
High-Throughput Sequencing of Phage Display Libraries Reveals Parasitic Enrichment of Indel Mutants Caused by Amplification Bias


Abstract
:1. Introduction
2. Results and Discussion
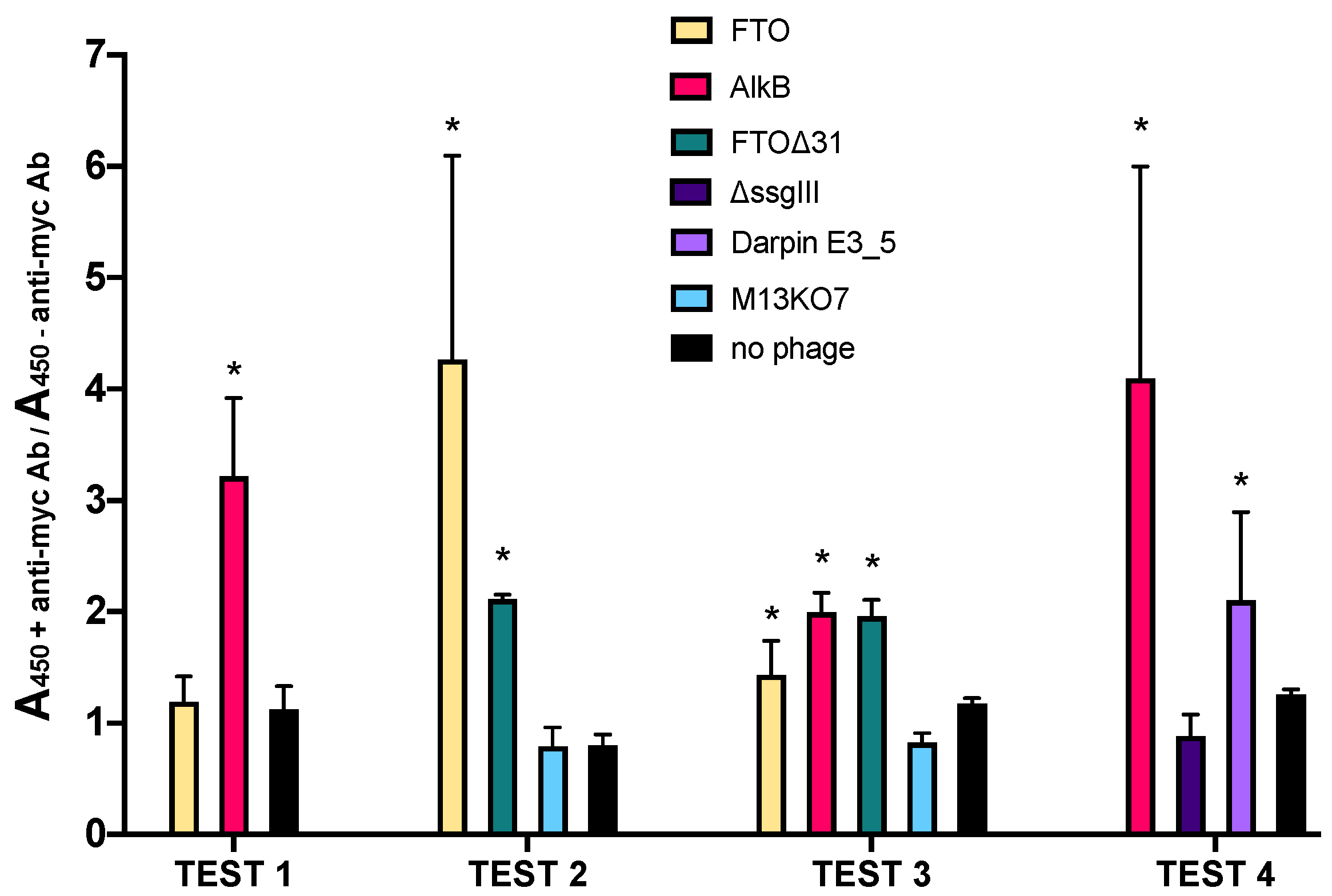
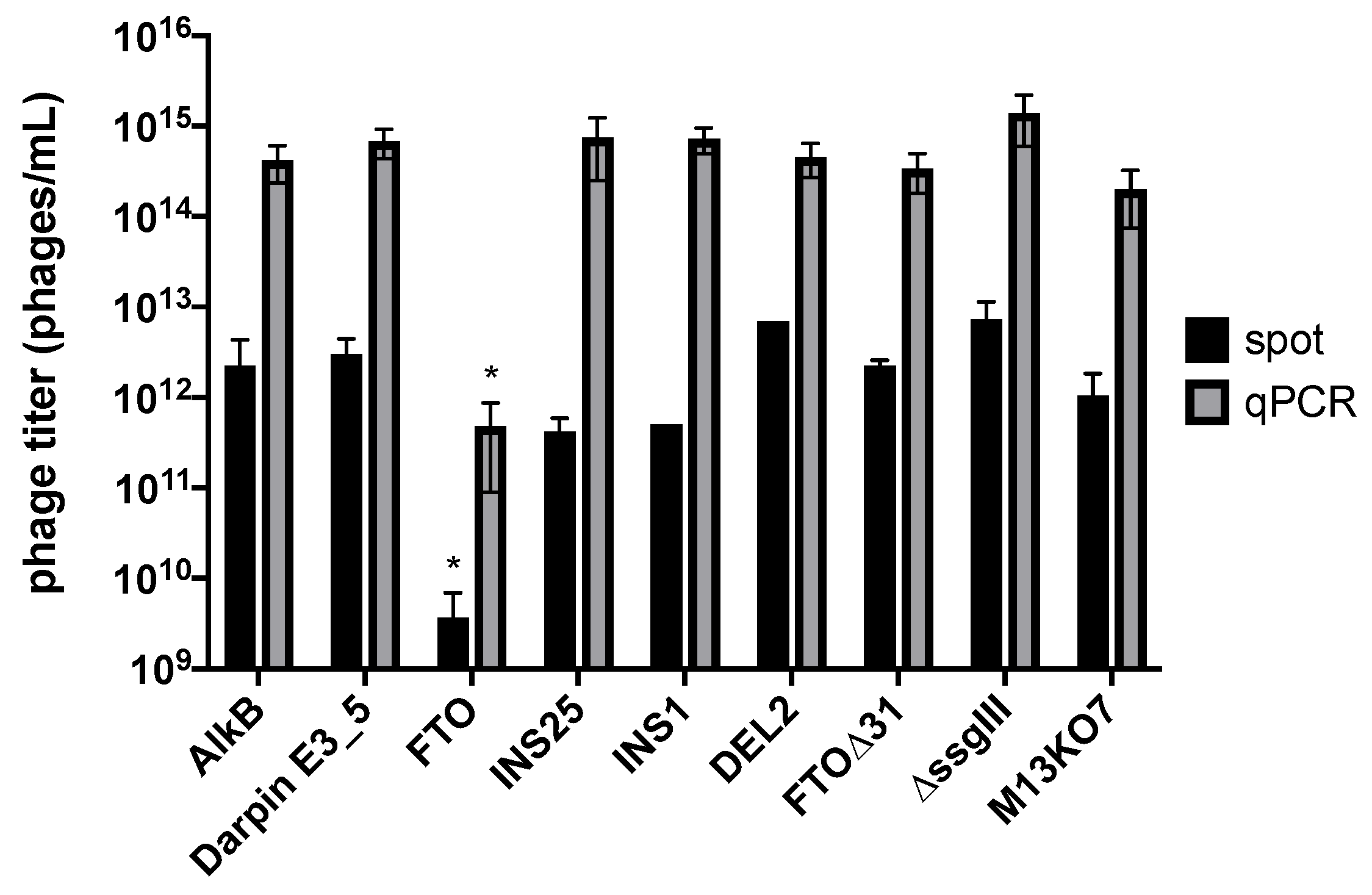
2.1. AlkB and FTO Are Amenable to Phage Display
2.2. High-Throughput Sequencing Reveals Enrichment of Parasitic FTO Genotypes
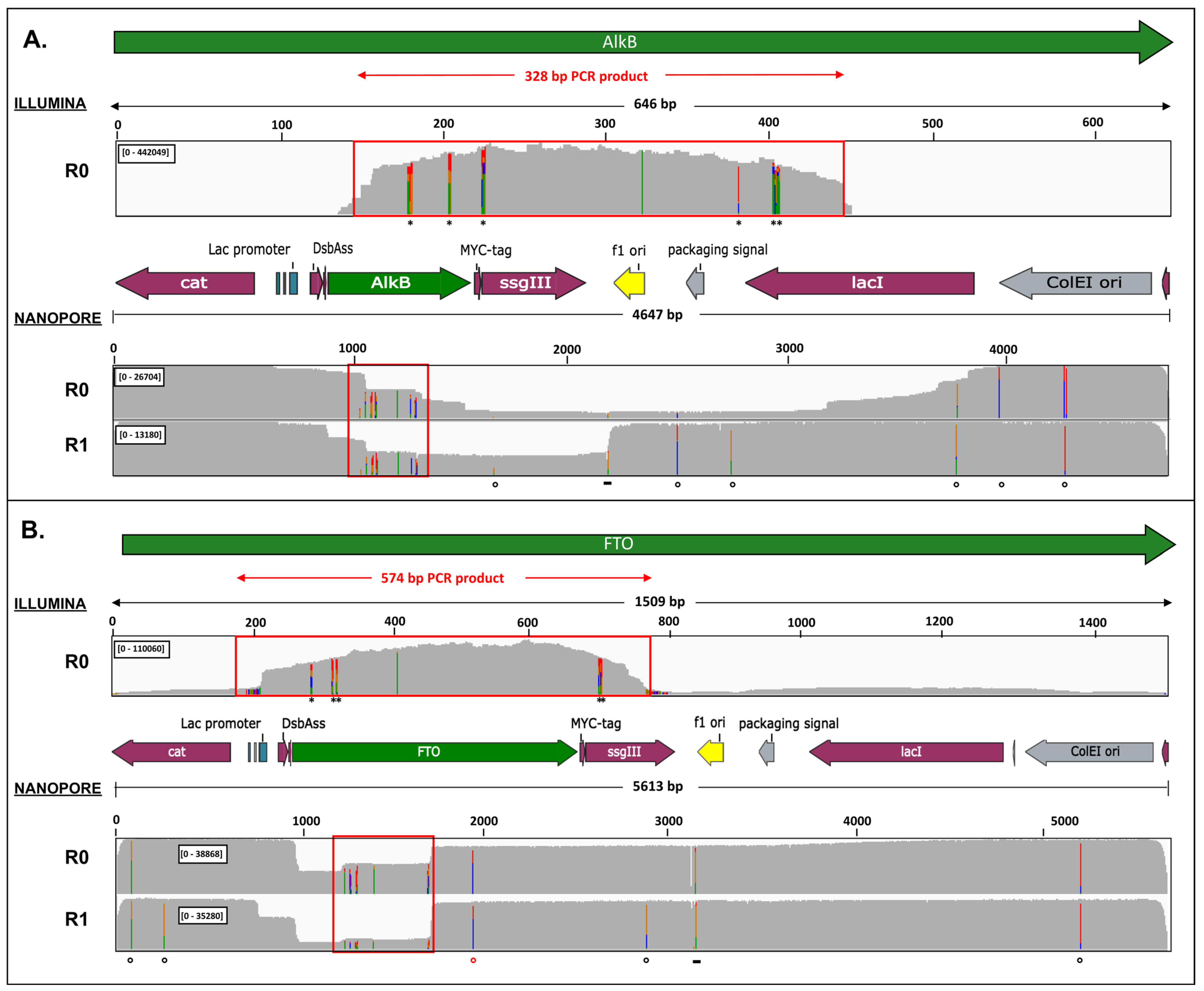
2.3. Nanopore Sequencing Allows Detection of Genotypes Not Observed by Illumina Sequencing
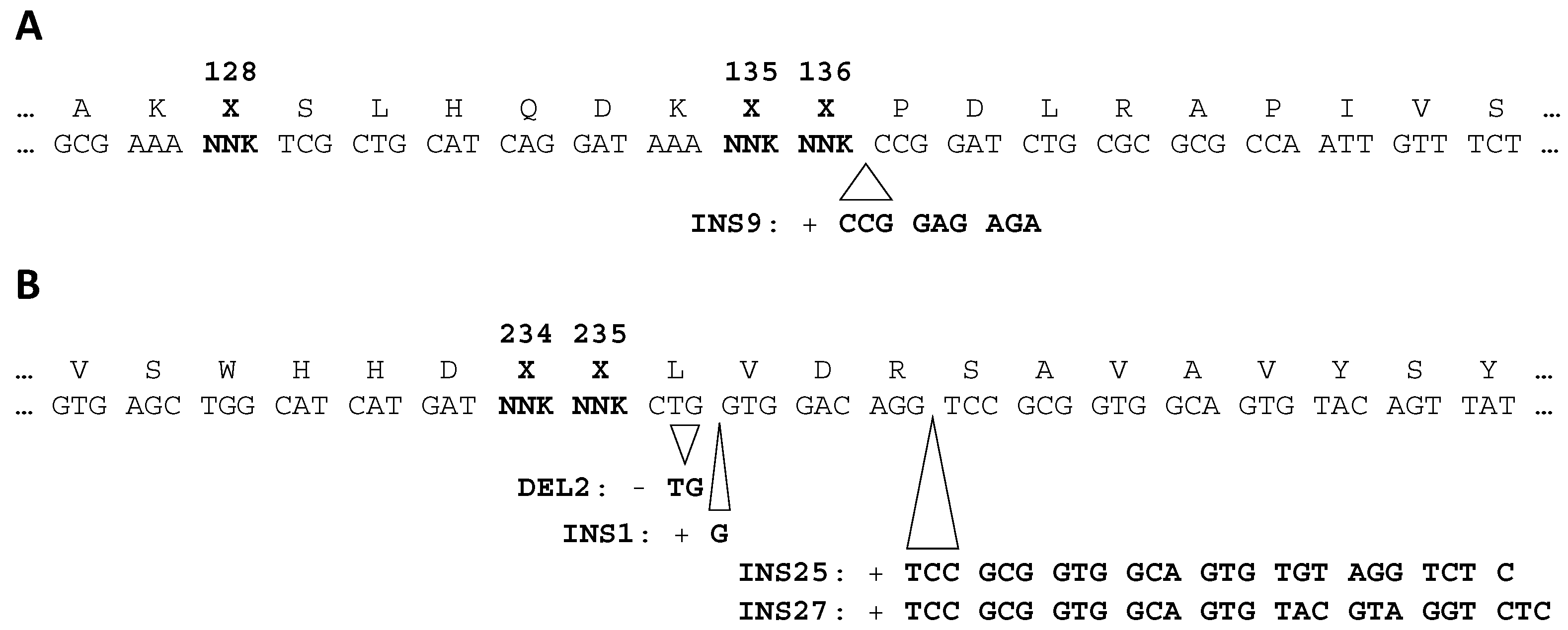
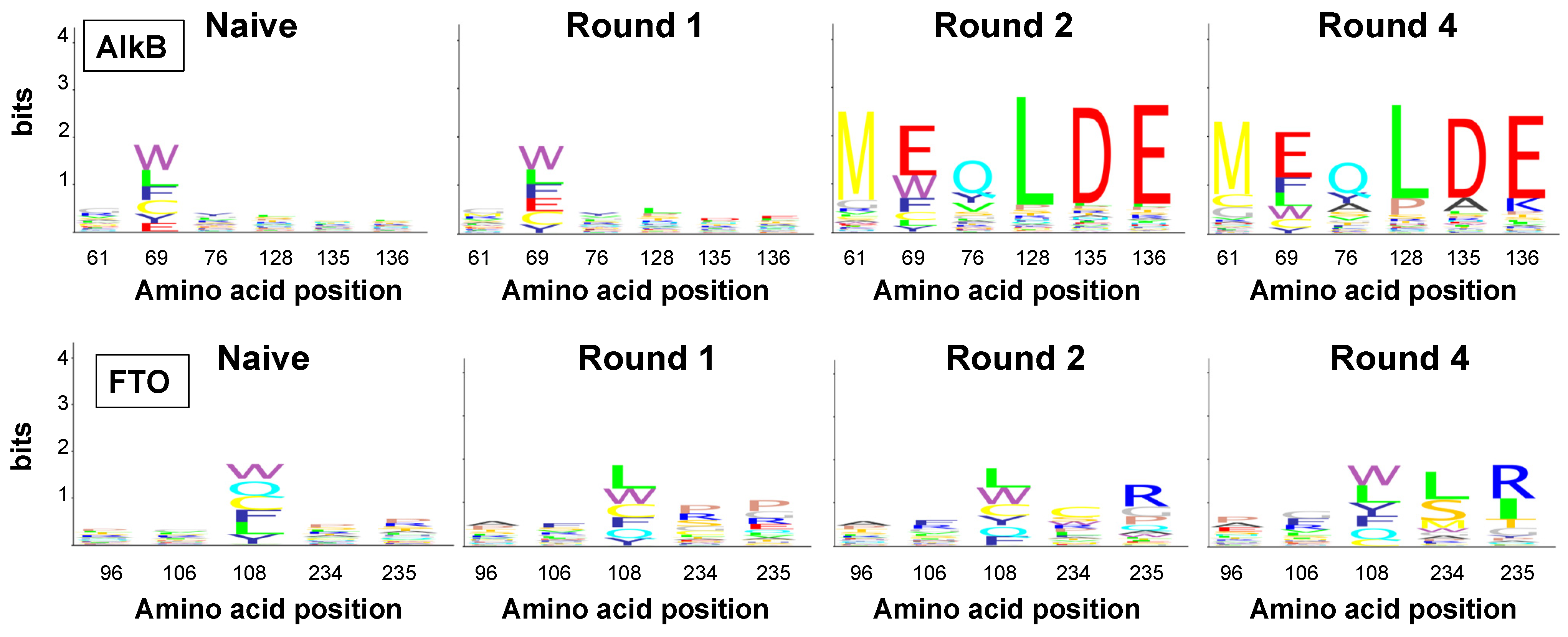
2.4. Amino Acid Patterns Deduced from Enriched Genotypes Can Be False Positives
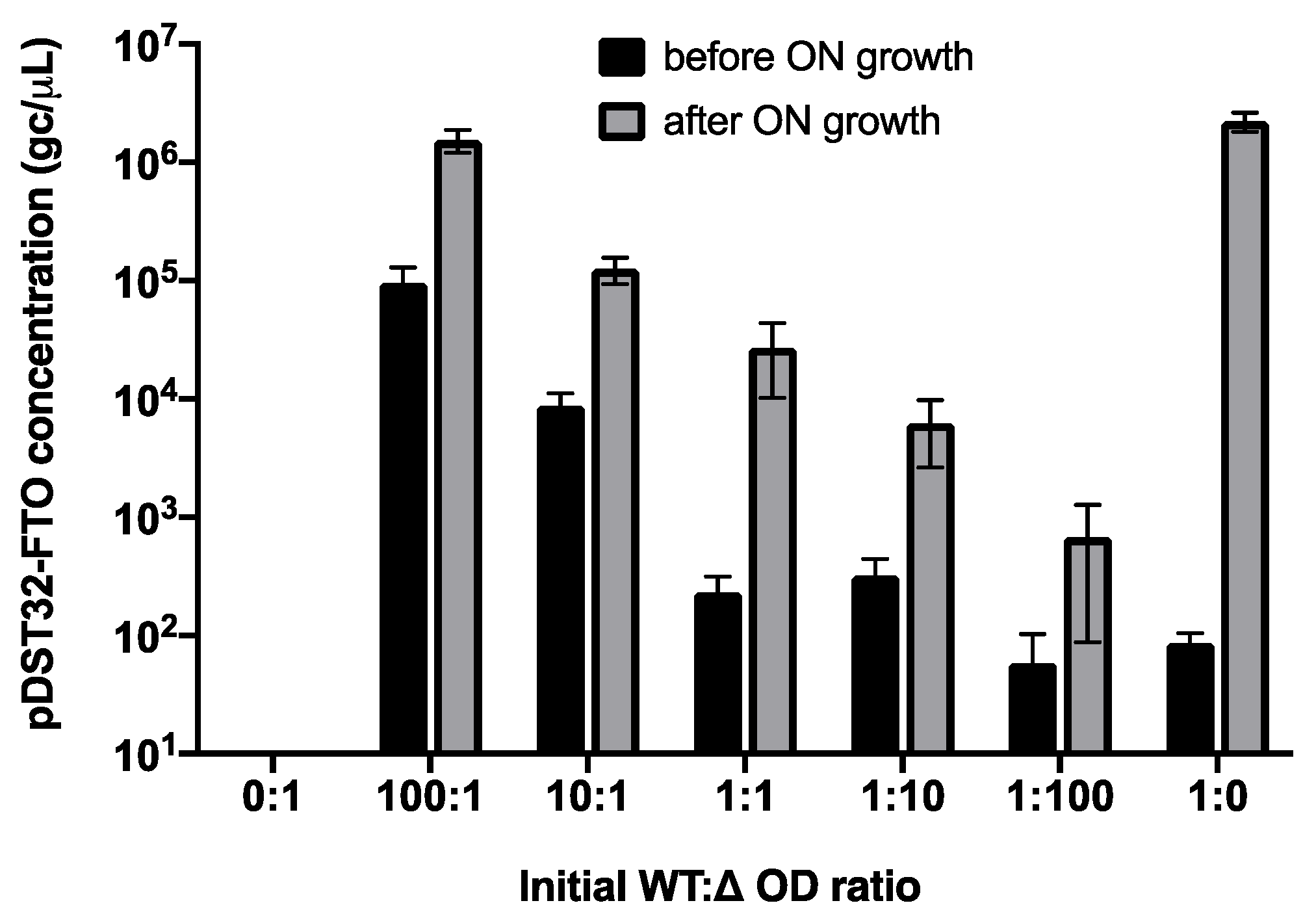
2.5. Parasitic Enrichment of FTO Variants Is Enhanced by Amplification Bias
3. Materials and Methods
3.1. AlkB and FTO Phage Production
3.2. Phage Titers
3.3. Anti-C-Myc Phage ELISA
3.4. Library Generation and Biopanning
3.5. Illumina Sequencing
3.6. Oxford Nanopore Sequencing
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Smith, G.P.; Petrenko, V.A. Phage Display. Chem. Rev. 1997, 97, 391–410. [Google Scholar] [CrossRef] [PubMed]
- Ravn, U.; Gueneau, F.; Baerlocher, L.; Osteras, M.; Desmurs, M.; Malinge, P.; Magistrelli, G.; Farinelli, L.; Kosco-Vilbois, M.H.; Fischer, N. By-passing in vitro screening--next generation sequencing technologies applied to antibody display and in silico candidate selection. Nucleic Acids Res. 2010, 38, e193. [Google Scholar] [CrossRef] [PubMed]
