
Neuroinflammation and Neuronal Loss in the Hippocampus Are Associated with Immediate Posttraumatic Seizures and Corticosterone Elevation in Rats


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
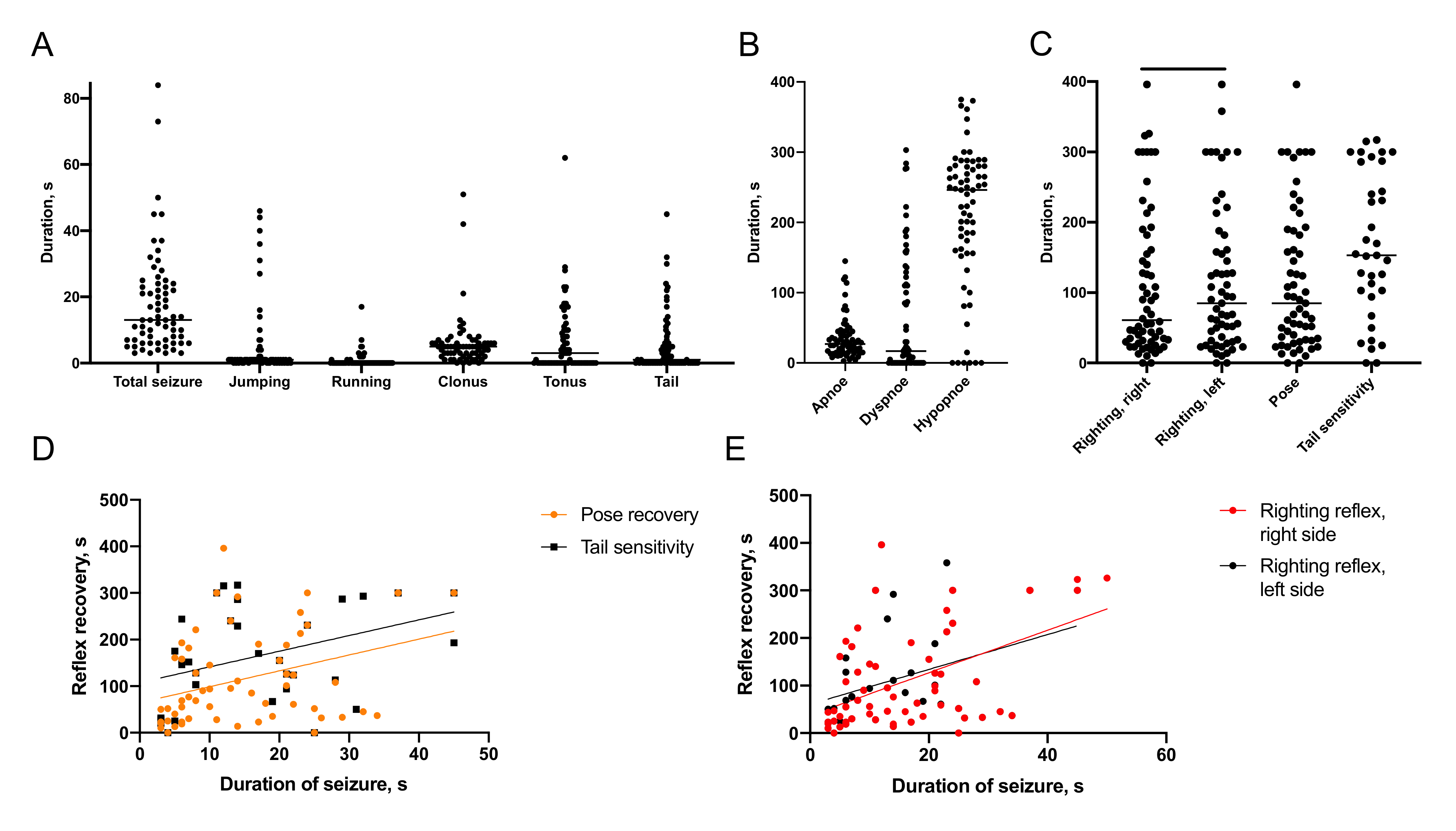
2.1. Characteristics of Immediate Posttraumatic Seizures in Rats
2.1.1. Semiology of Immediate Posttraumatic Seizures
2.1.2. Recovery of Breathing
2.1.3. Recovery of Reflexes
2.1.4. Association of Immediate Seizures with Recovery after TBI
2.1.5. Predictors of Acute Mortality
2.2. Distant Hippocampal Damage
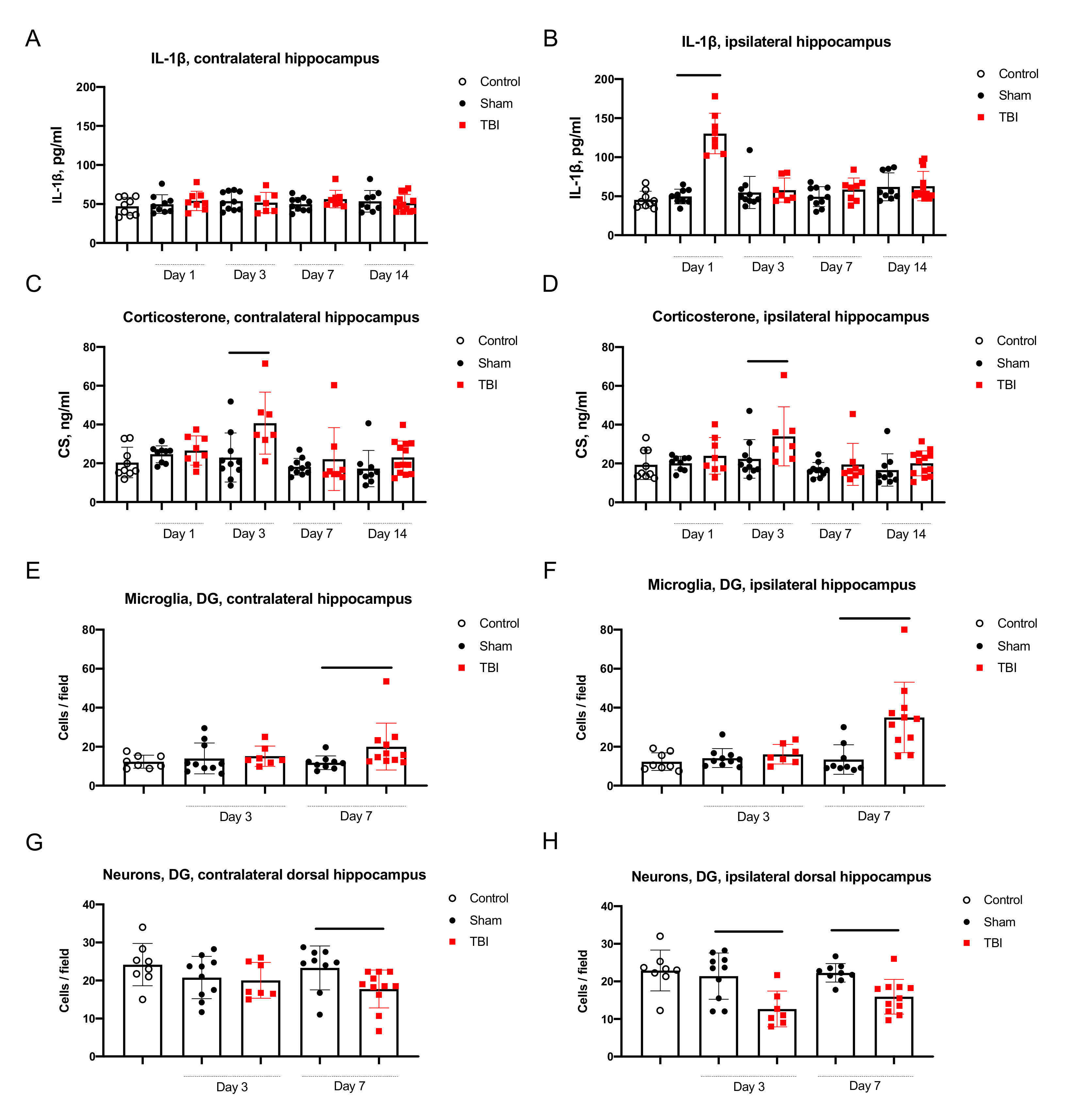
2.2.1. Neuroinflammation in the Hippocampus
