
Neuroprotective and Immunomodulatory Action of the Endocannabinoid System under Neuroinflammation


Abstract
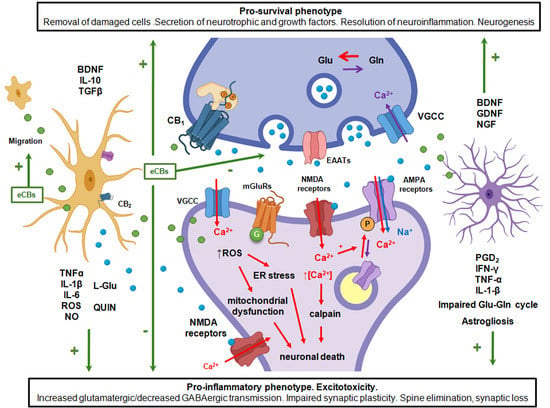
:
1. Highlights
- Retrograde endocannabinoid signaling provides a mechanism by which neurons can rapidly regulate the strength of their synaptic inputs.
- Stimulation of postsynaptic neurotransmitter receptors and sustained Ca2+ influx is a potent trigger for the production of endocannabinoids (eCBs) and their congeners.
- Neuroinflammation and alterations in endocannabinoid signaling is implicated in multiple neurological disorders.
- The activity-dependent flow of glutamate and eCBs from synapses controls microglial attraction, secretion of pro-inflammatory and pro-survival factors, and defines the synapse stability under inflammation and excitotoxicity.
- The pharmacological inhibition of eCB degradation exerts a primary effect in injured sites, where these mediators are actively produced de novo.
- The endocannabinoid system mediates communication within the tripartite synapse during the development and resolution of neuroinflammation.
2. Introduction
3. The Endocannabinoid System
3.1. Metabolism of Endocannabinoids and Related Compounds
3.2. Molecular Targets of the Endocannabinoid System
3.3. Involvement of Endocannabinoid System in Response to Neuropathology
4. Glutamate Receptor-Mediated Neurotoxicity
4.1. Glutamate as a Major Excitatory Neurotransmitter in Mammals and Potential Neurotoxin
4.2. Excitotoxicity as a Prerequisite and Consequence of Neuroinflammation and Neurodegeneration
5. The Role of Retrograde Endocannabinoid Signaling in the Tuning of Synaptic Strength
5.1. Synaptic Plasticity in Glutamatergic Synapses
5.2. Endocannabinoid-Mediated Synaptic Plasticity
6. Neuroinflammation-Induced Synaptopathy and Neurodegenerative Diseases
6.1. Microglia
6.2. Astrocytes
6.3. Cytokines Involved in Neuroinflammation
6.4. Effects of Endocannabinoids and Related Compounds on Neurodegenerative Diseases
7. The Role of the Blood–Brain Barrier Integrity in Restriction of Systemic Inflammation
7.1. Structure of the Blood–Brain Barrier
7.2. The BBB in Systemic Inflammation
7.3. The Role of the ECS in the Maintenance of the Blood Brain Barrier
7.4. Leukocyte Recruitment
8. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Basavarajappa, B.S.; Shivakumar, M.; Joshi, V.; Subbanna, S. Endocannabinoid system in neurodegenerative disorders. J. Neurochem. 2017, 142, 624–648. [Google Scholar] [CrossRef] [PubMed]
- Schurman, L.D.; Lichtman, A.H. Endocannabinoids: A Promising Impact for Traumatic Brain Injury. Front. Pharmacol. 2017, 8, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ranieri, R.; Laezza, C.; Bifulco, M.; Marasco, D.; Malfitano, A.M. Endocannabinoid System in Neurological Disorders. Recent Patents CNS Drug Discov. 2016, 10, 90–112. [Google Scholar] [CrossRef] [PubMed]
- Tsuboi, K.; Uyama, T.; Okamoto, Y.; Ueda, N. Endocannabinoids and related N-acylethanolamines: Biological activities and metabolism. Inflamm. Regen. 2018, 38, 28. [Google Scholar] [CrossRef] [PubMed]
- Bisogno, T.; Berrendero, F.; Ambrosino, G.; Cebeira, M.; Ramos, J.; Fernandez-Ruiz, J.; Di Marzo, V. Brain Regional Distribution of Endocannabinoids: Implications for Their Biosynthesis and Biological Function. Biochem. Biophys. Res. Commun. 1999, 256, 377–380. [Google Scholar] [CrossRef]