- Matochko, W.L.; Chu, K.; Jin, B.; Lee, S.W.; Whitesides, G.M.; Derda, R. Deep sequencing analysis of phage libraries using Illumina platform. Methods 2012, 58, 47–55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodwin, S.; McPherson, J.D.; McCombie, W.R. Coming of age: Ten years of next-generation sequencing technologies. Nat. Rev. Genet. 2016, 17, 333–351. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, J.A.; Jiang, N.; White, R.A.; Fisher, D.S.; Quake, S.R. High-throughput sequencing of the zebrafish antibody repertoire. Science 2009, 324, 807–810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeKosky, B.J.; Ippolito, G.C.; Deschner, R.P.; Lavinder, J.J.; Wine, Y.; Rawlings, B.M.; Varadarajan, N.; Giesecke, C.; Dörner, T.; Andrews, S.F.; et al. High-throughput sequencing of the paired human immunoglobulin heavy and light chain repertoire. Nat. Biotechnol. 2013, 31, 166–169. [Google Scholar] [CrossRef] [Green Version]
- Doria-Rose, N.A.; Schramm, C.A.; Gorman, J.; Moore, P.L.; Bhiman, J.N.; DeKosky, B.J.; Ernandes, M.J.; Georgiev, I.S.; Kim, H.J.; Pancera, M.; et al. Developmental pathway for potent V1V2-directed HIV-neutralizing antibodies. Nature 2014, 509, 55–62. [Google Scholar] [CrossRef]
- Robins, H.S.; Campregher, P.V.; Srivastava, S.K.; Wacher, A.; Turtle, C.J.; Kahsai, O.; Riddell, S.R.; Warren, E.H.; Carlson, C.S. Comprehensive assessment of T-cell receptor beta-chain diversity in alphabeta T cells. Blood 2009, 114, 4099–4107. [Google Scholar] [CrossRef]