2.2.2. Neurodegeneration in the Hippocampus
2.2.3. Corticosterone Elevation in Blood and the Hippocampus
2.3. Involvement of Immediate Seizure and Corticosterone Elevation in the Distant Hippocampal Damage
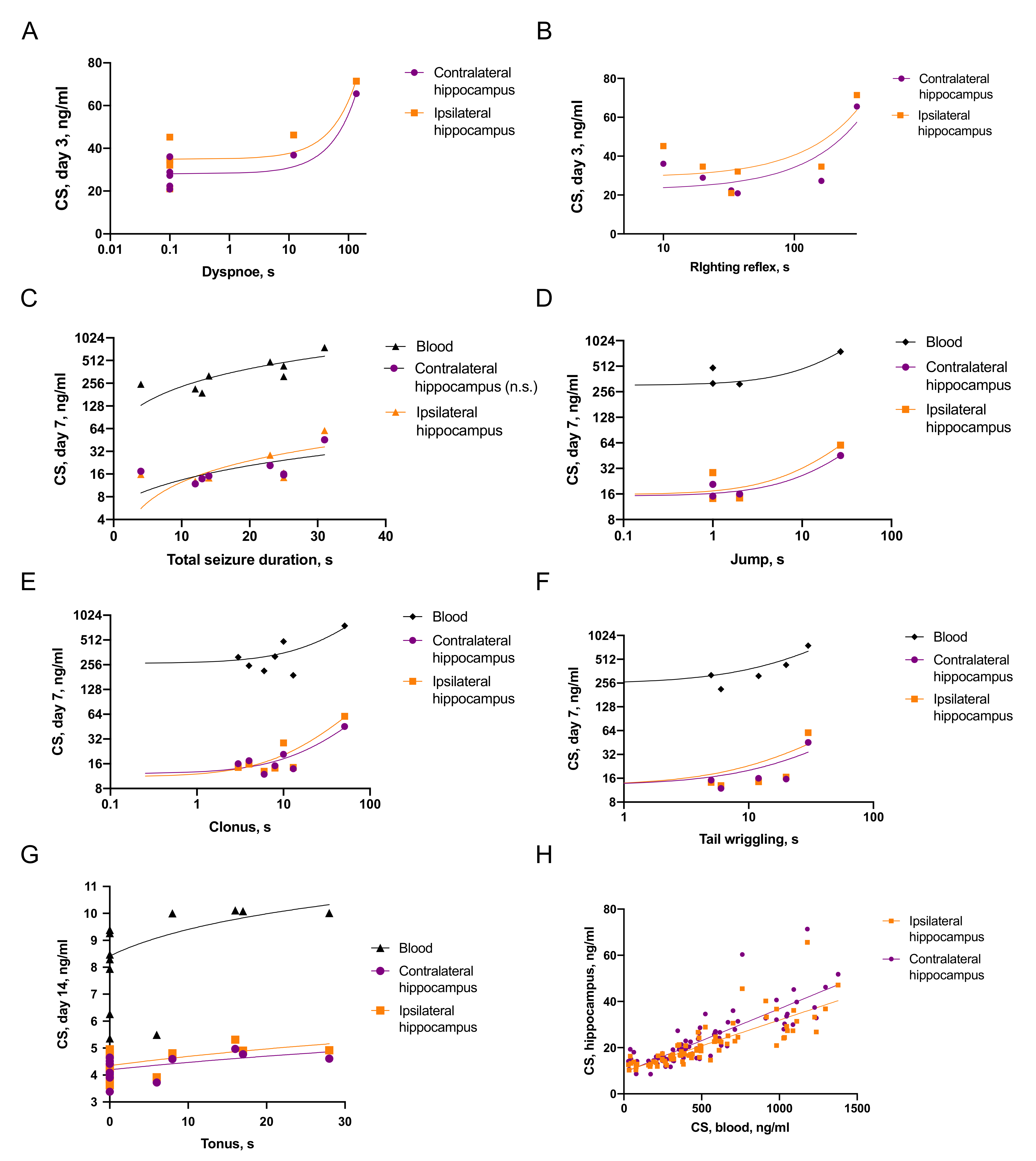
2.3.1. Associations of Seizures and CS Increase
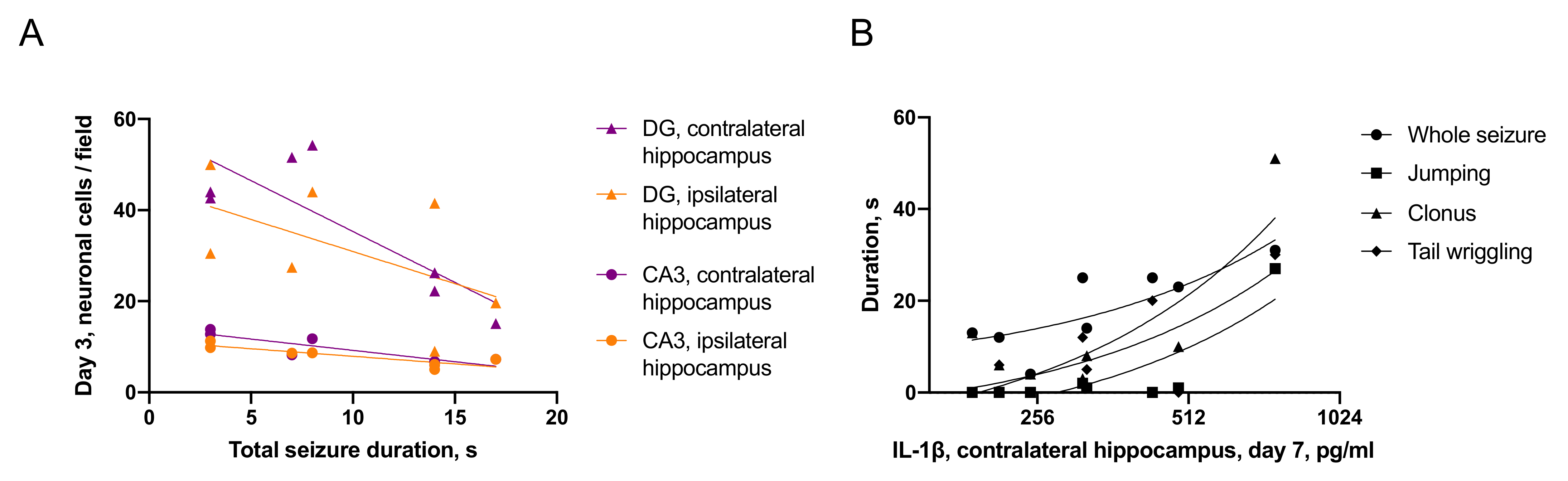
2.3.2. Associations between Seizures and Distant Hippocampal Damage
2.3.3. Associations between CS and Distant Hippocampal Damage
3. Discussion
3.1. TBI and Its Late Consequences
3.2. Seizure Semiology in Humans and in Animal Models
3.3. First Minutes after TBI: Immediate Posttraumatic Seizures and Reflex Recovery