- Felder, C.C.; Nielsen, A.; Briley, E.M.; Palkovits, M.; Priller, J.; Axelrod, J.; Nguyen, D.N.; Richardson, J.M.; Riggin, R.M.; Koppel, G.A.; et al. Isolation and measurement of the endogenous cannabinoid receptor agonist, anandamide, in brain and peripheral tissues of human and rat. FEBS Lett. 1996, 393, 231–235. [Google Scholar] [CrossRef] [Green Version]
- Schmid, H.H.; Schmid, P.C.; Berdyshev, E.V. Cell signaling by endocannabinoids and their congeners: Questions of selectivity and other challenges. Chem. Phys. Lipids 2002, 121, 111–134. [Google Scholar] [CrossRef]
- Natarajan, V.; Reddy, P.; Schmid, P.; Schmid, H. On the biosynthesis and metabolism of N-acylethanolamine phospholipids in infarcted dog heart. Biochim. Biophys. Acta BBA Lipids Lipid Metab. 1981, 664, 445–448. [Google Scholar] [CrossRef]
- Gulaya, N.M.; Volkov, G.L.; Klimashevsky, V.M.; Govseeva, N.N.; Melnik, A.A. Changes in lipid composition of neuro-blastoma C1300 N18 cell during differentiation. Neuroscience 1989, 30, 153–164. [Google Scholar] [CrossRef]
- Kasatkina, L.A.; Tarasenko, A.S.; Krupko, O.O.; Kuchmerovska, T.M.; Lisakovska, O.O.; Trikash, I.O. Vitamin D deficiency induces the excitation/inhibition brain imbalance and the proinflammatory shift. Int. J. Biochem. Cell Biol. 2020, 119, 105665. [Google Scholar] [CrossRef]
- Murataeva, N.; Straiker, A.; Mackie, K. Parsing the players: 2-arachidonoylglycerol synthesis and degradation in the CNS. Br. J. Pharmacol. 2014, 171, 1379–1391. [Google Scholar] [CrossRef] [Green Version]
- Kozak, K.; Marnett, L. Oxidative metabolism of endocannabinoids. Prostaglandins Leukot. Essent. Fat. Acids 2002, 66, 211–220. [Google Scholar] [CrossRef] [PubMed]
- Rouzer, C.A.; Marnett, L.J. Non-redundant Functions of Cyclooxygenases: Oxygenation of Endocannabinoids. J. Biol. Chem. 2008, 283, 8065–8069. [Google Scholar] [CrossRef] [Green Version]
- Tsuboi, K.; Sun, Y.-X.; Okamoto, Y.; Araki, N.; Tonai, T.; Ueda, N. Molecular Characterization of N-Acylethanolamine-hydrolyzing Acid Amidase, a Novel Member of the Choloylglycine Hydrolase Family with Structural and Functional Similarity to Acid Ceramidase. J. Biol. Chem. 2005, 280, 11082–11092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iannotti, F.A.; Di Marzo, V.; Petrosino, S. Endocannabinoids and endocannabinoid-related mediators: Targets, metabolism and role in neurological disorders. Prog. Lipid Res. 2016, 62, 107–128. [Google Scholar] [CrossRef]
- Solorzano, C.; Zhu, C.; Battista, N.; Astarita, G.; Lodola, A.; Rivara, S.; Mor, M.; Russo, R.; Maccarrone, M.; Antonietti, F.; et al. Selective N-acylethanolamine-hydrolyzing acid amidase inhibition reveals a key role for endogenous palmitoylethanolamide in inflammation. Proc. Natl. Acad. Sci. USA 2009, 106, 20966–20971. [Google Scholar] [CrossRef] [Green Version]
- Urquhart, P.; Nicolaou, A.; Woodward, D. Endocannabinoids and their oxygenation by cyclo-oxygenases, lipoxygenases and other oxygenases. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2015, 1851, 366–376. [Google Scholar] [CrossRef] [PubMed]
- McDougle, D.R.; Watson, J.E.; Abdeen, A.A.; Adili, R.; Caputo, M.P.; Krapf, J.E.; Johnson, R.W.; Kilian, K.A.; Holinstat, M.; Das, A. Anti-inflammatory omega-3 endocannabinoid epoxides. Proc. Natl. Acad. Sci. USA 2017, 114, E6034–E6043. [Google Scholar] [CrossRef] [Green Version]