- Glanville, J.; Zhai, W.; Berka, J.; Telman, D.; Huerta, G.; Mehta, G.R.; Ni, I.; Mei, L.; Sundar, P.D.; Day, G.M.; et al. Precise determination of the diversity of a combinatorial antibody library gives insight into the human immunoglobulin repertoire. Proc. Natl. Acad. Sci. USA 2009, 106, 20216–20221. [Google Scholar] [CrossRef] [Green Version]
- Rouet, R.; Jackson, K.J.L.; Langley, D.B.; Christ, D. Next-Generation Sequencing of Antibody Display Repertoires. Front. Immunol. 2018, 9, 118. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, N.; Malmborg, A.C.; Borrebaeck, C.A. The phage infection process: A functional role for the distal linker region of bacteriophage protein 3. J. Virol. 2000, 74, 4229–4235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinis, C.; Huber, A.; Demartis, S.; Bertschinger, J.; Melkko, S.; Lozzi, L.; Neri, P.; Neri, D. Selection of catalytically active biotin ligase and trypsin mutants by phage display. Protein Eng. 2001, 14, 1043–1052. [Google Scholar] [CrossRef]
- Bothmann, H.; Plückthun, A. Selection for a periplasmic factor improving phage display and functional periplasmic expression. Nat. Biotechnol. 1998, 16, 376–380. [Google Scholar] [CrossRef] [PubMed]
- Schimmele, B.; Gräfe, N.; Plückthun, A. Ribosome display of mammalian receptor domains. Protein Eng. Des. Sel. 2005, 18, 285–294. [Google Scholar] [CrossRef] [Green Version]