3.4. Hours and Days after TBI: Mechanisms of Distant Hippocampal Damage
3.5. Months and Years after TBI: Late Posttraumatic Pathology
4. Materials and Methods
4.1. Environment and Housing
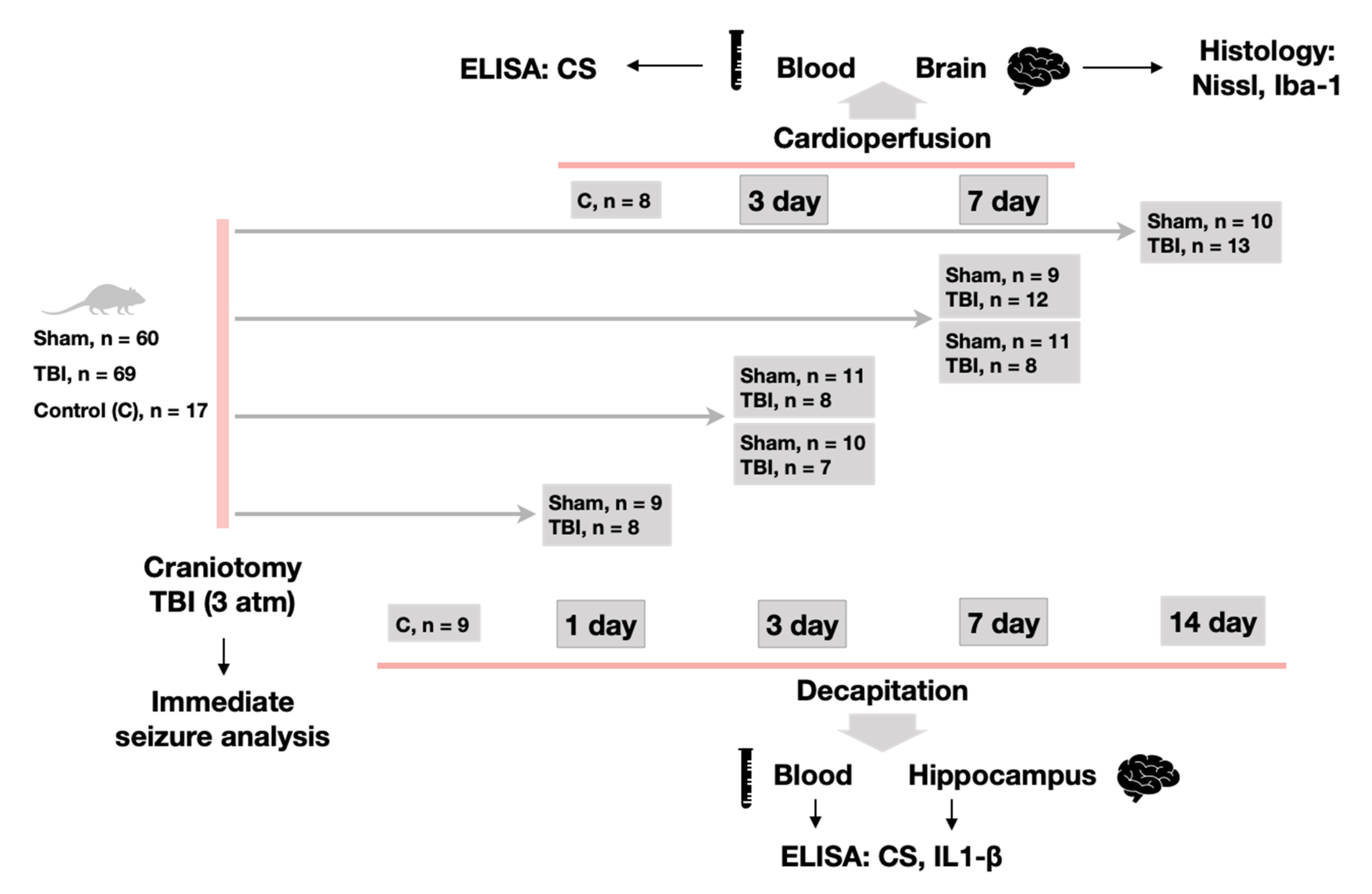
4.2. Experimental Design
4.3. Immediate Seizure Analysis
4.4. Histological Analysis
4.5. Biochemical Analysis
4.6. Statistical Analysis
5. Conclusions
- We report a first detailed description of seizure episodes immediately after lateral fluid percussion brain injury in rats and correlations between duration of immediate seizures and duration of reflexes recovery after TBI.
- IL-1β was elevated only in the ipsilateral hippocampus on day 1 after trauma. Microglial activation was evident in the ipsilateral and contralateral hippocampus 7 days after TBI. The duration of immediate seizures correlated with levels of IL-1β on day 7 after TBI.
- Neuronal cell loss was detected bilaterally, in the ipsilateral hippocampus it started earlier than in the contralateral. The duration of immediate seizures correlated with neuronal cell loss in the hippocampus on day 3 after TBI.
- CS was elevated on day 3 after TBI and the duration of immediate seizures correlated with CS levels.