- Hu, S.S.-J.; Bradshaw, H.B.; Benton, V.M.; Chen, J.S.-C.; Huang, S.M.; Minassi, A.; Bisogno, T.; Masuda, K.; Tan, B.; Roskoski, R.; et al. The biosynthesis of N-arachidonoyl dopamine (NADA), a putative endocannabinoid and endovanilloid, via conjugation of arachidonic acid with dopamine. Prostaglandins Leukot. Essent. Fat. Acids 2009, 81, 291–301. [Google Scholar] [CrossRef] [Green Version]
- Bouaboula, M.; Poinot-Chazel, C.; Bourrié, B.; Canat, X.; Calandra, B.; Rinaldi-Carmona, M.; Le Fur, G.; Casellas, P. Activation of mitogen-activated protein kinases by stimulation of the central cannabinoid receptor CB1. Biochem. J. 1995, 312, 637–641. [Google Scholar] [CrossRef] [Green Version]
- Gao, F.; Zhang, L.H.; Su, T.F.; Li, L.; Zhou, R.; Peng, M.; Wu, C.H.; Yuan, X.C.; Sun, N.; Meng, X.F.; et al. Signaling Mechanism of Cannabinoid Receptor-2 Activation-Induced beta-Endorphin Release. Mol. Neurobiol. 2016, 53, 3616–3625. [Google Scholar] [CrossRef]
- Glass, M.; Felder, C.C. Concurrent Stimulation of Cannabinoid CB1 and Dopamine D2 Receptors Augments cAMP Accumulation in Striatal Neurons: Evidence for a Gs Linkage to the CB1 Receptor. J. Neurosci. 1997, 17, 5327–5333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauckner, J.E.; Hille, B.; Mackie, K. The cannabinoid agonist WIN55,212-2 increases intracellular calcium via CB1 receptor coupling to Gq/11 G proteins. Proc. Natl. Acad. Sci. USA 2005, 102, 19144–19149. [Google Scholar] [CrossRef] [Green Version]
- Navarrete, M.; Araque, A. Endocannabinoids Mediate Neuron-Astrocyte Communication. Neuron 2008, 57, 883–893. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.; Ikeda, S.R. Endocannabinoids modulate N-type calcium channels and G-protein-coupled inwardly rectifying potas-sium channels via CB1 cannabinoid receptors heterologously expressed in mammalian neurons. Mol. Pharmacol. 2004, 65, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Ibsen, M.S.; Finlay, D.B.; Patel, M.; Javitch, J.A.; Glass, M.; Grimsey, N.L. Cannabinoid CB1 and CB2 Receptor-Mediated Arrestin Translocation: Species, Subtype, and Agonist-Dependence. Front. Pharmacol. 2019, 10, 350. [Google Scholar] [CrossRef] [PubMed]
- McGuinness, D.; Malikzay, A.; Visconti, R.; Lin, K.; Bayne, M.; Monsma, F.; Lunn, C.A. Characterizing cannabinoid CB2 receptor ligands using DiscoveRx PathHunter beta-arrestin assay. J. Biomol. Screen 2009, 14, 49–58. [Google Scholar] [CrossRef] [Green Version]
- Jin, W.; Brown, S.; Roche, J.P.; Hsieh, C.; Celver, J.P.; Kovoor, A.; Chavkin, C.; Mackie, K. Distinct Domains of the CB1 Cannabinoid Receptor Mediate Desensitization and Internalization. J. Neurosci. 1999, 19, 3773–3780. [Google Scholar] [CrossRef] [Green Version]
- Van Sickle, M.D.; Duncan, M.; Kingsley, P.J.; Mouihate, A.; Urbani, P.; Mackie, K.; Stella, N.; Makriyannis, A.; Piomelli, D.; Davison, J.S.; et al. Identification and functional characterization of brainstem cannabinoid CB2 receptors. Science 2005, 310, 329–332. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.-Y.; Gao, M.; Liu, Q.-R.; Bi, G.-H.; Li, X.; Yang, H.-J.; Gardner, E.L.; Wu, J.; Xi, Z.-X. Cannabinoid CB2 receptors modulate midbrain dopamine neuronal activity and dopamine-related behavior in mice. Proc. Natl. Acad. Sci. USA 2014, 111, E5007–E5015. [Google Scholar] [CrossRef] [Green Version]
