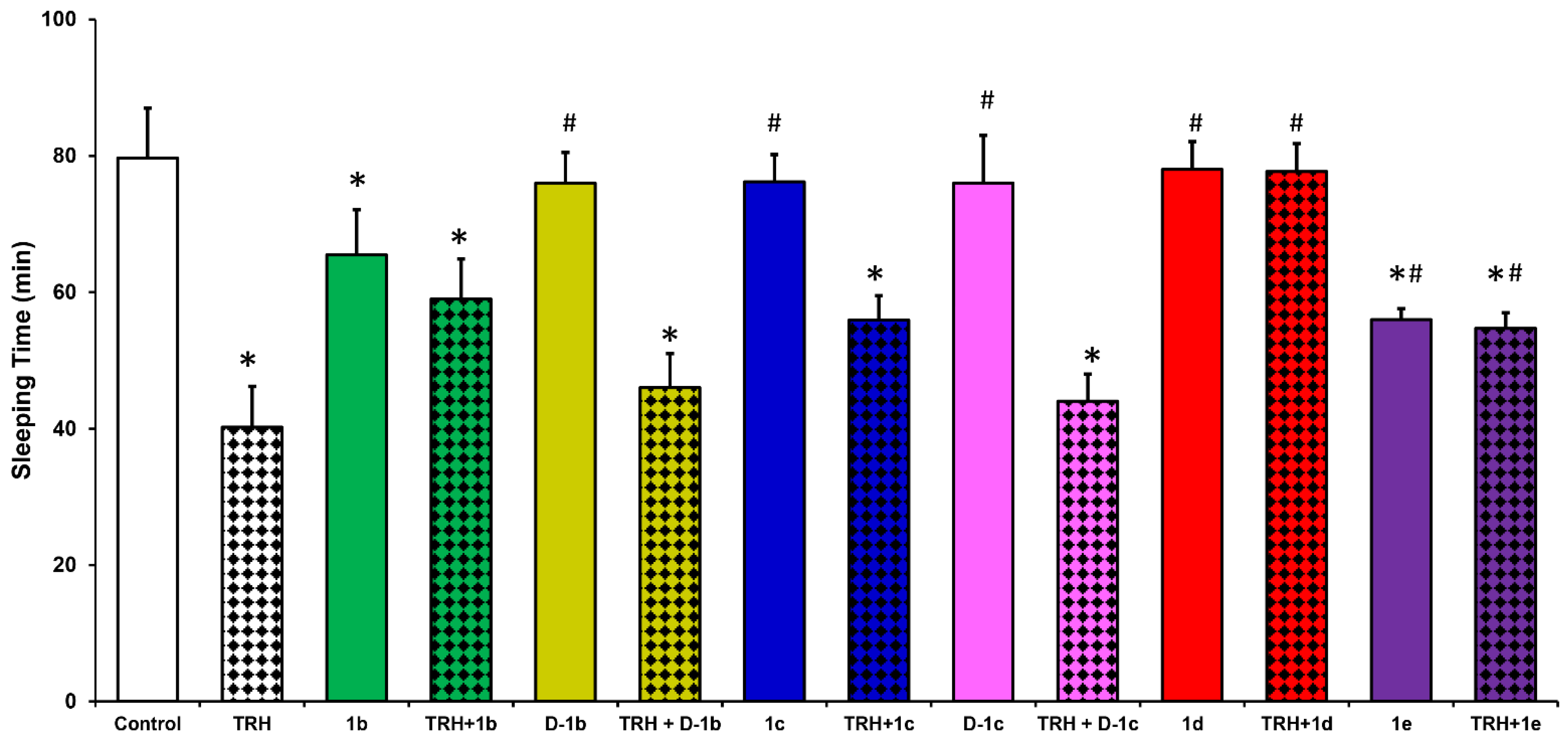
2.1. Analeptic Effects
2.2. Porsolt Swim Tests
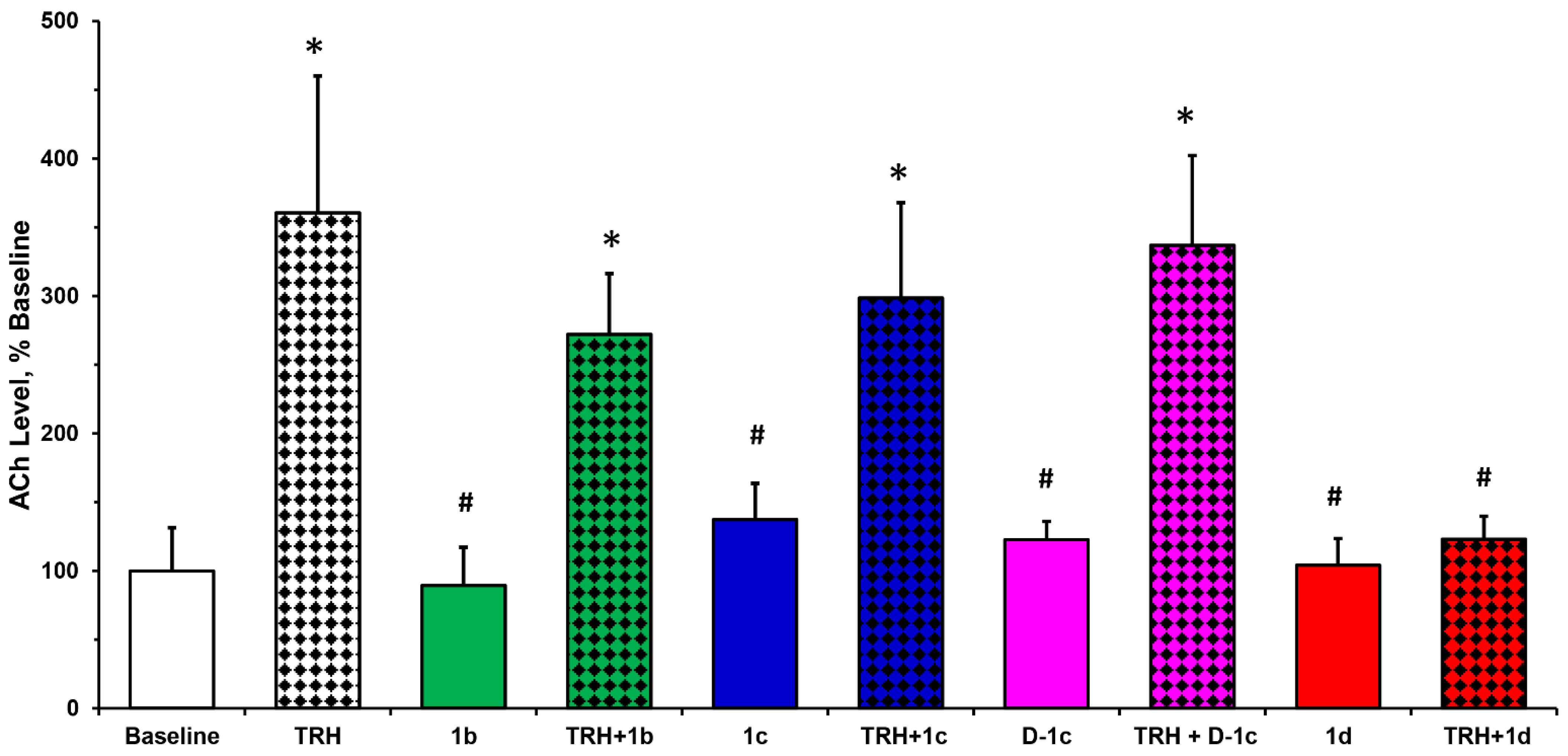
2.3. Hippocampal ACh Turnover
3. Materials and Methods
3.1. Materials
3.2. Animals
3.3. Instruments
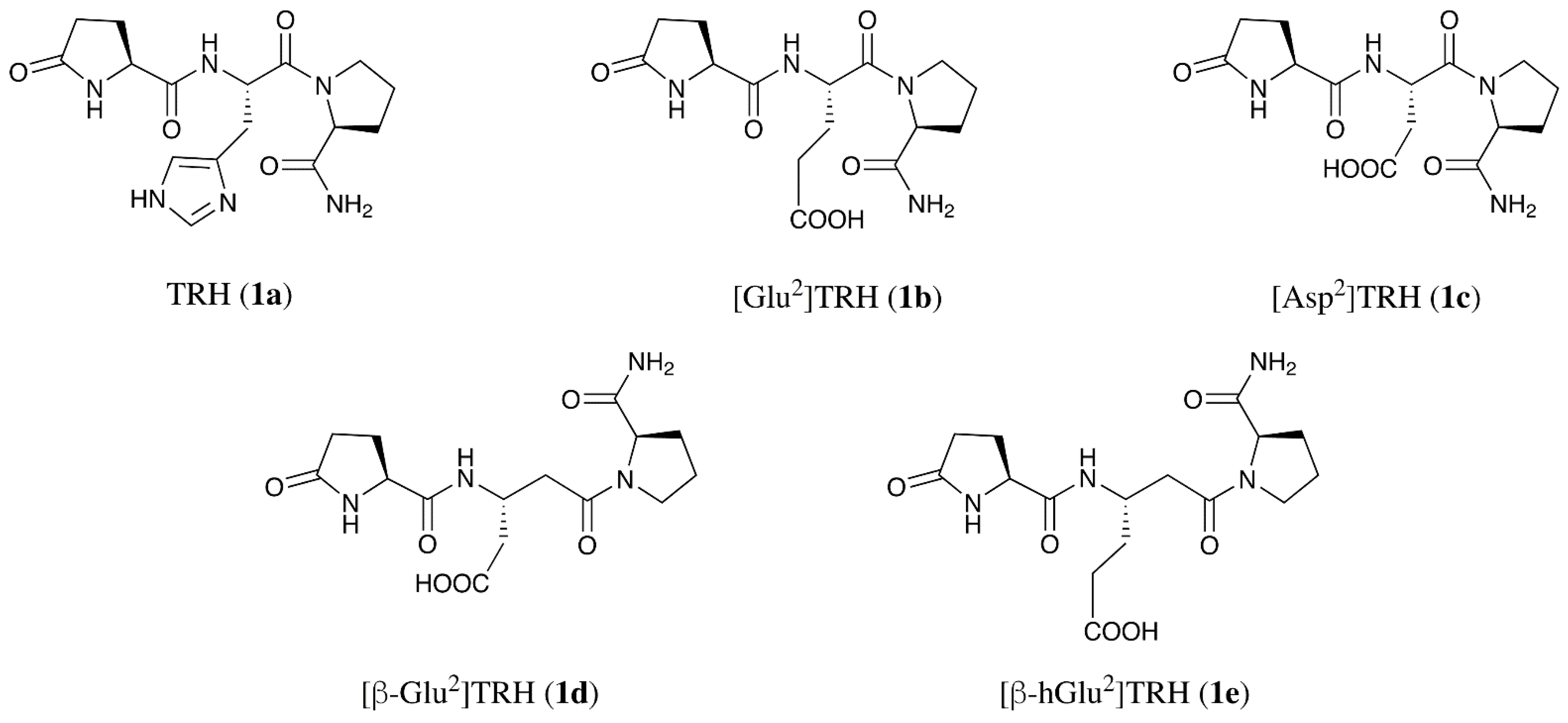
3.4. Synthesis
- [Asp2]TRH (1c): MS (ESI) m/z 340; Combustion analysis for C14H20N4O6 × 2 H2O: Calc.: C, 44.68, H, 6.43; N, 14.89. Found C, 44.74; H, 6.14; N, 14.90.
- [D-Asp2]TRH (D-1c): MS (ESI) m/z 340; combustion analysis for C14H20N4O6 × 2 H2O: C, 44.68, H, 6.43; N, 14.89. Found C, 44.96; H, 6.84; N, 14.61.
- [β-Glu2]TRH (1d): MS (ESI) m/z 354; Combustion analysis for C15H22N4O6 × 0.5 H2O, Calc.: C, 49.58, H, 6.38; N, 15.42. Found C, 49.53; H, 6.26; N, 15.58.
- [β-hGlu2]TRH (1e): MS (ESI) m/z 368; Combustion analysis for C16H24N4O6 × H2O × 0.5 TFA, Calc.: C, 46.05, H, 6.02; N, 12.64. Found C, 46.13; H, 5.73; N, 12.36.
