
Inflammasome NLRP3 Potentially Links Obesity-Associated Low-Grade Systemic Inflammation and Insulin Resistance with Alzheimer’s Disease


Abstract
:1. Introduction
2. Alzheimer’s Disease
3. Insulin Signaling Pathway in the Brain
3.1. Insulin in the Brain
3.2. Insulin Receptors
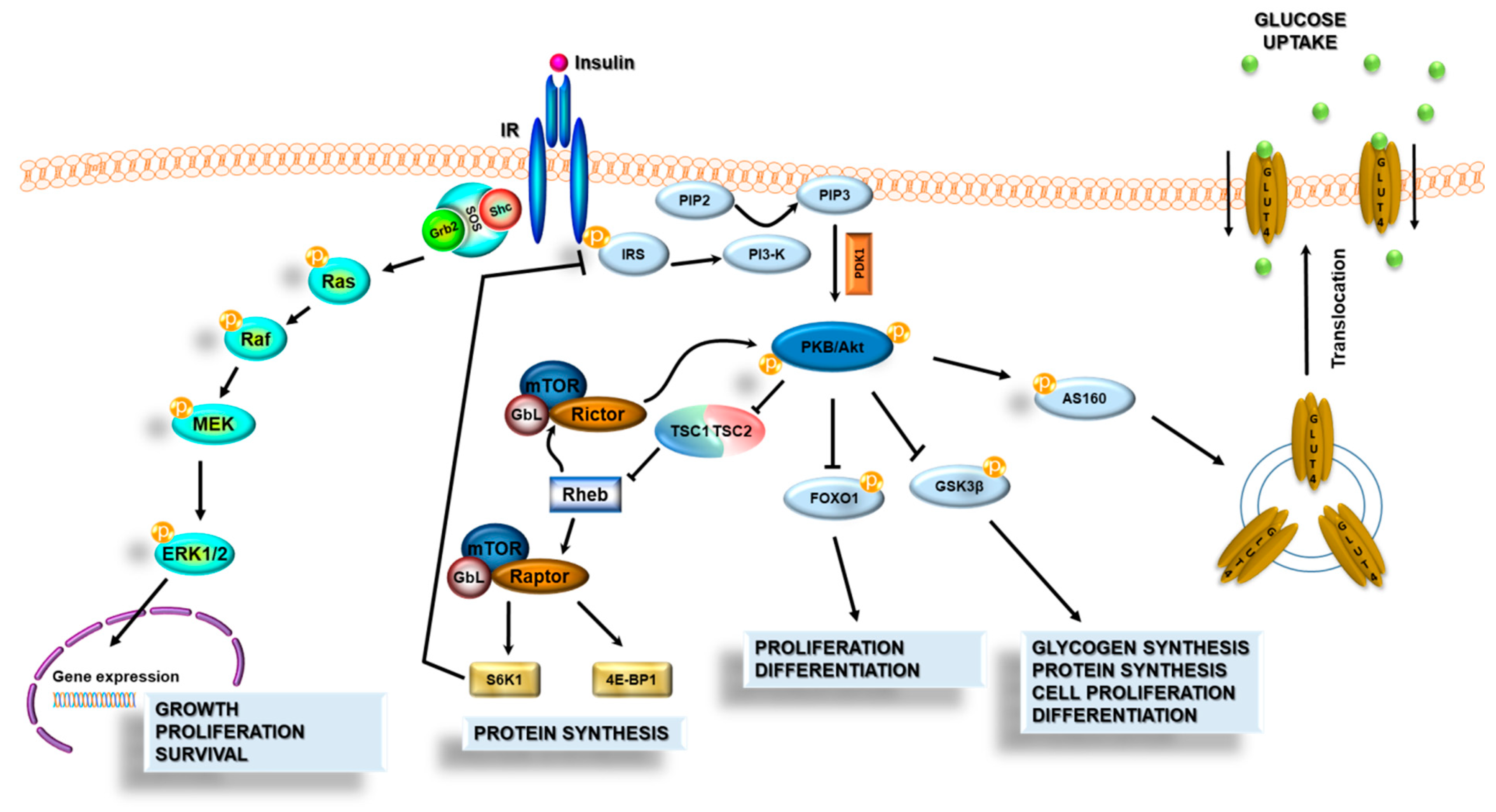
3.3. Insulin Signal Transduction
3.4. The Role of AKT and Glycogen Synthase Kinase-3 (GSK-3) in AD
4. Glucose Metabolism in the Brain
4.1. Physiological Brain Glucose Metabolism
4.2. Disturbed Glucose Metabolism in AD
5. Insulin Resistance as a Common Factor Linking Obesity, T2DM, and AD
5.1. Malfunction of Insulin Signaling
5.2. Insulin Resistance and AD
6. Inflammasomes as a Possible Link between Inflammation-Associated Peripheral and Brain Insulin Resistance in Metabolic Disease and AD
6.1. Inflammation and AD
6.2. Adiposity-Related Inflammation
6.3. The Role of Inflammasomes in Adiposity-Related Inflammation and Insulin Resistance
6.4. Inflammasomes and Metabolic Syndrome
6.5. NLRP3 Inflammasome and Aging
6.6. Inflammasomes and AD
6.7. Peripheral Inflammasome Activation and Microglia
6.8. AD Pathology Stimulates Central NLRP3 Activation
6.9. The Possibilities of Pharmacological Modulation of NLRP3 Inflammasome Activity
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| 18FDG-PET | 18F deoxyglucose positron emission tomography |
| AD | Alzheimer’s disease |
| AIM 2 | Absent in melanoma 2 |
| Akt | Protein kinase B |
| AMPK | 5’AMP-activated protein kinase |
| APP | Amyloid precursor protein |
| AS160 | Akt substrate 160 |
| Aβ | Amyloid β |
| BBB | Blood–brain barrier |
| Bcl-2 | B-cell lymphoma 2 |
| BMI | Body mass index |
| CARD | Caspase recruitment domain |
| CNS | Central nervous system |
| CX3CR1 | CX3C chemokine receptor 1 |
| DAMPs | Danger-associated molecular patterns |
| EOAD | Early-onset AD |
| FAD | Familial type of AD |
| FFA | Free fatty acid |
| GLUT1 | Glucose transporter 1 |
| GLUT3 | Glucose transporter 3 |
| GLUT4 | Glucose transporter 4 |
| GSDMD | Gasdermin D |
| GSK-3α | Glycogen synthase kinase-3α |
| GSK-3β | Glycogen synthase kinase-3β |
| HFD | High-fat diet |
| IDF | International Diabetes Federation |
| IGF-1R | Insulin-like growth factor 1 receptor |
| IKK | IκBα kinase |
| IL-10 | Interleukin-10 |
| IL-18 | Interleukin-18 |
| IL1r1 | Interleukin 1 Receptor Type 1 |
| IL-1β | Interleukin-1β |
| IL-6 | Interleukin-6 |
| IR | Insulin receptor |
| IRS | Insulin receptor substrate |
| ITG | Inferior temporal gyrus |
| JNK | c-Jun N-terminal kinase |
| LOAD | Late-onset AD |
| LRR | Leucine-rich repeats domain |
| LTD | Long-term depression |
| LTP | Long-term potentiation |
| MAPK | Mitogen-activated protein kinase |
| MCP-1 | Monocyte chemoattractant protein 1 |
| MetS | Metabolic syndrome |
| MFG | Middle frontal gyrus |
| MS | Multiple Sclerosis |
| mtDNA | Mitochondrial DNA |
| NBD | Nucleotide-binding oligomerization domain |
| NFT | Neurofibrillary tangles formation |
| NF-κβ | Nuclear factor kappa-light-chain-enhancer of activated β cells |
| NLRC4 | CARD domain-containing protein 4, also called ICE-protease activating factor (IPAF) |
| NLRP1 | NOD-like receptor protein 1 |
| NLRP2 | NOD-like receptor protein 2 |
| NLRP3 | NOD-like receptor protein 3 |
| NLRP3 | Nucleotide-binding oligomerization domain (NOD)-, leucine-rich repeat- (LRR)-, and pyrin domain-containing protein 3 (NLRP3) Inflammasome |
| NLRs | Nucleotide-binding oligomerization domain-like receptors (NOD-like receptors) |
| NO | Nitric oxide |
| OXPHOS | Oxidative phosphorylation |
| PAMPs | Pathogen-associated molecular patterns |
| PBMCs | Peripheral blood mononuclear cells |
| PD | Parkinson’s disease |
| PDK1 | Phosphoinositide-dependent protein kinase 1 |
| PI3K | Phosphatidylinositol 3-kinase |
| PIP2 | Phosphatidylinositol 4,5-bisphosphate |
| PIP3 | Phosphatidylinositol 3,4,5-trisphosphate |
| PRRs | Pattern-recognition receptors |
| PSEN 1 | Presenilin 1 |
| PSEN 2 | Presenilin 2 |
| PYD | Pyrin domain |
| RLHs | Retinoic acid-inducible gene I-like helicases |
| ROS | Reactive oxygen species |
| SAT | Subcutaneous adipose tissue |
| T2DM | Type 2 diabetes mellitus |
| TLRs | Toll-like receptors |
| TNFα | Tumor necrosis factor α |
| VAT | Visceral adipose tissue |
References
- Wimo, A.; Ali, G.-C.; Guerchet, M.; Prince, M.; Prina, M.; Wu, Y.-T.; World Alzheimer Report 2015. The Global Impact of Dementia. An Analysis of Prevalence, Incidence, Cost and Trends. Available online: https://www.alzint.org/resource/world-alzheimer-report-2015/ (accessed on 30 January 2021).