- Onaivi, E.S.; Ishiguro, H.; Gong, J.-P.; Patel, S.; Meozzi, P.A.; Myers, L.; Perchuk, A.; Mora, Z.; Tagliaferro, P.A.; Gardner, E.; et al. Brain Neuronal CB2 Cannabinoid Receptors in Drug Abuse and Depression: From Mice to Human Subjects. PLoS ONE 2008, 3, e1640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, L.K.; Devi, L.A. The Highs and Lows of Cannabinoid Receptor Expression in Disease: Mechanisms and Their Therapeutic Implications. Pharmacol. Rev. 2011, 63, 461–470. [Google Scholar] [CrossRef]
- Haspula, D.; Clark, M.A. Cannabinoid Receptors: An Update on Cell Signaling, Pathophysiological Roles and Therapeutic Opportunities in Neurological, Cardiovascular, and Inflammatory Diseases. Int. J. Mol. Sci. 2020, 21, 7693. [Google Scholar] [CrossRef] [PubMed]
- Stempel, A.V.; Stumpf, A.; Zhang, H.-Y.; Özdoğan, T.; Pannasch, U.; Theis, A.-K.; Otte, D.-M.; Wojtalla, A.; Rácz, I.; Ponomarenko, A.; et al. Cannabinoid Type 2 Receptors Mediate a Cell Type-Specific Plasticity in the Hippocampus. Neuron 2016, 90, 795–809. [Google Scholar] [CrossRef] [Green Version]
- Sharir, H.; Console-Bram, L.; Mundy, C.; Popoff, S.N.; Kapur, A.; Abood, M.E. The Endocannabinoids Anandamide and Virodhamine Modulate the Activity of the Candidate Cannabinoid Receptor GPRJ. Neuroimmune Pharmacol. 2012, 7, 856–865. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Irving, A.; Abdulrazzaq, G.; Chan, S.L.; Penman, J.; Harvey, J.; Alexander, S.P. Cannabinoid Receptor-Related Orphan G Protein-Coupled Receptors. Rapid Acting Antidepress. 2017, 80, 223–247. [Google Scholar] [CrossRef]
- Brown, R.C.; Cascio, C.; Papadopoulos, V. Pathways of neurosteroid biosynthesis in cell lines from human brain: Regulation of dehydroepiandrosterone formation by oxidative stress and beta-amyloid peptide. J. Neurochem. 2000, 74, 847–859. [Google Scholar] [CrossRef]
- O’Sullivan, S.E. An update on PPAR activation by cannabinoids. Br. J. Pharmacol. 2016, 173, 1899–1910. [Google Scholar] [CrossRef] [Green Version]
- D’Agostino, G.; La Rana, G.; Russo, R.; Sasso, O.; Iacono, A.; Esposito, E.; Mattace Raso, G.; Cuzzocrea, S.; Loverme, J.; Piomelli, D.; et al. Central administration of palmitoylethanolamide reduces hyperalgesia in mice via inhibition of NF-kappaB nuclear signalling in dorsal root ganglia. Eur. J. Pharmacol. 2009, 613, 54–59. [Google Scholar] [CrossRef]
- Beggiato, S.; Tomasini, M.C.; Cassano, T.; Ferraro, L. Chronic Oral Palmitoylethanolamide Administration Rescues Cognitive Deficit and Reduces Neuroinflammation, Oxidative Stress, and Glutamate Levels in A Transgenic Murine Model of Alzheimer’s Disease. J. Clin. Med. 2020, 9, 428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rankin, L.; Fowler, C.J. The basal pharmacology of palmitoylethanolamide. Int. J. Mol. Sci. 2020, 21, 7942. [Google Scholar] [CrossRef] [PubMed]
- Ambrosino, P.; Soldovieri, M.V.; Russo, C.; Taglialatela, M. Activation and desensitization of TRPV1 channels in sensory neurons by the PPARalpha agonist palmitoylethanolamide. Br. J. Pharmacol. 2013, 168, 1430–1444. [Google Scholar] [CrossRef] [Green Version]