- Steiner, D.; Forrer, P.; Stumpp, M.T.; Plückthun, A. Signal sequences directing cotranslational translocation expand the range of proteins amenable to phage display. Nat. Biotechnol. 2006, 24, 823–831. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.J.; Kim, H.S.; Ryu, A.J.; Jeong, K.J. Enhanced production of full-length immunoglobulin G via the signal recognition particle (SRP)-dependent pathway in Escherichia coli. J. Biotechnol. 2013, 165, 102–108. [Google Scholar] [CrossRef]
- Speck, J.; Arndt, K.M.; Müller, K.M. Efficient phage display of intracellularly folded proteins mediated by the TAT pathway. Protein Eng. Des. Sel. 2011, 24, 473–484. [Google Scholar] [CrossRef] [Green Version]
- Matochko, W.L.; Ng, S.; Jafari, M.R.; Romaniuk, J.; Tang, S.K.; Derda, R. Uniform amplification of phage display libraries in monodisperse emulsions. Methods 2012, 58, 18–27. [Google Scholar] [CrossRef]
- Løset, G.; Kristinsson, S.G.; Sandlie, I. Reliable titration of filamentous bacteriophages independent of pIII fusion moiety and genome size by using trypsin to restore wild-type pIII phenotype. Biotechniques 2008, 44, 551–554. [Google Scholar] [CrossRef] [Green Version]
- Matochko, W.L.; Li, S.C.; Tang, S.K.; Derda, R. Prospective identification of parasitic sequences in phage display screens. Nucleic Acids Res. 2014, 42, 1784–1798. [Google Scholar] [CrossRef] [Green Version]