- de Bem, A.F.; Krolow, R.; Farias, H.R.; de Rezende, V.L.; Gelain, D.P.; Moreira, J.C.F.; Duarte, J.M. das N.; de Oliveira, J. Animal Models of Metabolic Disorders in the Study of Neurodegenerative Diseases: An Overview. Front. Neurosci. 2021, 14. [Google Scholar] [CrossRef]
- Sun, Y.; Ma, C.; Sun, H.; Wang, H.; Peng, W.; Zhou, Z.; Wang, H.; Pi, C.; Shi, Y.; He, X. Metabolism: A Novel Shared Link between Diabetes Mellitus and Alzheimer’s Disease. J. Diabetes Res. 2020, 2020, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Bharadwaj, P.; Wijesekara, N.; Liyanapathirana, M.; Newsholme, P.; Ittner, L.; Fraser, P.; Verdile, G. The Link between Type 2 Diabetes and Neurodegeneration: Roles for Amyloid-β, Amylin, and Tau Proteins. J. Alzheimer’s Dis. 2017, 59, 421–432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laiteerapong, N.; Ham, S.A.; Gao, Y.; Moffet, H.H.; Liu, J.Y.; Huang, E.S.; Karter, A.J. The Legacy Effect in Type 2 Diabetes: Impact of Early Glycemic Control on Future Complications (The Diabetes & Aging Study). Diabetes Care 2018, 42, 416–426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carranza-Naval, M.J.; Vargas-Soria, M.; Hierro-Bujalance, C.; Baena-Nieto, G.; Garcia-Alloza, M.; Infante-Garcia, C.; del Marco, A. Alzheimer’s Disease and Diabetes: Role of Diet, Microbiota and Inflammation in Preclinical Models. Biomolecules 2021, 11, 262. [Google Scholar] [CrossRef] [PubMed]
- Klöting, N.; Blüher, M. Adipocyte dysfunction, inflammation and metabolic syndrome. Rev. Endocr. Metab. Disord. 2014, 15, 277–287. [Google Scholar] [CrossRef]
- Wang, B.; Trayhurn, P. Acute and prolonged effects of TNF-α on the expression and secretion of inflammation-related adipokines by human adipocytes differentiated in culture. Pflügers Arch. Eur. J. Physiol. 2006, 452, 418–427. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-C.; Chang, T.-J.; Lee, W.-J.; Chuang, L.-M. The relationship of visfatin/pre–B-cell colony-enhancing factor/nicotinamide phosphoribosyltransferase in adipose tissue with inflammation, insulin resistance, and plasma lipids. Metabolism 2010, 59, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Lee, J. Adipose tissue macrophages in the development of obesity-induced inflammation, insulin resistance and type 2 Diabetes. Arch. Pharmacal Res. 2013, 36, 208–222. [Google Scholar] [CrossRef] [Green Version]
- Pedra, J.H.; Cassel, S.L.; Sutterwala, F.S. Sensing pathogens and danger signals by the inflammasome. Curr. Opin. Immunol. 2009, 21, 10–16. [Google Scholar] [CrossRef] [Green Version]
- Amarante-Mendes, G.P.; Adjemian, S.; Branco, L.M.; Zanetti, L.C.; Weinlich, R.; Bortoluci, K.R. Pattern Recognition Receptors and the Host Cell Death Molecular Machinery. Front. Immunol. 2018, 9, 2379. [Google Scholar] [CrossRef] [Green Version]
- Zheng, D.; Liwinski, T.; Elinav, E. Inflammasome activation and regulation: Toward a better understanding of complex mechanisms. Cell Discov. 2020, 6, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Lukens, J.R.; Dixit, V.D.; Kanneganti, T.-D. Inflammasome activation in obesity-related inflammatory diseases and autoimmunity. Discov. Med. 2011, 12, 65–74. [Google Scholar] [PubMed]
- Scheltens, P.; Blennow, K.; Breteler, M.M.B.; de Strooper, B.; Frisoni, G.B.; Salloway, S.; van der Flier, W. Alzheimer’s disease. Lancet 2016, 388, 505–517. [Google Scholar] [CrossRef]
- Tarawneh, R.; Holtzman, D.M. The Clinical Problem of Symptomatic Alzheimer Disease and Mild Cognitive Impairment. Cold Spring Harb. Perspect. Med. 2012, 2, a006148. [Google Scholar] [CrossRef] [PubMed]
- Montenegro, J.M.F.; Villarini, B.; Angelopoulou, A.; Kapetanios, E.; Garcia-Rodriguez, J.; Argyriou, V. A Survey of Alzheimer’s Disease Early Diagnosis Methods for Cognitive Assessment. Sensors 2020, 20, 7292. [Google Scholar] [CrossRef] [PubMed]
- Bird, T.D. Alzheimer Disease Overview. GeneReviews® [Internet] 2018. Available online: https://www.ncbi.nlm.nih.gov/books/NBK1161/ (accessed on 30 January 2021).