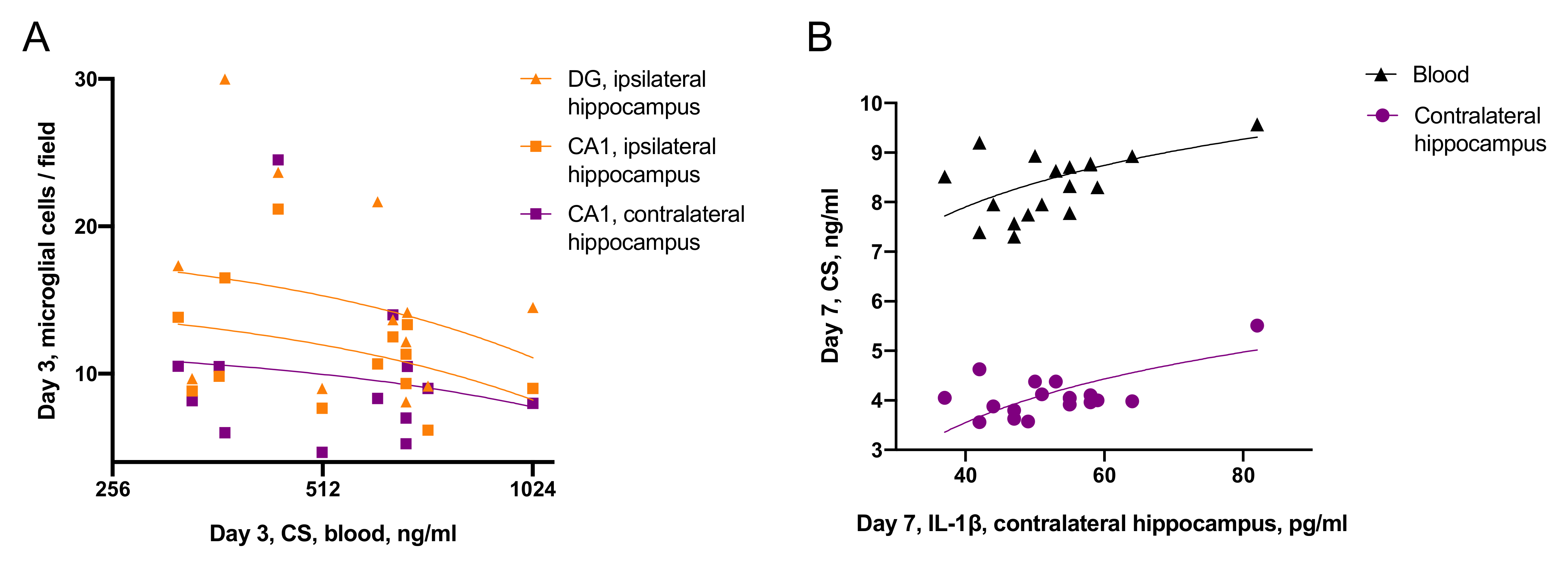
- Effects of CS on neuroinflammation was time-dependent: on day 3 blood CS negatively correlated with microglial activation, while on day 7, blood CS positively correlated with IL-1β level in the contralateral hippocampus.
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| CA1 | cornu ammonis 1, hippocampal field |
| CA3 | cornu ammonis 1, hippocampal field |
| CS | corticosterone |
| DG | dentate gyrus |
| ELISA | enzyme-linked immunosorbent assay |
| GABA | gamma-aminobutyric acid |
| GCs | glucocorticoids |
| Iba1 | ionized calcium-binding adaptor molecule 1 |
| IL-1β | interleukin-1β |
| LFPI | lateral fluid percussion injury |
| NMDA | N-methyl-D-aspartate |
| PBS | phosphate buffer saline |
References
- Reilly, P. The impact of neurotrauma on society: An international perspective. In Progress in Brain Research; Elsevier: Amsterdam, The Netherlands, 2007; Volume 161, pp. 3–9. ISBN 9780444530172. [Google Scholar]
- Bramlett, H.M.; Dietrich, W.D. Long-Term Consequences of Traumatic Brain Injury: Current Status of Potential Mechanisms of Injury and Neurological Outcomes. J. Neurotrauma 2015, 32, 1834–1848. [Google Scholar] [CrossRef]
- Thompson, H.J.; Lifshitz, J.; Marklund, N.; Grady, M.S.; Graham, D.I.; Hovda, D.A.; McIntosh, T.K. Lateral Fluid Percussion Brain Injury: A 15-Year Review and Evaluation. J. Neurotrauma 2005, 22, 42–75. [Google Scholar] [CrossRef] [PubMed]
- Gulyaeva, N.V. Functional Neurochemistry of the Ventral and Dorsal Hippocampus: Stress, Depression, Dementia and Remote Hippocampal Damage. Neurochem. Res. 2019, 44, 1306–1322. [Google Scholar] [CrossRef] [PubMed]
- Gulyaeva, N.V. Biochemical Mechanisms and Translational Relevance of Hippocampal Vulnerability to Distant Focal Brain Injury: The Price of Stress Response. Biochem 2019, 84, 1306–1328. [Google Scholar] [CrossRef]
- Gulyaeva, N.V. Ventral hippocampus, Stress and Psychopathology: Translational implications. Neurochem. J. 2015, 9, 85–94. [Google Scholar] [CrossRef]
- Prager, E.M.; Johnson, L.R. Stress at the Synapse: Signal Transduction Mechanisms of Adrenal Steroids at Neuronal Membranes. Sci. Signal. 2009, 2, re5. [Google Scholar] [CrossRef]
- Sorrells, S.F.; Caso, J.R.; Munhoz, C.D.; Sapolsky, R.M. The Stressed CNS: When Glucocorticoids Aggravate Inflammation. Neuron 2009, 64, 33–39. [Google Scholar] [CrossRef] [Green Version]
- Sorrells, S.F.; Munhoz, C.D.; Manley, N.C.; Yen, S.; Sapolsky, R.M. Glucocorticoids Increase Excitotoxic Injury and Inflammation in the Hippocampus of Adult Male Rats. Neuroendocrinology 2014, 100, 129–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben Assayag, E.; Tene, O.; Korczyn, A.D.; Shopin, L.; Auriel, E.; Molad, J.; Hallevi, H.; Kirschbaum, C.; Bornstein, N.M.; Shenhar-Tsarfaty, S.; et al. High hair cortisol concentrations predict worse cognitive outcome after stroke: Results from the TABASCO prospective cohort study. Psychoneuroendocrinology 2017, 82, 133–139. [Google Scholar] [CrossRef] [PubMed]
- Kwon, S.K.C.; Kovesdi, E.; Gyorgy, A.B.; Wingo, D.; Kamnaksh, A.; Walker, J.; Long, J.B.; Agoston, D.V. Stress and traumatic brain injury: A behavioral, proteomics, and histological study. Front. Neurol. 2011, 2, 12. [Google Scholar] [CrossRef] [Green Version]
- Capizzi, A.; Woo, J.; Verduzco-Gutierrez, M. Traumatic Brain Injury. Med. Clin. North Am. 2020, 104, 213–238. [Google Scholar] [CrossRef] [PubMed]
- Roozenbeek, B.; Maas, A.I.R.; Menon, D.K. Changing patterns in the epidemiology of traumatic brain injury. Nat. Rev. Neurol. 2013, 9, 231–236. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Marmarou, A.; Choi, S.; Maas, A.; Murray, G.; Steyerberg, E.W. Mortality from traumatic brain injury. Acta Neurochir Suppl. 2005, 95, 281–285. [Google Scholar] [CrossRef] [PubMed]