- Kuzmicheva, G.A.; Jayanna, P.K.; Sorokulova, I.B.; Petrenko, V.A. Diversity and censoring of landscape phage libraries. Protein Eng. Des. Sel. 2009, 22, 9–18. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Koch, H.; Dübel, S. Mutations in the N-terminus of the major coat protein (pVIII, gp8) of filamentous bacteriophage affect infectivity. J. Mol. Microbiol Biotechnol 2003, 6, 57–66. [Google Scholar] [CrossRef] [PubMed]
- Rodi, D.J.; Soares, A.S.; Makowski, L. Quantitative assessment of peptide sequence diversity in M13 combinatorial peptide phage display libraries. J. Mol. Biol. 2002, 322, 1039–1052. [Google Scholar] [CrossRef]
- Brammer, L.A.; Bolduc, B.; Kass, J.L.; Felice, K.M.; Noren, C.J.; Hall, M.F. A target-unrelated peptide in an M13 phage display library traced to an advantageous mutation in the gene II ribosome-binding site. Anal. Biochem. 2008, 373, 88–98. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, K.T.; Adamkiewicz, M.A.; Hebert, L.E.; Zygiel, E.M.; Boyle, H.R.; Martone, C.M.; Meléndez-Ríos, C.B.; Noren, K.A.; Noren, C.J.; Hall, M.F. Identification and characterization of mutant clones with enhanced propagation rates from phage-displayed peptide libraries. Anal. Biochem. 2014, 462, 35–43. [Google Scholar] [CrossRef] [Green Version]
- Thomas, W.D.; Golomb, M.; Smith, G.P. Corruption of phage display libraries by target-unrelated clones: Diagnosis and countermeasures. Anal. Biochem. 2010, 407, 237–240. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Falnes, P.; Johansen, R.F.; Seeberg, E. AlkB-mediated oxidative demethylation reverses DNA damage in Escherichia coli. Nature 2002, 419, 178–182. [Google Scholar] [CrossRef]