- Bekris, L.M.; Yu, C.-E.; Bird, T.D.; Tsuang, D.W. Review Article: Genetics of Alzheimer Disease. J. Geriatr. Psychiatry Neurol. 2010, 23, 213–227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dorszewska, J.; Prendecki, M.; Oczkowska, A.; Dezor, M.; Kozubski, W. Molecular Basis of Familial and Sporadic Alzheimer’s Disease. Curr. Alzheimer Res. 2016, 13, 952–963. [Google Scholar] [CrossRef]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef]
- De La Monte, S.M.; Wands, J.R. Alzheimer’s Disease is Type 3 Diabetes—Evidence Reviewed. J. Diabetes Sci. Technol. 2008, 2, 1101–1113. [Google Scholar] [CrossRef] [Green Version]
- Pugazhenthi, S.; Qin, L.; Reddy, P.H. Common neurodegenerative pathways in obesity, diabetes, and Alzheimer’s disease. Biochim. et Biophys. Acta (BBA) Mol. Basis Dis. 2017, 1863, 1037–1045. [Google Scholar] [CrossRef] [PubMed]
- Walker, J.M.; Harrison, F.E. Shared Neuropathological Characteristics of Obesity, Type 2 Diabetes and Alzheimer’s Disease: Impacts on Cognitive Decline. Nutrition 2015, 7, 7332–7357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tabassum, S.; Misrani, A.; Yang, L. Exploiting Common Aspects of Obesity and Alzheimer’s Disease. Front. Hum. Neurosci. 2020, 14, 14. [Google Scholar] [CrossRef] [PubMed]
- Schrijvers, E.; Elisabeth, M.C.; Witteman, J.; Sijbrands, E.; Hofman, A.; Koudstaal, P.J.; Breteler, M. Insulin metabolism and the risk of Alzheimer disease: The Rotterdam Study. Neurol. 2010, 75, 1982–1987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, X.; Liu, G.; Guo, J.; Su, Z. The PI3K/AKT pathway in obesity and type 2 diabetes. Int. J. Biol. Sci. 2018, 14, 1483–1496. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Litwiniuk, A.; Domańska, A.; Chmielowska, M.; Martyńska, L.; Bik, W.; Kalisz, M. The Effects of Alpha-Linolenic Acid on the Secretory Activity of Astrocytes and β Amyloid-Associated Neurodegeneration in Differentiated SH-SY5Y Cells: Alpha-Linolenic Acid Protects the SH-SY5Y cells against β Amyloid Toxicity. Oxidative Med. Cell. Longev. 2020, 2020, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Takano, K.; Koarashi, K.; Kawabe, K.; Itakura, M.; Nakajima, H.; Moriyama, M.; Nakamura, Y. Insulin expression in cultured astrocytes and the decrease by amyloid β. Neurochem. Int. 2018, 119, 119. [Google Scholar] [CrossRef]
- Lee, C.-C.; Huang, C.-C.; Hsu, K.-S. Insulin promotes dendritic spine and synapse formation by the PI3K/Akt/mTOR and Rac1 signaling pathways. Neuropharmacol. 2011, 61, 867–879. [Google Scholar] [CrossRef]
- Ziegler, A.N.; Levison, S.W.; Wood, T.L. Insulin and IGF receptor signalling in neural-stem-cell homeostasis. Nat. Rev. Endocrinol. 2015, 11, 161–170. [Google Scholar] [CrossRef] [Green Version]
- Baranowska-Bik, A.; Bik, W. Vascular Dysfunction and Insulin Resistance in Aging. Curr. Vasc. Pharmacol. 2019, 17, 465–475. [Google Scholar] [CrossRef]
- Pomytkin, I.; Costa-Nunes, J.P.; Kasatkin, V.; Veniaminova, E.; Demchenko, A.; Lyundup, A.; Lesch, K.-P.; Ponomarev, E.D.; Strekalova, T. Insulin receptor in the brain: Mechanisms of activation and the role in the CNS pathology and treatment. CNS Neurosci. Ther. 2018, 24, 763–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boucher, J.; Kleinridders, A.; Kahn, C.R. Insulin Receptor Signaling in Normal and Insulin-Resistant States. Cold Spring Harb. Perspect. Biol. 2014, 6, a009191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Urban, M.J.; Dobrowsky, R.T.; Blagg, B.S. Heat shock response and insulin-associated neurodegeneration. Trends Pharmacol. Sci. 2012, 33, 129–137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zakharova, I.O.; Sokolova, T.V.; Bayunova, L.V.; Zorina, I.I.; Rychkova, M.P.; Shpakov, A.O.; Avrova, N.F. The Protective Effect of Insulin on Rat Cortical Neurons in Oxidative Stress and Its Dependence on the Modulation of Akt, GSK-3beta, ERK1/2, and AMPK Activities. Int. J. Mol. Sci. 2019, 20, 3702. [Google Scholar] [CrossRef] [Green Version]
- Duarte, A.I.; Santos, P.; Oliveira, C.R.; Santos, M.S.; Rego, A.C. Insulin neuroprotection against oxidative stress is mediated by Akt and GSK-3β signaling pathways and changes in protein expression. Biochim. et Biophys. Acta (BBA) Bioenerg. 2008, 1783, 994–1002. [Google Scholar] [CrossRef] [Green Version]
- Arnold, S.E.; Arvanitakis, Z.; Macauley-Rambach, S.L.; Koenig, A.M.; Wang, H.-Y.; Ahima, R.S.; Craft, S.; Gandy, S.; Buettner, C.; Stoeckel, L.E.; et al. Brain insulin resistance in type 2 diabetes and Alzheimer disease: Concepts and conundrums. Nat. Rev. Neurol. 2018, 14, 168–181. [Google Scholar] [CrossRef]
- Bradley, C.A.; Peineau, S.; Taghibiglou, C.; Nicolas, C.S.; Whitcomb, D.J.; Bortolotto, Z.A.; Kaang, B.-K.; Cho, K.; Wang, Y.T.; Collingridge, G.L. A pivotal role of GSK-3 in synaptic plasticity. Front. Mol. Neurosci. 2012, 5, 13. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.; Chen, Z.; Gao, J.; Shi, W.; Li, L.; Jiang, S.; Hu, H.; Liu, Z.; Xu, D.; Wu, L. The Key Roles of GSK-3β in Regulating Mitochondrial Activity. Cell. Physiol. Biochem. 2017, 44, 1445–1459. [Google Scholar] [CrossRef]
- Zhou, H.; Li, X.-M.; Meinkoth, J.; Pittman, R.N. Akt Regulates Cell Survival and Apoptosis at a Postmitochondrial Level. J. Cell Biol. 2000, 151, 483–494. [Google Scholar] [CrossRef] [Green Version]
- Steiner, P. Brain Fuel Utilization in the Developing Brain. Ann. Nutr. Metab. 2019, 75, 8–18. [Google Scholar] [CrossRef]
- Mergenthaler, P.; Lindauer, U.; Dienel, G.A.; Meisel, A. Sugar for the brain: The role of glucose in physiological and pathological brain function. Trends Neurosci. 2013, 36, 587–597. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bélanger, M.; Allaman, I.; Magistretti, P.J. Brain Energy Metabolism: Focus on Astrocyte-Neuron Metabolic Cooperation. Cell Metab. 2011, 14, 724–738. [Google Scholar] [CrossRef] [Green Version]
- Dienel, G.A. Brain Glucose Metabolism: Integration of Energetics with Function. Physiol. Rev. 2019, 99, 949–1045. [Google Scholar] [CrossRef] [PubMed]
- Falkowska, A.; Gutowska, I.; Goschorska, M.; Nowacki, P.; Chlubek, D.; Baranowska-Bosiacka, I. Energy Metabolism of the Brain, Including the Cooperation between Astrocytes and Neurons, Especially in the Context of Glycogen Metabolism. Int. J. Mol. Sci. 2015, 16, 25959–25981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koepsell, H. Glucose transporters in brain in health and disease. Pflügers Arch. Eur. J. Physiol. 2020, 472, 1299–1343. [Google Scholar] [CrossRef] [PubMed]