- Simon, D.W.; McGeachy, M.J.; Bayır, H.; Clark, R.S.B.; Loane, D.J.; Kochanek, P.M. The far-reaching scope of neuroinflammation after traumatic brain injury. Nat. Rev. Neurol. 2017, 13, 171–191. [Google Scholar] [CrossRef] [Green Version]
- Aronica, E.; Mühlebner, A.; van Vliet, E.A.; Gorter, J.A. Characterization of Pathology. In Models of Seizures and Epilepsy: Second Edition; Academic Press: Cambridge, MA, USA, 2017; ISBN 9780128040669. [Google Scholar]
- McIntosh, T.K.; Vink, R.; Noble, L.; Yamakami, I.; Fernyak, S.; Soares, H.; Faden, A.L. Traumatic brain injury in the rat: Characterization of a lateral fluid-percussion model. Neuroscience 1989, 28, 233–244. [Google Scholar] [CrossRef]
- McIntosh, T.K.; Noble, L.; Andrews, B.; Faden, A.I. Traumatic brain injury in the rat: Characterization of a midline fluid-percussion model. Cent. Nerv. Syst. Trauma 1987, 4, 119–134. [Google Scholar] [CrossRef] [PubMed]
- Pitkänen, A.; Kyyriäinen, J.; Andrade, P.; Pasanen, L.; Ndode-Ekane, X.E. Epilepsy After Traumatic Brain Injury. In Models of Seizures and Epilepsy; Elsevier: Amsterdam, The Netherlands, 2017; pp. 661–681. ISBN 9780128040669. [Google Scholar]
- D’Ambrosio, R.; Fender, J.S.; Fairbanks, J.P.; Simon, E.A.; Born, D.E.; Doyle, D.L.; Miller, J.W. Progression from frontal-parietal to mesial-temporal epilepsy after fluid percussion injury in the rat. Brain 2005, 128, 174–188. [Google Scholar] [CrossRef] [Green Version]
- D’Ambrosio, R.; Fairbanks, J.P.; Fender, J.S.; Born, D.E.; Doyle, D.L.; Miller, J.W. Post-traumatic epilepsy following fluid percussion injury in the rat. Brain 2004, 127, 304–314. [Google Scholar] [CrossRef] [Green Version]
- Kharatishvili, I.; Nissinen, J.P.; McIntosh, T.K.; Pitkänen, A. A model of posttraumatic epilepsy induced by lateral fluid-percussion brain injury in rats. Neuroscience 2006, 140, 685–697. [Google Scholar] [CrossRef]
- Smith, D.; Rau, T.; Poulsen, A.; MacWilliams, Z.; Patterson, D.; Kelly, W.; Poulsen, D. Convulsive seizures and EEG spikes after lateral fluid-percussion injury in the rat. Epilepsy Res. 2018, 147, 87–94. [Google Scholar] [CrossRef]
- Jones, N.C.; Cardamone, L.; Williams, J.P.; Salzberg, M.R.; Myers, D.; O’Brien, T.J. Experimental traumatic brain injury induces a pervasive hyperanxious phenotype in rats. J. Neurotrauma 2008, 25, 1367–1374. [Google Scholar] [CrossRef] [PubMed]
- Gurkoff, G.G.; Gahan, J.D.; Ghiasvand, R.T.; Hunsaker, M.R.; Van, K.; Feng, J.-F.; Shahlaie, K.; Berman, R.F.; Lyeth, B.G.; Folkerts, M.M. Evaluation of Metric, Topological, and Temporal Ordering Memory Tasks after Lateral Fluid Percussion Injury. J. Neurotrauma 2013, 30, 292–300. [Google Scholar] [CrossRef]
- Noachtar, S.; Peters, A.S. Semiology of epileptic seizures: A critical review. Epilepsy Behav. 2009, 15, 2–9. [Google Scholar] [CrossRef]
- Beniczky, S.; Neufeld, M.; Diehl, B.; Dobesberger, J.; Trinka, E.; Mameniskiene, R.; Rheims, S.; Gil-Nagel, A.; Craiu, D.; Pressler, R.; et al. Testing patients during seizures: A European consensus procedure developed by a joint taskforce of the ILAE—Commission on European Affairs and the European Epilepsy Monitoring Unit Association. Epilepsia 2016, 57, 1363–1368. [Google Scholar] [CrossRef] [PubMed]
- Hamer, H.M. Electrophysiology of focal clonic seizures in humans: A study using subdural and depth electrodes. Brain 2003, 126, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Lu, X.-C.M.; Browning, J.; Liao, Z.; Cao, Y.; Yang, W.; Shear, D.A. Post-Traumatic Epilepsy and Seizure Susceptibility in Rat Models of Penetrating and Closed-Head Brain Injury. J. Neurotrauma 2020, 37, 236–247. [Google Scholar] [CrossRef] [PubMed]
- Racine, R.J. Modification of seizure activity by electrical stimulation: II. Motor seizure. Electroencephalogr. Clin. Neurophysiol. 1972, 32, 281–294. [Google Scholar] [CrossRef]