- Falnes, P.; Bjørås, M.; Aas, P.A.; Sundheim, O.; Seeberg, E. Substrate specificities of bacterial and human AlkB proteins. Nucleic Acids Res. 2004, 32, 3456–3461. [Google Scholar] [CrossRef]
- Jia, G.; Fu, Y.; Zhao, X.; Dai, Q.; Zheng, G.; Yang, Y.; Yi, C.; Lindahl, T.; Pan, T.; Yang, Y.G.; et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 2011, 7, 885–887. [Google Scholar] [CrossRef]
- Mauer, J.; Luo, X.; Blanjoie, A.; Jiao, X.; Grozhik, A.V.; Patil, D.P.; Linder, B.; Pickering, B.F.; Vasseur, J.J.; Chen, Q.; et al. Reversible methylation of m6Am in the 5′ cap controls mRNA stability. Nature 2017, 541, 371–375. [Google Scholar] [CrossRef] [Green Version]
- Goldman, E.; Korus, M.; Mandecki, W. Efficiencies of translation in three reading frames of unusual non-ORF sequences isolated from phage display. FASEB J. 2000, 14, 603–611. [Google Scholar] [CrossRef] [PubMed]
- Baranov, P.V.; Gurvich, O.L.; Hammer, A.W.; Gesteland, R.F.; Atkins, J.F. RECODE 2003. Nucleic Acids Res. 2003, 31, 87–89. [Google Scholar] [CrossRef] [PubMed]
- Singaravelan, B.; Roshini, B.R.; Munavar, M.H. Evidence that the supE44 mutation of Escherichia coli is an amber suppressor allele of glnX and that it also suppresses ochre and opal nonsense mutations. J. Bacteriol. 2010, 192, 6039–6044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Wei, L.H.; Wang, Y.; Xiao, Y.; Liu, J.; Zhang, W.; Yan, N.; Amu, G.; Tang, X.; Zhang, L.; et al. Structural insights into FTO’s catalytic mechanism for the demethylation of multiple RNA substrates. Proc. Natl. Acad. Sci. USA 2019, 116, 2919–2924. [Google Scholar] [CrossRef] [Green Version]