- Pearson-Leary, J.; McNay, E.C. Novel Roles for the Insulin-Regulated Glucose Transporter-4 in Hippocampally Dependent Memory. J. Neurosci. 2016, 36, 11851–11864. [Google Scholar] [CrossRef] [PubMed]
- Berlanga-Acosta, J.; Guillén-Nieto, G.; Rodríguez-Rodríguez, N.; Bringas-Vega, M.L.; García-Del-Barco-Herrera, D.; Berlanga-Saez, J.O.; García-Ojalvo, A.; Valdés-Sosa, M.J.; Valdés-Sosa, P.A. Insulin Resistance at the Crossroad of Alzheimer Disease Pathology: A Review. Front. Endocrinol. 2020, 11, 11. [Google Scholar] [CrossRef]
- Chen, Z.; Zhong, C. Decoding Alzheimer’s disease from perturbed cerebral glucose metabolism: Implications for diagnostic and therapeutic strategies. Prog. Neurobiol. 2013, 108, 21–43. [Google Scholar] [CrossRef] [Green Version]
- Landau, S.M.; Harvey, D.; Madison, C.M.; Koeppe, R.A.; Reiman, E.M.; Foster, N.L.; Weiner, M.W.; Jagust, W.J. Associations between cognitive, functional, and FDG-PET measures of decline in AD and MCI. Neurobiol. Aging 2011, 32, 1207–1218. [Google Scholar] [CrossRef] [Green Version]
- Calsolaro, V.; Edison, P. Alterations in Glucose Metabolism in Alzheimer’s Disease. Recent Pat. Endocr. Metab. Immune Drug Discov. 2016, 10, 31–39. [Google Scholar] [CrossRef]
- An, Y.; Varma, V.R.; Varma, S.; Casanova, R.; Dammer, E.; Pletnikova, O.; Chia, C.W.; Egan, J.M.; Ferrucci, L.; Troncoso, J.; et al. Evidence for brain glucose dysregulation in Alzheimer’s disease. Alzheimer’s Dement. 2018, 14, 318–329. [Google Scholar] [CrossRef]
- Biessels, G.J.; Strachan, M.W.J.; Visseren, F.; Kappelle, L.J.; Whitmer, R.A. Dementia and cognitive decline in type 2 diabetes and prediabetic stages: Towards targeted interventions. Lancet Diabetes Endocrinol. 2014, 2, 246–255. [Google Scholar] [CrossRef]
- De La Monte, S.M. Relationships between Diabetes and Cognitive Impairment. Endocrinol. Metab. Clin. N. Am. 2014, 43, 245–267. [Google Scholar] [CrossRef] [Green Version]
- Femminella, G.D.; Livingston, N.R.; Raza, S.; van der Doef, T.; Frangou, E.; Love, S.; Busza, G.; Calsolaro, V.; Carver, S.; Holmes, C.; et al. Does insulin resistance influence neurodegeneration in non-diabetic Alzheimer’s subjects? Alzheimer’s Res. Ther. 2021, 13, 1–11. [Google Scholar] [CrossRef]
- Mielke, J.G.; Taghibiglou, C.; Liu, L.; Zhang, Y.; Jia, Z.; Adeli, K.; Wang, Y.T. A biochemical and functional characterization of diet-induced brain insulin resistance. J. Neurochem. 2005, 93, 1568–1578. [Google Scholar] [CrossRef]
- Kulas, J.A.; Weigel, T.K.; Ferris, H.A. Insulin resistance and impaired lipid metabolism as a potential link between diabetes and Alzheimer’s disease. Drug Dev. Res. 2020, 81, 194–205. [Google Scholar] [CrossRef]
- Gabbouj, S.; Ryhänen, S.; Marttinen, M.; Wittrahm, R.; Takalo, M.; Kemppainen, S.; Martiskainen, H.; Tanila, H.; Haapasalo, A.; Hiltunen, M.; et al. Altered Insulin Signaling in Alzheimer’s Disease Brain—Special Emphasis on PI3K-Akt Pathway. Front. Neurosci. 2019, 13, 629. [Google Scholar] [CrossRef]
- Capurso, C.; Capurso, A. From excess adiposity to insulin resistance: The role of free fatty acids. Vasc. Pharmacol. 2012, 57, 91–97. [Google Scholar] [CrossRef]
- Pal, M.; Febbraio, M.A.; Lancaster, G.I. The roles of c-Jun NH2-terminal kinases (JNKs) in obesity and insulin resistance. J. Physiol. 2016, 594, 267–279. [Google Scholar] [CrossRef] [Green Version]
- Hegde, V.; Dhurandhar, N.V.; Reddy, P.H. Hyperinsulinemia or Insulin Resistance: What Impacts the Progression of Alzheimer’s Disease? J. Alzheimer’s Dis. 2019, 72, S71–S79. [Google Scholar] [CrossRef]
- Ma, Q.-L.; Yang, F.; Rosario, E.R.; Ubeda, O.J.; Beech, W.; Gant, D.J.; Chen, P.P.; Hudspeth, B.; Chen, C.; Zhao, Y.; et al. Amyloid Oligomers Induce Phosphorylation of Tau and Inactivation of Insulin Receptor Substrate via c-Jun N-Terminal Kinase Signaling: Suppression by Omega-3 Fatty Acids and Curcumin. J. Neurosci. 2009, 29, 9078–9089. [Google Scholar] [CrossRef]
- Bomfim, T.R.; Forny-Germano, L.; Sathler, L.B.; Brito-Moreira, J.; Houzel, J.-C.; Decker, H.; Silverman, M.A.; Kazi, H.; Melo, H.M.; McClean, P.L.; et al. An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease–associated Aβ oligomers. J. Clin. Investig. 2012, 122, 1339–1353. [Google Scholar] [CrossRef] [PubMed]
- Savage, M.J.; Lin, Y.-G.; Ciallella, J.R.; Flood, D.G.; Scott, R.W. Activation of c-Jun N-Terminal Kinase and p38 in an Alzheimer’s Disease Model Is Associated with Amyloid Deposition. J. Neurosci. 2002, 22, 3376–3385. [Google Scholar] [CrossRef] [Green Version]
- Stafeev, I.S.; Vorotnikov, A.V.; Ratner, E.I.; Menshikov, M.Y.; Parfyonova, Y.V. Latent Inflammation and Insulin Resistance in Adipose Tissue. Int. J. Endocrinol. 2017, 2017, 1–12. [Google Scholar] [CrossRef]
- Yau, P.L.; Kluger, A.; Borod, J.C.; Convit, A. Neural substrates of verbal memory impairments in adults with type 2 diabetes mellitus. J. Clin. Exp. Neuropsychol. 2014, 36, 74–87. [Google Scholar] [CrossRef] [Green Version]
- Hsu, F.-C.; Yuan, M.; Bowden, N.W.; Xu, J.; Smith, S.C.; Wagenknecht, L.E.; Langefeld, C.D.; Divers, J.; Register, T.C.; Carr, J.J.; et al. Adiposity is inversely associated with hippocampal volume in African Americans and European Americans with diabetes. J. Diabetes Complicat. 2016, 30, 1506–1512. [Google Scholar] [CrossRef] [Green Version]
- De La Monte, S.M. The Full Spectrum of Alzheimer’s Disease Is Rooted in Metabolic Derangements That Drive Type 3 Diabetes. Adv. Exp. Med. Biol. 2019, 1128, 45–83. [Google Scholar] [CrossRef]
- Gaspar, J.; Baptista, F.I.; Macedo, M.P.; Ambrósio, A.F. Inside the Diabetic Brain: Role of Different Players Involved in Cognitive Decline. ACS Chem. Neurosci. 2015, 7, 131–142. [Google Scholar] [CrossRef]
- Garcia-Serrano, A.M.; Duarte, J.M.N. Brain Metabolism Alterations in Type 2 Diabetes: What Did We Learn From Diet-Induced Diabetes Models? Front. Neurosci. 2020, 14, 229. [Google Scholar] [CrossRef] [Green Version]
- Santiago, J.A.; Potashkin, J.A. The Impact of Disease Comorbidities in Alzheimer’s Disease. Front. Aging Neurosci. 2021, 13, 13. [Google Scholar] [CrossRef]
- Rotermund, C.; Truckenmüller, F.M.; Schell, H.; Kahle, P.J. Diet-induced obesity accelerates the onset of terminal phenotypes in α-synuclein transgenic mice. J. Neurochem. 2014, 131, 848–858. [Google Scholar] [CrossRef] [PubMed]