3.5. Assessment of Analeptic Activity
3.6. Assessment of Antidepressent-Like Activity
3.7. Measurement of Hippocampal ACh Levels
3.8. Data Analysis
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Breese, G.R.; Mueller, R.A.; Mailman, R.B.; Frye, G.D. Effects of TRH on central nervous system function. Review Prog. Clin. Biol. Res. 1981, 68, 99–116. [Google Scholar]
- Shibusawa, N.; Hashimoto, K.; Yamada, M. Thyrotropin-releasing hormone (TRH) in the cerebellum. Cerebellum 2008, 7, 84–95. [Google Scholar] [CrossRef]
- Yarbrough, G.G. Thyrotropin releasing hormone and CNS cholinergic neurons. Life Sci. 1983, 33, 111–118. [Google Scholar] [CrossRef]
- Yarbrough, G.G. TRH interactions with cholinergic mechanisms and consequent therapeutic implications. In Psychoneuroendocrine Dysfunction; Ryall, R., Kelly, J.S., Eds.; Springer: Boston, MA, USA, 1984; pp. 73–81. [Google Scholar]
- Marangell, L.B.; George, M.S.; Callahan, A.M.; Ketter, T.A.; Pazzaglia, P.J.; L’Herrou, T.A.; Leverich, G.S.; Post, R.M. Effects of intrathecal thyrotropin-releasing hormone (Protirelin) in refractory depressed patients. Arch. Gen. Psych. 1997, 54, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Ushijima, I.; Yamada, K.; Furukawa, T. Acute and long-term effects of thyrotropin releasing hormone on behavior mediated by dopaminergic and cholinergic activities in mice. Psychopharmacology 1984, 82, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Duval, F. Thyroid hormone treatment of mood disorders. Curr. Treat. Options Psych. 2018, 5, 363–376. [Google Scholar] [CrossRef]
- Lehrer, S. Inhaled thyrotropin-releasing hormone for treatment of neuropsychiatric disorders. J. Clin. Psychopharmacol. 2014, 34, 288–290. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamath, J. Cancer-related fatigue, inflammation and thyrotropin-releasing hormone. Curr. Aging Sci. 2012, 5, 195–202. [Google Scholar] [CrossRef] [PubMed]
- Fröhlich, E.; Wahl, R. The forgotten effects of thyrotropin-releasing hormone: Metabolic functions and medical applications. Front. Neuroendocrin. 2019, 52, 29–43. [Google Scholar] [CrossRef]
- Takeuchi, Y.; Takano, T.; Abe, J.; Takikita, S.; Ohno, M. Thyrotropin-releasing hormone: Role in the treatment of West syndrome and related epileptic encephalopathies. Brain Dev. 2001, 23, 662–667. [Google Scholar] [CrossRef]
- Prokai, L. Central nervous system effects of thyrotropin-releasing hormone and its analogues: Opportunities and perspectives for drug discovery and development. In Progress in Drug Research; Jucker, J.M., Ed.; Birkhauser: Basel, Switzerland, 2002; Volume 59, pp. 133–170. [Google Scholar]
- Khomane, K.S.; Meena, C.L.; Jain, R.; Bansal, A.K. Novel thyrotropin-releasing hormone analogs: A patent review. Expert Opin. Ther. Pat. 2011, 21, 1673–1691. [Google Scholar] [CrossRef]
- Sun, Y.; Lu, X.; Gershengorn, M.C. Thyrotropin-releasing hormone receptors—Similarities and differences. J. Mol. Endocrinol. 2003, 30, 87–97. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hogan, N.; O’Boyle, K.M.; Hinkle, P.M.; Kelly, J.A. A novel TRH analog, Glp-Asn-Pro-D-Tyr-D-TrpNH2, binds to [3H][3-Me-His2]TRH-labelled sites in rat hippocampus and cortex but not pituitary or heterologous cells expressing TRHR1 or TRHR2. Neurosci Lett. 2008, 431, 26–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matre, V.; Karlsen, H.E.; Wright, M.S.; Lundell, I.; Fjeldheim, Å.K.; Gabrielsen, O.S.; Larhammar, D.; Gautvik, K.M. Molecular cloning of a functional human thyrotropin-releasing hormone receptor. Biochem. Biophys. Res. Commun. 1993, 195, 179–185. [Google Scholar] [CrossRef] [PubMed]
- Thirunarayanan, N.; Nir, E.A.; Raaka, B.M.; Gershengorn, M.C. Thyrotropin-releasing hormone receptor type 1 (TRH-R1), not TRH-R2, primarily mediates taltirelin actions in the CNS of mice. Neuropsychopharmacology 2013, 38, 950–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neumann, S.; Raaka, B.M.; Gershengorn, M.C. Constitutively active thyrotropin and thyrotropin-releasing hormone receptors and their inverse agonists. Methods Enzymol. 2010, 485, 147–160. [Google Scholar] [PubMed]
- Horita, A. An update on the CNS actions of TRH and its analogs. Life Sci. 1998, 17–18, 1443–1448. [Google Scholar] [CrossRef]