- Knappik, A.; Ge, L.; Honegger, A.; Pack, P.; Fischer, M.; Wellnhofer, G.; Hoess, A.; Wölle, J.; Plückthun, A.; Virnekäs, B. Fully synthetic human combinatorial antibody libraries (HuCAL) based on modular consensus frameworks and CDRs randomized with trinucleotides. J. Mol. Biol. 2000, 296, 57–86. [Google Scholar] [CrossRef] [PubMed]
- Binz, H.K.; Stumpp, M.T.; Forrer, P.; Amstutz, P.; Plückthun, A. Designing repeat proteins: Well-expressed, soluble and stable proteins from combinatorial libraries of consensus ankyrin repeat proteins. J. Mol. Biol. 2003, 332, 489–503. [Google Scholar] [CrossRef]
- Peng, X.; Nguyen, A.; Ghosh, D. Quantification of M13 and T7 bacteriophages by TaqMan and SYBR green qPCR. J. Virol. Methods 2018, 252, 100–107. [Google Scholar] [CrossRef]
- Van Deuren, V.; Plessers, S.; Robben, J. Structural determinants of nucleobase modification recognition in the AlkB family of dioxygenases. DNA Repair. (Amst.) 2020, 96, 102995. [Google Scholar] [CrossRef]
- Han, Z.; Niu, T.; Chang, J.; Lei, X.; Zhao, M.; Wang, Q.; Cheng, W.; Wang, J.; Feng, Y.; Chai, J. Crystal structure of the FTO protein reveals basis for its substrate specificity. Nature 2010, 464, 1205–1209. [Google Scholar] [CrossRef]
- Felix, K. Trim Galore. 2012. Available online: https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ (accessed on 9 March 2020).