- Kimura, N. Diabetes Mellitus Induces Alzheimer’s Disease Pathology: Histopathological Evidence from Animal Models. Int. J. Mol. Sci. 2016, 17, 503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Wise, L.; Fukuchi, K.-I. TLR4 Cross-Talk With NLRP3 Inflammasome and Complement Signaling Pathways in Alzheimer’s Disease. Front. Immunol. 2020, 11, 724. [Google Scholar] [CrossRef] [PubMed]
- Scott, X.O.; Stephens, M.E.; Desir, M.C.; Dietrich, W.D.; Keane, R.W.; Vaccari, J.P.D.R. The Inflammasome Adaptor Protein ASC in Mild Cognitive Impairment and Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 4674. [Google Scholar] [CrossRef]
- Fernandez-Lizarbe, S.; Pascual, M.; Guerri, C. Critical Role of TLR4 Response in the Activation of Microglia Induced by Ethanol. J. Immunol. 2009, 183, 4733–4744. [Google Scholar] [CrossRef] [Green Version]
- Rezai-Zadeh, K.; Gate, D.; Gowing, G.; Town, T. How to get from here to there: Macrophage recruitment in Alzheimer’s disease. Curr. Alzheimer Res. 2011, 8, 156–163. [Google Scholar] [CrossRef] [Green Version]
- Bhaskar, K.; Maphis, N.; Xu, G.; Varvel, N.H.; Kokiko-Cochran, O.N.; Weick, J.P.; Staugaitis, S.M.; Cardona, A.; Ransohoff, R.M.; Herrup, K.; et al. Microglial derived tumor necrosis factor-α drives Alzheimer’s disease-related neuronal cell cycle events. Neurobiol. Dis. 2014, 62, 273–285. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calsolaro, V.; Edison, P. Neuroinflammation in Alzheimer’s disease: Current evidence and future directions. Alzheimer’s Dement. 2016, 12, 719–732. [Google Scholar] [CrossRef]
- Britschgi, M.; Wyss-Coray, T. Systemic and Acquired Immune Responses in Alzheimer’s Disease. Int. Rev. Neurobiol. 2007, 82, 205–233. [Google Scholar] [CrossRef]
- Zenaro, E.; Piacentino, G.; Constantin, G. The blood-brain barrier in Alzheimer’s disease. Neurobiol. Dis. 2017, 107, 41–56. [Google Scholar] [CrossRef] [Green Version]
- Van Dyken, P.; Lacoste, B. Impact of Metabolic Syndrome on Neuroinflammation and the Blood–Brain Barrier. Front. Neurosci. 2018, 12, 930. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, J.C.D.; Killcross, A.S.; Jenkins, T.A. Obesity and cognitive decline: Role of inflammation and vascular changes. Front. Neurosci. 2014, 8, 375. [Google Scholar] [CrossRef]
- Latz, E.; Duewell, P. NLRP3 inflammasome activation in inflammaging. Semin. Immunol. 2018, 40, 61–73. [Google Scholar] [CrossRef]
- Guo, D.-H.; Yamamoto, M.; Hernandez, C.M.; Khodadadi, H.; Baban, B.; Stranahan, A.M. Visceral adipose NLRP3 impairs cognition in obesity via IL-1R1 on CX3CR1+ cells. J. Clin. Investig. 2020, 130, 1961–1976. [Google Scholar] [CrossRef] [Green Version]
- Guan, Y.; Han, F. Key Mechanisms and Potential Targets of the NLRP3 Inflammasome in Neurodegenerative Diseases. Front. Integr. Neurosci. 2020, 14, 37. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Dixit, V.M. Mechanisms and Functions of Inflammasomes. Cell 2014, 157, 1013–1022. [Google Scholar] [CrossRef] [Green Version]
- Assmann, T.S.; Brondani, L.D.A.; Bouças, A.P.; Canani, L.H.; Crispim, D. Toll-like receptor 3 (TLR3) and the development of type 1 diabetes mellitus. Arch. Endocrinol. Metab. 2015, 59, 4–12. [Google Scholar] [CrossRef] [Green Version]
- Ezhong, Y.; Ekinio, A.; Esaleh, M. Functions of NOD-Like Receptors in Human Diseases. Front. Immunol. 2013, 4, 333. [Google Scholar] [CrossRef] [Green Version]
- De Zoete, M.R.; Palm, N.W.; Zhu, S.; Flavell, R.A. Inflammasomes. Cold Spring Harb. Perspect. Biol. 2014, 6, a016287. [Google Scholar] [CrossRef]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Minkiewicz, J.; Vaccari, J.P.D.R.; Keane, R.W. Human astrocytes express a novel NLRP2 inflammasome. Glia 2013, 61, 1113–1121. [Google Scholar] [CrossRef]
- Von Herrmann, K.M.; Salas, L.A.; Martinez, E.M.; Young, A.L.; Howard, J.M.; Feldman, M.S.; Christensen, B.C.; Wilkins, O.M.; Lee, S.L.; Hickey, W.F.; et al. NLRP3 expression in mesencephalic neurons and characterization of a rare NLRP3 polymorphism associated with decreased risk of Parkinson’s disease. npj Park. Dis. 2018, 4, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Xia, M.; Boini, K.M.; Abais, J.M.; Xu, M.; Zhang, Y.; Li, P.-L. Endothelial NLRP3 Inflammasome Activation and Enhanced Neointima Formation in Mice by Adipokine Visfatin. Am. J. Pathol. 2014, 184, 1617–1628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, H.; Zhu, F.; Xu, Z.; Xiong, J. Role of Inflammasomes in Kidney Diseases via Both Canonical and Non-canonical Pathways. Front. Cell Dev. Biol. 2020, 8, 106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, R.; Tardivel, A.; Thorens, B.; Choi, I.; Tschopp, J. Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat. Immunol. 2010, 11, 136–140. [Google Scholar] [CrossRef]
- Vandanmagsar, B.; Youm, Y.-H.; Ravussin, A.; Galgani, J.E.; Stadler, K.; Mynatt, R.L.; Ravussin, E.; Stephens, J.M.; Dixit, V.D. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat. Med. 2011, 17, 179–188. [Google Scholar] [CrossRef]
- Cañadas-Lozano, D.; Marín-Aguilar, F.; Castejón-Vega, B.; Ryffel, B.; Navarro-Pando, J.M.; Ruiz-Cabello, J.; Alcocer-Gómez, E.; Bullón, P.; Cordero, M.D. Blockade of the NLRP3 inflammasome improves metabolic health and lifespan in obese mice. GeroScience 2020, 42, 715–725. [Google Scholar] [CrossRef]
- Pahwa, R.; Singh, A.; Adams-Huet, B.; Devaraj, S.; Jialal, I. Increased inflammasome activity in subcutaneous adipose tissue of patients with metabolic syndrome. Diabetes/Metabolism Res. Rev. 2021, 37. [Google Scholar] [CrossRef]
- Kursawe, R.; Dixit, V.D.; Scherer, P.E.; Santoro, N.; Narayan, D.; Gordillo, R.; Giannini, C.; Lopez, X.; Pierpont, B.; Nouws, J.; et al. A Role of the Inflammasome in the Low Storage Capacity of the Abdominal Subcutaneous Adipose Tissue in Obese Adolescents. Diabetes 2016, 65, 610–618. [Google Scholar] [CrossRef] [Green Version]
- Gao, D.; Madi, M.; Ding, C.; Fok, M.; Steele, T.; Ford, C.; Hunter, L.; Bing, C. Interleukin-1β mediates macrophage-induced impairment of insulin signaling in human primary adipocytes. Am. J. Physiol. Metab. 2014, 307, E289–E304. [Google Scholar] [CrossRef]
- Owyang, A.M.; Maedler, K.; Gross, L.; Yin, J.; Esposito, L.; Shu, L.; Jadhav, J.; Domsgen, E.; Bergemann, J.; Lee, S.; et al. XOMA 052, an Anti-IL-1β Monoclonal Antibody, Improves Glucose Control and β-Cell Function in the Diet-Induced Obesity Mouse Model. Endocrinol. 2010, 151, 2515–2527. [Google Scholar] [CrossRef]