- Bílek, R.; Bičíková, M.; Šafařík, L. TRH-like peptides. Physiol. Res. 2011, 60, 207–215. [Google Scholar] [CrossRef]
- Pekary, A.E.; Sattin, A. TRH and TRH-like peptide levels co-vary with reproductive and metabolic rhythms. Horm. Metab. Res. 2017, 49, 86–94. [Google Scholar] [CrossRef]
- Pekary, E.A.; Faull, K.F.; Paulson, M.; Lloyd, R.L.; Sattin, A. TRH-like antidepressant peptide, pyroglutamyltyroslyprolineamide, occurs in rat brain. J. Mass Spectrom. 2005, 40, 1232–1236. [Google Scholar] [CrossRef]
- Prokai-Tatrai, K.; Nguyen, V.; Szarka, S.; Konya, K.; Prokai, L. Design and exploratory neuropharmacological evaluation of novel thyrotropin-releasing hormone analogs and their brain-targeting bioprecursor prodrugs. Pharmaceutics 2013, 5, 318–328. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, V.; Zharikova, A.D.; Prokai, L. Evidence for interplay between thyrotropin-releasing hormone (TRH) and its structural analogue pGlu-Glu-Pro-NH2 ([Glu2]TRH) in the brain: An in vivo microdialysis study. Neurosci. Lett. 2007, 415, 64–67. [Google Scholar] [CrossRef]
- Nguyen, V.; Zharikova, A.D.; Prokai-Tatrai, K.; Prokai, L. [Glu2]TRH dose-dependently attenuates TRH-evoked analeptic effect in mice. Brain Res. Bull. 2010, 82, 83–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Borgo, M.P.; Kulkarni, K.; Aguilar, M.I. Using β-amino acids and β-peptide templates to create bioactive ligands and biomaterials. Curr. Pharm. Design 2017, 26, 3772–3785. [Google Scholar] [CrossRef]
- Hinkle, P.M.; Pekary, E.A.; Senanayaki, S.; Sattin, A. Role of TRH receptors as possible mediators of analeptic actions of TRH-like peptides. Brain Res. 2002, 935, 59–64. [Google Scholar] [CrossRef]
- Lloyd, R.L.; Pekary, A.E.; Sattin, A.; Amundson, T. Antidepressant effects of thyrotropin-releasing hormone analogues using a rodent model of depression. Pharmacol. Biochem. Behav. 2001, 70, 15–22. [Google Scholar] [CrossRef]
- Prokai-Tatrai, K.; Teixido, M.; Nguyen, V.; Zharikova, A.D.; Prokai, L. A pyridinium-substituted analog of the TRH-like tripeptide pGlu-Glu-Pro-NH2 and its prodrugs as central nervous system agents. Med. Chem. 2005, 1, 141–152. [Google Scholar] [CrossRef] [PubMed]
- Prokai-Tatrai, K.; Prokai, L. Prodrugs of thyrotropin-releasing hormone and related peptides as central nervous system agents. Molecules 2009, 14, 633–654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prokai-Tatrai, K.; De La Cruz, D.L.; Nguyen, V.; Ross, B.R.; Toth, I.; Prokai, L. Brain delivery of thyrotropin-releasing hormone via a novel prodrug approach. Pharmaceutics 2019, 11, 349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castagné, V.; Moser, P.; Porsolt, R.D. Behavioral assessment of antidepressant activity in rodents. In Methods of Behavior Analysis in Neuroscience, 2nd ed.; Buccafusco, J.J., Ed.; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2009; Chapter 6; pp. 103–117. [Google Scholar]
- Zant, J.C.; Kim, T.; Prokai, L.; Szarka, S.; McNally, J.; McKenna, J.T.; Shukla, C.; Yang, C.; Kalinchuk, A.V.; McCarley, R.W.; et al. Cholinergic neurons in the basal forebrain promote wakefulness by actions on neighboring non-cholinergic neurons: An opto-dialysis study. J. Neurosci. 2016, 36, 2057–2067. [Google Scholar] [CrossRef] [Green Version]
- Cheng, Y.; Prusoff, W.H. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 percent inhibition (IC50) of an enzymatic reaction. Biochem. Pharmacol. 1973, 22, 3099–3108. [Google Scholar]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Prokai-Tatrai, K.; Nguyen, V.; Prokai, L. [β-Glu2]TRH Is a Functional Antagonist of Thyrotropin-Releasing Hormone (TRH) in the Rodent Brain. Int. J. Mol. Sci. 2021, 22, 6230. https://doi.org/10.3390/ijms22126230
Prokai-Tatrai K, Nguyen V, Prokai L. [β-Glu2]TRH Is a Functional Antagonist of Thyrotropin-Releasing Hormone (TRH) in the Rodent Brain. International Journal of Molecular Sciences. 2021; 22(12):6230. https://doi.org/10.3390/ijms22126230
Chicago/Turabian StyleProkai-Tatrai, Katalin, Vien Nguyen, and Laszlo Prokai. 2021. "[β-Glu2]TRH Is a Functional Antagonist of Thyrotropin-Releasing Hormone (TRH) in the Rodent Brain" International Journal of Molecular Sciences 22, no. 12: 6230. https://doi.org/10.3390/ijms22126230