- Greg, H. FASTX-Toolkit. 2010. Available online: http://hannonlab.cshl.edu/fastx_toolkit/ (accessed on 9 March 2020).
- Brian, B. BBMap. 2013. Available online: https://jgi.doe.gov/data-and-tools/bbtools/bb-tools-user-guide/bbmap-guide/ (accessed on 9 March 2020).
- Marcel, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10–12. [Google Scholar] [CrossRef]
- De Coster, W.; D’Hert, S.; Schultz, D.T.; Cruts, M.; van Broeckhoven, C. NanoPack: Visualizing and processing long-read sequencing data. Bioinformatics 2018, 34, 2666–2669. [Google Scholar] [CrossRef]
- Sedlazeck, F.J.; Rescheneder, P.; Smolka, M.; Fang, H.; Nattestad, M.; von Haeseler, A.; Schatz, M.C. Accurate detection of complex structural variations using single-molecule sequencing. Nat. Methods 2018, 15, 461–468. [Google Scholar] [CrossRef] [Green Version]
- Lowden, M.J.; Henry, K.A. Oxford nanopore sequencing enables rapid discovery of single-domain antibodies from phage display libraries. Biotechniques 2018, 65, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Tang, L. Circular consensus sequencing with long reads. Nat. Methods 2019, 16, 958. [Google Scholar] [CrossRef] [PubMed]
- Nannini, F.; Senicar, L.; Parekh, F.; Kong, K.J.; Kinna, A.; Bughda, R.; Sillibourne, J.; Hu, X.; Ma, B.; Bai, Y.; et al. Combining phage display with SMRTbell next-generation sequencing for the rapid discovery of functional scFv fragments. MAbs 2021, 13, 1864084. [Google Scholar] [CrossRef]
- Villequey, C.; Kong, X.D.; Heinis, C. Bypassing bacterial infection in phage display by sequencing DNA released from phage particles. Protein Eng. Des. Sel. 2017, 30, 761–768. [Google Scholar] [CrossRef]
- Marquez, H.; Koch, M.; Kestemont, D.; Arangundy-Franklin, S.; Pinheiro, V.B. Antha-guided Automation of Darwin Assembly for the Construction of Bespoke Gene Libraries. BioRxiv 2019, 12.17.8794486. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| alkB | FTO | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Library | Total Paired Reads | CAN (%) | INS9 (%) | Total Paired Reads | CAN (%) | INS25 (%) | INS27 (%) | INS1 (%) | DEL2 (%) |
| Naive | 761,541 | 77.09 | 17.22 | 115,191 | 67.69 | 1.06 | 0.14 | 0.01 | 0 |
| Round 1 | 151,953 | 82.26 | 14.14 | 106,962 | 28.59 | 19.88 | 0.61 | 0.76 | 0.06 |
| Round 2 | 140,577 | 86.26 | 3.71 | 64,156 | 16.7 | 25.24 | 0.71 | 2.6 | 1.67 |
| Round 4 | 128,364 | 85.79 | 4.28 | 9453 | 5.74 | 28.49 | 0.51 | 8.05 | 7.39 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Plessers, S.; Van Deuren, V.; Lavigne, R.; Robben, J. High-Throughput Sequencing of Phage Display Libraries Reveals Parasitic Enrichment of Indel Mutants Caused by Amplification Bias. Int. J. Mol. Sci. 2021, 22, 5513. https://doi.org/10.3390/ijms22115513
Plessers S, Van Deuren V, Lavigne R, Robben J. High-Throughput Sequencing of Phage Display Libraries Reveals Parasitic Enrichment of Indel Mutants Caused by Amplification Bias. International Journal of Molecular Sciences. 2021; 22(11):5513. https://doi.org/10.3390/ijms22115513
Chicago/Turabian StylePlessers, Sander, Vincent Van Deuren, Rob Lavigne, and Johan Robben. 2021. "High-Throughput Sequencing of Phage Display Libraries Reveals Parasitic Enrichment of Indel Mutants Caused by Amplification Bias" International Journal of Molecular Sciences 22, no. 11: 5513. https://doi.org/10.3390/ijms22115513