- Chang, Y.-W.; Hung, L.-C.; Chen, Y.-C.; Wang, W.-H.; Lin, C.-Y.; Tzeng, H.-H.; Suen, J.-L.; Chen, Y.-H. Insulin Reduces Inflammation by Regulating the Activation of the NLRP3 Inflammasome. Front. Immunol. 2021, 11. [Google Scholar] [CrossRef]
- Murphy, A.J.; Kraakman, M.J.; Kammoun, H.L.; Dragoljevic, D.; Lee, M.K.; Lawlor, K.E.; Wentworth, J.M.; Vasanthakumar, A.; Gerlic, M.; Whitehead, L.W.; et al. IL-18 Production from the NLRP1 Inflammasome Prevents Obesity and Metabolic Syndrome. Cell Metab. 2016, 23, 155–164. [Google Scholar] [CrossRef] [Green Version]
- Unamuno, X.; Gómez-Ambrosi, J.; Ramírez, B.; Rodríguez, A.; Becerril, S.; Valentí, V.; Moncada, R.; Silva, C.; Salvador, J.; Frühbeck, G.; et al. NLRP3 inflammasome blockade reduces adipose tissue inflammation and extracellular matrix remodeling. Cell. Mol. Immunol. 2021, 18, 1045–1057. [Google Scholar] [CrossRef]
- Jialal, I.; Rajamani, U.; Adams-Huet, B.; Kaur, H. Circulating pathogen associated molecular pattern—Binding proteins and High Mobility Group Box protein 1 in nascent metabolic syndrome: Implications for cellular Toll-like receptor activity. Atheroscler 2014, 236, 182–187. [Google Scholar] [CrossRef]
- Yin, Z.; Deng, T.; Peterson, L.E.; Yu, R.; Lin, J.; Hamilton, D.J.; Reardon, P.R.; Sherman, V.; Winnier, G.E.; Zhan, M.; et al. Transcriptome analysis of human adipocytes implicates the NOD-like receptor pathway in obesity-induced adipose inflammation. Mol. Cell. Endocrinol. 2014, 394, 80–87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, H.; Youm, Y.-H.; Vandanmagsar, B.; Rood, J.; Kumar, K.G.; Butler, A.; Dixit, V.D. Obesity accelerates thymic aging. Blood 2009, 114, 3803–3812. [Google Scholar] [CrossRef] [Green Version]
- Camell, C.D.; Günther, P.; Lee, A.; Goldberg, E.L.; Spadaro, O.; Youm, Y.-H.; Bartke, A.; Hubbard, G.B.; Ikeno, Y.; Ruddle, N.H.; et al. Aging Induces an Nlrp3 Inflammasome-Dependent Expansion of Adipose B Cells That Impairs Metabolic Homeostasis. Cell Metab. 2019, 30, 1024–1039. [Google Scholar] [CrossRef]
- Meyers, A.K.; Zhu, X. The NLRP3 Inflammasome: Metabolic Regulation and Contribution to Inflammaging. Cells 2020, 9, 1808. [Google Scholar] [CrossRef]
- Sebastian-Valverde, M.; Pasinetti, G.M. The NLRP3 Inflammasome as a Critical Actor in the Inflammaging Process. Cells 2020, 9, 1552. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T. The metabolic regulation of aging. Nat. Med. 2015, 21, 1416–1423. [Google Scholar] [CrossRef]
- Wen, H.; Gris, D.; Lei, Y.; Jha, S.; Zhang, L.; Huang, M.T.-H.; Brickey, W.J.; Ting, J.P.-Y. Fatty acid–induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat. Immunol. 2011, 12, 408–415. [Google Scholar] [CrossRef] [Green Version]
- Stienstra, R.; van Diepen, J.A.; Tack, C.J.; Zaki, M.H.; van de Veerdonk, F.L.; Perera, D.; Neale, G.A.; Hooiveld, G.J.; Hijmans, A.; Vroegrijk, I.; et al. Inflammasome is a central player in the induction of obesity and insulin resistance. Proc. Natl. Acad. Sci. USA 2011, 108, 15324–15329. [Google Scholar] [CrossRef] [Green Version]
- Duewell, P.; Kono, H.; Rayner, K.; Sirois, C.M.; Vladimer, G.; Bauernfeind, F.G.; Abela, G.S.; Franchi, L.; Nuñez, G.; Schnurr, M.; et al. NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nat. Cell Biol. 2010, 464, 1357–1361. [Google Scholar] [CrossRef] [Green Version]
- Marín-Aguilar, F.; Lechuga-Vieco, A.V.; Alcocer-Gómez, E.; Castejón-Vega, B.; Lucas, J.; Garrido, C.; Peralta-Garcia, A.; Pérez-Pulido, A.J.; Varela-López, A.; Quiles, J.L.; et al. NLRP3 inflammasome suppression improves longevity and prevents cardiac aging in male mice. Aging Cell 2020, 19, e13050. [Google Scholar] [CrossRef] [Green Version]
- Bullón, P.; Cano-García, F.J.; Alcocer-Gómez, E.; Varela-López, A.; Roman-Malo, L.; Ruiz-Salmerón, R.J.; Quiles, J.L.; Navarro-Pando, J.M.; Battino, M.; Ruiz-Cabello, J.; et al. Could NLRP3–Inflammasome Be a Cardiovascular Risk Biomarker in Acute Myocardial Infarction Patients? Antioxidants Redox Signal. 2017, 27, 269–275. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Zeng, X.; Li, X.; Mehta, J.L.; Wang, X. Role of NLRP3 inflammasome in the pathogenesis of cardiovascular diseases. Basic Res. Cardiol. 2018, 113, 5. [Google Scholar] [CrossRef]
- Youm, Y.-H.; Kanneganti, T.-D.; Vandanmagsar, B.; Zhu, X.; Ravussin, A.; Adijiang, A.; Owen, J.S.; Thomas, M.J.; Francis, J.; Parks, J.S.; et al. The NLRP3 Inflammasome Promotes Age-Related Thymic Demise and Immunosenescence. Cell Rep. 2012, 1, 56–68. [Google Scholar] [CrossRef] [Green Version]
- Broz, P.; Ruby, T.; Belhocine, K.; Bouley, D.M.; Kayagaki, N.; Dixit, V.M.; Monack, D.M. Caspase-11 increases susceptibility to Salmonella infection in the absence of caspase-1. Nat. Cell Biol. 2012, 490, 288–291. [Google Scholar] [CrossRef]
- Fang, R.; Tsuchiya, K.; Kawamura, I.; Shen, Y.; Hara, H.; Sakai, S.; Yamamoto, T.; Fernandes-Alnemri, T.; Yang, R.; Hernandez-Cuellar, E.; et al. Critical Roles of ASC Inflammasomes in Caspase-1 Activation and Host Innate Resistance toStreptococcus pneumoniaeInfection. J. Immunol. 2011, 187, 4890–4899. [Google Scholar] [CrossRef] [Green Version]
- Marín-Aguilar, F.; Castejón-Vega, B.; Alcocer-Gómez, E.; Lendines-Cordero, D.; Cooper, M.A.; De La Cruz, P.; Andujar, E.; Pérez-Alegre, M.; Muntané, J.; Pérez-Pulido, A.J.; et al. NLRP3 Inflammasome Inhibition by MCC950 in Aged Mice Improves Health via Enhanced Autophagy and PPARα Activity. Journals Gerontol. Ser. A: Boil. Sci. Med Sci. 2019, 75, 1457–1464. [Google Scholar] [CrossRef]
- Luo, H.; Mu, W.-C.; Karki, R.; Chiang, H.-H.; Mohrin, M.; Shin, J.J.; Ohkubo, R.; Ito, K.; Kanneganti, T.-D.; Chen, D. Mitochondrial Stress-Initiated Aberrant Activation of the NLRP3 Inflammasome Regulates the Functional Deterioration of Hematopoietic Stem Cell Aging. Cell Rep. 2019, 26, 945–954.e4. [Google Scholar] [CrossRef] [Green Version]
- Youm, Y.-H.; Grant, R.; McCabe, L.R.; Albarado, D.C.; Nguyen, K.Y.; Ravussin, A.; Pistell, P.; Newman, S.; Carter, R.; Laque, A.; et al. Canonical Nlrp3 Inflammasome Links Systemic Low-Grade Inflammation to Functional Decline in Aging. Cell Metab. 2013, 18, 519–532. [Google Scholar] [CrossRef] [Green Version]
- Guo, H.; Callaway, J.B.; Ting, J.P.Y. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef] [Green Version]