- Van Erum, J.; Van Dam, D.; De Deyn, P.P. PTZ-induced seizures in mice require a revised Racine scale. Epilepsy Behav. 2019, 95, 51–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jefferys, J.G.R.; Jiruska, P.; Curtis, M.; De Avoli, M. Limbic Network Synchronization and Temporal Lobe Epilepsy. In Jasper’s Basic Mechanisms of the Epilepsies; Oxford University Press: Oxford, UK, 2012; pp. 1–18. ISBN 978-0199746545. [Google Scholar]
- Shivacharan, R.S.; Chiang, C.-C.; Zhang, M.; Gonzalez-Reyes, L.E.; Durand, D.M. Self-propagating, non-synaptic epileptiform activity recruits neurons by endogenous electric fields. Exp. Neurol. 2019, 317, 119–128. [Google Scholar] [CrossRef]
- Lowenstein, D.H.; Thomas, M.J.; Smith, D.H.; McIntosh, T.K. Selective vulnerability of dentate hilar neurons following traumatic brain injury: A potential mechanistic link between head trauma and disorders of the hippocampus. J. Neurosci. 1992, 12, 4846–4853. [Google Scholar] [CrossRef]
- Tupper, D.E.; Wallace, R.B. Utility of the neurological examination in rats. Acta Neurobiol. Exp. (Wars). 1980, 40, 999–1003. [Google Scholar]
- Dampney, R.A.L. Central neural control of the cardiovascular system: Current perspectives. Adv. Physiol. Educ. 2016, 40, 283–296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feldman, J.L.; Del Negro, C.A.; Gray, P.A. Understanding the Rhythm of Breathing: So Near, Yet So Far. Annu. Rev. Physiol. 2013, 75, 423–452. [Google Scholar] [CrossRef] [Green Version]
- Dixon, K.J. Pathophysiology of Traumatic Brain Injury. Phys. Med. Rehabil. Clin. N. Am. 2017, 28, 215–225. [Google Scholar] [CrossRef] [PubMed]
- Taş, Y.Ç.; Solaroğlu, İ.; Gürsoy-Özdemir, Y. Spreading Depolarization Waves in Neurological Diseases: A Short Review about its Pathophysiology and Clinical Relevance. Curr. Neuropharmacol. 2018, 17, 151–164. [Google Scholar] [CrossRef]
- Grady, M.S.; Charleston, J.S.; Maris, D.; Witgen, B.M.; Lifshitz, J. Neuronal and glial cell number in the hippocampus after experimental traumatic brain injury: Analysis by stereological estimation. J. Neurotrauma 2003, 20, 929–941. [Google Scholar] [CrossRef]
- Komol’tsev, I.G.; Volkova, A.A.; Levshina, I.P.; Novikova, M.R.; Manolova, A.O.; Stepanichev, M.Y.; Gulyaeva, N.V. The Number of IgG-Positive Neurons in the Rat Hippocampus Increases after Dosed Traumatic Brain Injury. J. Neurochem. 2018, 12, 256–261. [Google Scholar] [CrossRef]
- Tran, L.D.; Lifshitz, J.; Witgen, B.M.; Schwarzbach, E.; Cohen, A.S.; Grady, M.S. Response of the Contralateral Hippocampus to Lateral Fluid Percussion Brain Injury. J. Neurotrauma 2006, 23, 1330–1342. [Google Scholar] [CrossRef] [PubMed]
- Huusko, N.; Römer, C.; Ndode-Ekane, X.E.; Lukasiuk, K.; Pitkänen, A. Loss of hippocampal interneurons and epileptogenesis: A comparison of two animal models of acquired epilepsy. Brain Struct. Funct. 2015, 220, 153–191. [Google Scholar] [CrossRef] [PubMed]
- Ruth, R.E. Kainic-acid lesions of hippocampus produced iontophoretically: The problem of distant damage. Exp. Neurol. 1982, 76, 508–527. [Google Scholar] [CrossRef]
- Dubreuil, C.I.; Marklund, N.; Deschamps, K.; McIntosh, T.K.; McKerracher, L. Activation of Rho after traumatic brain injury and seizure in rats. Exp. Neurol. 2006, 198, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Rink, A.; Fung, K.; Trojanowski, J.Q.; Lee, V.M.; Neugebauer, E.; Mcintosh, T.K. Evidence of Apoptotic Cell Death after Experimental Traumatic Brain Injury in the Rat. Am. J. Pathol. 1995, 147, 1575. [Google Scholar] [CrossRef]
- Raghupathi, R. Cell Death Mechanisms Following Traumatic Brain Injury. Brain Pathol. 2004, 14, 215–222. [Google Scholar] [CrossRef]
- Gulyaeva, N.V. Glucocorticoid regulation of the glutamatergic synapse: Mechanisms of stress-dependent neuroplasticity. Ross. Fiziol. Zhurnal im. I. M. Sechenova (Sechenova Physiol. J.) 2021, 107, 518–523. [Google Scholar] [CrossRef]
- De Kloet, E.R.; Meijer, O.C.; de Nicola, A.F.; de Rijk, R.H.; Joëls, M. Importance of the brain corticosteroid receptor balance in metaplasticity, cognitive performance and neuro-inflammation. Front. Neuroendocrinol. 2018, 49, 124–145. [Google Scholar] [CrossRef]