- Song, L.; Pei, L.; Yao, S.; Wu, Y.; Shang, Y. NLRP3 Inflammasome in Neurological Diseases, from Functions to Therapies. Front. Cell. Neurosci. 2017, 11, 63. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.-C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nat. Cell Biol. 2013, 493, 674–678. [Google Scholar] [CrossRef]
- Dempsey, C.; Araiz, A.R.; Bryson, K.; Finucane, O.; Larkin, C.; Mills, E.; Robertson, A.; Cooper, M.; O’Neill, L.; Lynch, M. Inhibiting the NLRP3 inflammasome with MCC950 promotes non-phlogistic clearance of amyloid-β and cognitive function in APP/PS1 mice. Brain, Behav. Immun. 2017, 61, 306–316. [Google Scholar] [CrossRef] [Green Version]
- Kaushal, V.; Dye, R.; Pakavathkumar, P.; Foveau, B.; Flores, J.C.; Hyman, B.T.; Ghetti, B.; Koller, B.H.; Leblanc, A.C. Neuronal NLRP1 inflammasome activation of Caspase-1 coordinately regulates inflammatory interleukin-1-beta production and axonal degeneration-associated Caspase-6 activation. Cell Death Differ. 2015, 22, 1676–1686. [Google Scholar] [CrossRef] [Green Version]
- Freeman, L.; Guo, H.; David, C.N.; Brickey, W.J.; Jha, S.; Ting, J.P.-Y. NLR members NLRC4 and NLRP3 mediate sterile inflammasome activation in microglia and astrocytes. J. Exp. Med. 2017, 214, 1351–1370. [Google Scholar] [CrossRef] [Green Version]
- Gong, Z.; Pan, J.; Shen, Q.; Li, M.; Peng, Y. Mitochondrial dysfunction induces NLRP3 inflammasome activation during cerebral ischemia/reperfusion injury. J. Neuroinflamm. 2018, 15, 1–17. [Google Scholar] [CrossRef]
- McKenzie, B.A.; Mamik, M.K.; Saito, L.B.; Boghozian, R.; Monaco, M.C.; Major, E.O.; Lu, J.-Q.; Branton, W.G.; Power, C. Caspase-1 inhibition prevents glial inflammasome activation and pyroptosis in models of multiple sclerosis. Proc. Natl. Acad. Sci. USA 2018, 115, E6065–E6074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saresella, M.; La Rosa, F.; Piancone, F.; Zoppis, M.; Marventano, I.; Calabrese, E.; Rainone, V.; Nemni, R.; Mancuso, R.; Clerici, M. The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol. Neurodegener. 2016, 11, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Serena, C.; Keiran, N.; Ceperuelo-Mallafre, V.; Ejarque, M.; Fradera, R.; Roche, K.; Nuñez-Roa, C.; Vendrell, J.; Fernández-Veledo, S. Obesity and Type 2 Diabetes Alters the Immune Properties of Human Adipose Derived Stem Cells. Stem Cells 2016, 34, 2559–2573. [Google Scholar] [CrossRef] [PubMed]
- Ising, C.; Venegas, C.; Zhang, S.; Scheiblich, H.; Schmidt, S.V.; Vieira-Saecker, A.; Schwartz, S.; Albasset, S.; McManus, R.M.; Tejera, D.; et al. NLRP3 inflammasome activation drives tau pathology. Nature 2019, 575, 669–673. [Google Scholar] [CrossRef]
- Li, L.; Ismael, S.; Nasoohi, S.; Sakata, K.; Liao, F.-F.; McDonald, M.P.; Ishrat, T. Thioredoxin-Interacting Protein (TXNIP) Associated NLRP3 Inflammasome Activation in Human Alzheimer’s Disease Brain. J. Alzheimer’s Dis. 2019, 68, 255–265. [Google Scholar] [CrossRef]
- Griffin, W.S.T.; Liu, L.; Li, Y.; Mrak, R.E.; Barger, S.W. Interleukin-1 mediates Alzheimer and Lewy body pathologies. J. Neuroinflamm. 2006, 3, 5. [Google Scholar] [CrossRef] [Green Version]
- Bauernfeind, F.; Niepmann, S.; Knolle, P.A.; Hornung, V. Aging-Associated TNF Production Primes Inflammasome Activation and NLRP3-Related Metabolic Disturbances. J. Immunol. 2016, 197, 2900–2908. [Google Scholar] [CrossRef] [Green Version]
- He, X.-F.; Xu, J.-H.; Li, G.; Li, M.-Y.; Li, L.-L.; Pei, Z.; Zhang, L.-Y.; Hu, X.-Q. NLRP3-dependent microglial training impaired the clearance of amyloid-beta and aggravated the cognitive decline in Alzheimer’s disease. Cell Death Dis. 2020, 11, 1–11. [Google Scholar] [CrossRef]
- Ojala, J.; Alafuzoff, I.; Herukka, S.-K.; van Groen, T.; Tanila, H.; Pirttilä, T. Expression of interleukin-18 is increased in the brains of Alzheimer’s disease patients. Neurobiol. Aging 2009, 30, 198–209. [Google Scholar] [CrossRef]
- Parajuli, B.; Sonobe, Y.; Horiuchi, H.; Takeuchi, H.; Mizuno, T.; Suzumura, A. Oligomeric amyloid β induces IL-1β processing via production of ROS: Implication in Alzheimer’s disease. Cell Death Dis. 2013, 4, e975. [Google Scholar] [CrossRef] [Green Version]
- Venegas, C.; Kumar, S.; Franklin, B.S.; Dierkes, T.; Brinkschulte, R.; Tejera, D.; Vieira-Saecker, A.; Schwartz, S.; Santarelli, F.; Kummer, M.P.; et al. Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature 2017, 552, 355–361. [Google Scholar] [CrossRef]
- Lee, J.H.; Kim, H.J.; Kim, J.U.; Yook, T.H.; Kim, K.H.; Lee, J.Y.; Yang, G. A Novel Treatment Strategy by Natural Products in NLRP3 Inflammasome-Mediated Neuroinflammation in Alzheimer’s and Parkinson’s Disease. Int. J. Mol. Sci. 2021, 22, 1324. [Google Scholar] [CrossRef]
- Zahid, A.; Li, B.; Kombe, A.J.K.; Jin, T.; Tao, J. Pharmacological Inhibitors of the NLRP3 Inflammasome. Front. Immunol. 2019, 10, 2538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lamkanfi, M.; Mueller, J.L.; Vitari, A.C.; Misaghi, S.; Fedorova, A.; Deshayes, K.; Lee, W.P.; Hoffman, H.M.; Dixit, V.M. Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J. Cell Biol. 2009, 187, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Kuwar, R.; Rolfe, A.; Di, L.; Xu, H.; He, L.; Jiang, Y.; Zhang, S.; Sun, D. A novel small molecular NLRP3 inflammasome inhibitor alleviates neuroinflammatory response following traumatic brain injury. J. Neuroinflammation 2019, 16, 1–14. [Google Scholar] [CrossRef]
- Yin, J.; Zhao, F.; Chojnacki, J.E.; Fulp, J.; Klein, W.L.; Zhang, S.; Zhu, X. NLRP3 Inflammasome Inhibitor Ameliorates Amyloid Pathology in a Mouse Model of Alzheimer’s Disease. Mol. Neurobiol. 2018, 55, 1977–1987. [Google Scholar] [CrossRef]
- Coll, R.; Robertson, A.A.B.; Chae, J.J.; Higgins, S.C.; Muñoz-Planillo, R.; Inserra, M.C.; Vetter, I.; Dungan, L.S.; Monks, B.G.; Stutz, A.; et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat. Med. 2015, 21, 248–255. [Google Scholar] [CrossRef] [Green Version]
- Qi, Y.; Klyubin, I.; Cuello, A.C.; Rowan, M.J. NLRP3-dependent synaptic plasticity deficit in an Alzheimer’s disease amyloidosis model in vivo. Neurobiol. Dis. 2018, 114, 24–30. [Google Scholar] [CrossRef]
- Li, J.; Zhuang, L.; Luo, X.; Liang, J.; Sun, E.; He, Y. Protection of MCC950 against Alzheimer’s disease via inhibiting neuronal pyroptosis in SAMP8 mice. Exp. Brain Res. 2020, 238, 2603–2614. [Google Scholar] [CrossRef]
- Marchetti, C.; Swartzwelter, B.; Gamboni, F.; Neff, C.P.; Richter, K.; Azam, T.; Carta, S.; Tengesdal, I.; Nemkov, T.; D’Alessandro, A.; et al. OLT1177, a β-sulfonyl nitrile compound, safe in humans, inhibits the NLRP3 inflammasome and reverses the metabolic cost of inflammation. Proc. Natl. Acad. Sci. USA 2018, 115, E1530–E1539. [Google Scholar] [CrossRef] [Green Version]
- Lonnemann, N.; Hosseini, S.; Marchetti, C.; Skouras, D.B.; Stefanoni, D.; D’Alessandro, A.; Dinarello, C.A.; Korte, M. The NLRP3 inflammasome inhibitor OLT1177 rescues cognitive impairment in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2020, 117, 32145–32154. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhao, Y.; Zhang, J.; Yang, G. Mechanisms of NLRP3 Inflammasome Activation: Its Role in the Treatment of Alzheimer’s Disease. Neurochem. Res. 2020, 45, 2560–2572. [Google Scholar] [CrossRef]
- Daniels, M.J.D.; Rivers-Auty, J.; Schilling, T.; Spencer, N.G.; Watremez, W.; Fasolino, V.; Booth, S.J.; White, C.S.; Baldwin, A.G.; Freeman, S.; et al. Fenamate NSAIDs inhibit the NLRP3 inflammasome and protect against Alzheimer’s disease in rodent models. Nat. Commun. 2016, 7, 12504. [Google Scholar] [CrossRef] [Green Version]
- Shippy, D.C.; Wilhelm, C.; Viharkumar, P.A.; Raife, T.J.; Ulland, T.K. β-Hydroxybutyrate inhibits inflammasome activation to attenuate Alzheimer’s disease pathology. J. Neuroinflamm. 2020, 17, 1–12. [Google Scholar] [CrossRef]
- Lilamand, M.; Mouton-Liger, F.; Paquet, C. Ketogenic diet therapy in Alzheimer’s disease. Curr. Opin. Clin. Nutr. Metab. Care 2021, Publish Ah, 3892. [Google Scholar] [CrossRef]
- Wani, K.; Alharthi, H.; Alghamdi, A.; Sabico, S.; Al-Daghri, N.M. Role of NLRP3 Inflammasome Activation in Obesity-Mediated Metabolic Disorders. Int. J. Environ. Res. Public Heal. 2021, 18, 511. [Google Scholar] [CrossRef]
- Mccarty, M.F.; Assanga, S.B.I.; Luján, L.L.; O’Keefe, J.H.; DiNicolantonio, J.J. Nutraceutical Strategies for Suppressing NLRP3 Inflammasome Activation: Pertinence to the Management of COVID-19 and Beyond. Nutr. 2020, 13, 47. [Google Scholar] [CrossRef]
- Feng, Y.-S.; Tan, Z.-X.; Wu, L.-Y.; Dong, F.; Zhang, F. The involvement of NLRP3 inflammasome in the treatment of neurodegenerative diseases. Biomed. Pharmacother. 2021, 138, 111428. [Google Scholar] [CrossRef]
- Rosa, J.M.; Camargo, A.; Wolin, I.A.V.; Kaster, M.P.; Rodrigues, A.L.S. Physical exercise prevents amyloid β1−40-induced disturbances in NLRP3 inflammasome pathway in the hippocampus of mice. Metab. Brain Dis. 2021, 36, 351–359. [Google Scholar] [CrossRef]
- Liang, F.; Huang, T.; Li, B.; Zhao, Y.; Zhang, X.; Xu, B. High-intensity interval training and moderate-intensity continuous training alleviate β-amyloid deposition by inhibiting NLRP3 inflammasome activation in APPswe/PS1dE9 mice. NeuroReport 2020, 31, 425–432. [Google Scholar] [CrossRef]
- Camell, C.; Goldberg, E.L.; Dixit, V.D. Regulation of Nlrp3 inflammasome by dietary metabolites. Semin. Immunol. 2015, 27, 334–342. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Study | Samples | Results | Reference |
|---|---|---|---|
| Clinical study | VAT | ↑ expression of NLRP3, NLRP6, and ASC mRNA levels as well as the expression and release of IL-1β and IL-18 in VAT in patients with obesity and obesity-associated T2DM | [106] |
| SAT | ↑ expression of NLRP3, PYCARD, IL-1β, and IL-18 in adipocytes of obese postmenopausal women | [108] | |
| SAT | ↑ genes expression of TLR4, NLRP3, IL-1β, and caspase-1 in obese adolescents with high VAT/SAT fat depot | [101] | |
| ASCs | ↑ NLRP3 expression in ASCs from obese and/or T2DM patients | [135] | |
| SAT | ↑ expression of caspase-1, IL-1β, and IL-18 in SAT from MetS patients | [100] | |
| PBMC | ↑ genes expression of NLRP1, NLRP3, PYCARD, caspase-1, -5, -8, and downstream effectors IL-1β and IL-18 in monocytes from severe and mild AD | [134] | |
| The cortex of frontotemporal dementia (FTD) and AD patients | Elevated cleavage of caspase-1 and increased ASC levels and mature IL-1β | [136] | |
| The cortex of postmortem human brain of AD patients | ↑expression of cleaved caspase-1 and IL-1β in the cortex of AD brains | [137] | |
| Animal study | Old NLRP3−/− mice | Absence of NLRP3 diminished metabolic impairment induced by HFD during aging. | [99] |
| Nlrp3−/− mutant mice and Tg mice with inducible deletion of Il1r1 in CX3CR1-expressing cells | Nlrp3−/− mutant mice indicate a protective effect against obesity-induced neuroinflammation.Reduced level of IL-1β in VAT and hippocampal lysates in Nlrp3−/− mutant mice. | [86] | |
| Primary cultures of microglia and cortical neurons from Spraque-Dawley rats | IL-1β induces tau protein hyperphosphorylation. | [138] | |
| Female C57BL/6JRccHsd and C57BL/6JRccHsd aged mice | Aged mice show caspase-1 activation in myeloid cells within the adipose tissue, as well as enhanced serum IL-18 and impaired glucose tolerance. | [139] | |
| Wildtype and APP/PS1 mice | Inhibition of NLRP3 reduces Aβ accumulation in APP/PS1 mice. | [129] | |
| Caspase11−/−, Nlrp3−/−, Asc−/−, and IL1r−/− mice | Age-associated enrichment of NFκB, IL-1, and IL-8 signaling pro-inflammatory pathways was partially dependent on the Nlrp3 inflammasome. | [125] | |
| Mouse model of sporadic Alzheimer’s disease (SAD) induced by STZ | Increased levels of NLRP3 in the cortex and hippocampus in the STZ group compared with those in the sham group. | [140] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Litwiniuk, A.; Bik, W.; Kalisz, M.; Baranowska-Bik, A. Inflammasome NLRP3 Potentially Links Obesity-Associated Low-Grade Systemic Inflammation and Insulin Resistance with Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 5603. https://doi.org/10.3390/ijms22115603
Litwiniuk A, Bik W, Kalisz M, Baranowska-Bik A. Inflammasome NLRP3 Potentially Links Obesity-Associated Low-Grade Systemic Inflammation and Insulin Resistance with Alzheimer’s Disease. International Journal of Molecular Sciences. 2021; 22(11):5603. https://doi.org/10.3390/ijms22115603
Chicago/Turabian StyleLitwiniuk, Anna, Wojciech Bik, Małgorzata Kalisz, and Agnieszka Baranowska-Bik. 2021. "Inflammasome NLRP3 Potentially Links Obesity-Associated Low-Grade Systemic Inflammation and Insulin Resistance with Alzheimer’s Disease" International Journal of Molecular Sciences 22, no. 11: 5603. https://doi.org/10.3390/ijms22115603