- Roberts, A.J.; Donald Keith, L. Corticosteroids enhance convulsion susceptibility via central mineralocorticoid receptors. Psychoneuroendocrinology 1995, 20, 891–902. [Google Scholar] [CrossRef]
- Mattson, M.P. Excitotoxicity. In Stress: Physiology, Biochemistry, and Pathology; Elsevier: Amsterdam, The Netherlands, 2019; pp. 125–134. [Google Scholar]
- Virgin, C.E.; Ha, T.P.T.; Packan, D.R.; Tombaugh, G.C.; Yang, S.H.; Homer, H.C.; Sapolsky, R.M. Glucocorticoids Inhibit Glucose Transport and Glutamate Uptake in Hippocampal Astrocytes: Implications for Glucocorticoid Neurotoxicity. J. Neurochem. 1991, 57, 1422–1428. [Google Scholar] [CrossRef]
- Bolshakov, A.P.; Tret’yakova, L.V.; Kvichansky, A.A.; Gulyaeva, N.V. Glucocorticoids: Dr. Jekyll and Mr. Hyde of Hippocampal Neuroinflammation. Biochem 2021, 86, 156–167. [Google Scholar] [CrossRef]
- Gulyaeva, N.V. Stress-Associated Molecular and Cellular Hippocampal Mechanisms Common for Epilepsy and Comorbid Depressive Disorders. Biochem 2021, 86, 1–16. [Google Scholar] [CrossRef]
- Frank, M.G.; Miguel, Z.D.; Watkins, L.R.; Maier, S.F. Prior exposure to glucocorticoids sensitizes the neuroinflammatory and peripheral inflammatory responses to E. coli lipopolysaccharide. Brain. Behav. Immun. 2010, 24, 19–30. [Google Scholar] [CrossRef] [PubMed]
- Vezzani, A.; Balosso, S.; Ravizza, T. Inflammation and epilepsy. In Handbook of Clinical Neurology; Elsevier: Amsterdam, The Netherlands, 2012; Volume 107, pp. 163–175. ISBN 9780444528988. [Google Scholar]
- Komoltsev, I.G.; Sinkin, M.V.; Volkova, A.A.; Smirnova, E.A.; Novikova, M.R.; Kordonskaya, O.O.; Talypov, A.E.; Guekht, A.B.; Krylov, V.V.; Gulyaeva, N.V. A Translational Study on Acute Traumatic Brain Injury: High Incidence of Epileptiform Activity on Human and Rat Electrocorticograms and Histological Correlates in Rats. Brain Sci. 2020, 10, 570. [Google Scholar] [CrossRef]
- Parent, J.M.; Kron, M.M. Neurogenesis and epilepsy. Epilepsia 2010, 51, 45. [Google Scholar] [CrossRef]
- Kim, Y.K.; Na, K.S.; Myint, A.M.; Leonard, B.E. The role of pro-inflammatory cytokines in neuroinflammation, neurogenesis and the neuroendocrine system in major depression. Prog. Neuro Psychopharmacology Biol. Psychiatry 2016, 64, 277–284. [Google Scholar] [CrossRef] [PubMed]
- Gulyaeva, N.V. Aberrant neurogenesis in adult epileptic brain: Compensatory or pathologic. Neurochem. J. 2010, 4, 84–89. [Google Scholar] [CrossRef]
- Alder, J.; Fujioka, W.; Lifshitz, J.; Crockett, D.P.; Thakker-Varia, S. Lateral Fluid Percussion: Model of Traumatic Brain Injury in Mice. J. Vis. Exp. 2011, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Komoltsev, I.G.; Frankevich, S.O.; Shirobokova, N.I.; Volkova, A.A.; Onufriev, M.V.; Moiseeva, J.V.; Novikova, M.R.; Gulyaeva, N.V. Neuroinflammation and Neuronal Loss in the Hippocampus Are Associated with Immediate Posttraumatic Seizures and Corticosterone Elevation in Rats. Int. J. Mol. Sci. 2021, 22, 5883. https://doi.org/10.3390/ijms22115883
Komoltsev IG, Frankevich SO, Shirobokova NI, Volkova AA, Onufriev MV, Moiseeva JV, Novikova MR, Gulyaeva NV. Neuroinflammation and Neuronal Loss in the Hippocampus Are Associated with Immediate Posttraumatic Seizures and Corticosterone Elevation in Rats. International Journal of Molecular Sciences. 2021; 22(11):5883. https://doi.org/10.3390/ijms22115883
Chicago/Turabian StyleKomoltsev, Ilia G., Stepan O. Frankevich, Natalia I. Shirobokova, Aleksandra A. Volkova, Mikhail V. Onufriev, Julia V. Moiseeva, Margarita R. Novikova, and Natalia V. Gulyaeva. 2021. "Neuroinflammation and Neuronal Loss in the Hippocampus Are Associated with Immediate Posttraumatic Seizures and Corticosterone Elevation in Rats" International Journal of Molecular Sciences 22, no. 11: 5883. https://doi.org/10.3390/ijms22115883