- Ryberg, E.; Larsson, N.; Sjögren, S.; Hjorth, S.; Hermansson, N.-O.; Leonova, J.; Elebring, T.; Nilsson, K.; Drmota, T.; Greasley, P.J. The orphan receptor GPR55 is a novel cannabinoid receptor. Br. J. Pharmacol. 2007, 152, 1092–1101. [Google Scholar] [CrossRef] [PubMed]
- Di Marzo, V.; Melck, D.; Orlando, P.; Bisogno, T.; Zagoory, O.; Bifulco, M.; Vogel, Z.; De Petrocellis, L. Palmitoylethanolamide inhibits the expression of fatty acid amide hydrolase and enhances the anti-proliferative effect of anandamide in human breast cancer cells. Biochem. J. 2001, 358, 249–255. [Google Scholar] [CrossRef]
- TuTunchi, H.; Saghafi-Asl, M.; Ostadrahimi, A. A systematic review of the effects of oleoylethanolamide, a high-affinity endogenous ligand of PPAR-α, on the management and prevention of obesity. Clin. Exp. Pharmacol. Physiol. 2020, 47, 543–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- González-Aparicio, R.; Moratalla, R. Oleoylethanolamide reduces L-DOPA-induced dyskinesia via TRPV1 receptor in a mouse model of Parkinson´s disease. Neurobiol. Dis. 2014, 62, 416–425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasatkina, L.A.; Heinemann, A.; Hudz, Y.A.; Thomas, D.; Sturm, E.M. Stearoylethanolamide interferes with retrograde endocannabinoid signalling and supports the blood-brain barrier integrity under acute systemic inflammation. Biochem. Pharmacol. 2020, 174, 113783. [Google Scholar] [CrossRef]
- Maccarrone, M.; Cartoni, A.; Parolaro, D.; Margonelli, A.; Massi, P.; Bari, M.; Battista, N.; Finazzi-Agro, A. Cannabimimetic activity, binding, and degradation of stearoylethanolamide within the mouse central nervous system. Mol. Cell Neurosci. 2002, 21, 126–140. [Google Scholar] [CrossRef] [PubMed]
- Artmann, A.; Petersen, G.; Hellgren, L.I.; Boberg, J.; Skonberg, C.; Nellemann, C.; Hansen, S.H.; Hansen, H.S. Influence of dietary fatty acids on endocannabinoid and N-acylethanolamine levels in rat brain, liver and small intestine. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2008, 1781, 200–212. [Google Scholar] [CrossRef] [PubMed]
- Movahed, P.; Jönsson, B.A.G.; Birnir, B.; Wingstrand, J.A.; Jørgensen, T.D.; Ermund, A.; Sterner, O.; Zygmunt, P.M.; Högestätt, E.D. Endogenous Unsaturated C18 N-Acylethanolamines Are Vanilloid Receptor (TRPV1) Agonists. J. Biol. Chem. 2005, 280, 38496–38504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garg, P.; Duncan, R.S.; Kaja, S.; Zabaneh, A.; Chapman, K.D.; Koulen, P. Lauroylethanolamide and linoleoylethanolamide improve functional outcome in a rodent model for stroke. Neurosci. Lett. 2011, 492, 134–138. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.-Y.; Spector, A.A.; Xiong, Z.-M. A synaptogenic amide N-docosahexaenoylethanolamide promotes hippocampal development. Prostaglandins Other Lipid Mediat. 2011, 96, 114–120. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.-W.; Huang, B.X.; Kwon, H.; Rashid, A.; Kharebava, G.; Desai, A.; Patnaik, S.; Marugan, J.; Kim, H.-Y. Orphan GPR110 (ADGRF1) targeted by N-docosahexaenoylethanolamine in development of neurons and cognitive function. Nat. Commun. 2016, 7, 13123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, T.; Chen, H.; Kim, H.-Y. GPR110 (ADGRF1) mediates anti-inflammatory effects of N-docosahexaenoylethanolamine. J. Neuroinflammation 2019, 16, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Berman, P.; Sulimani, L.; Gelfand, A.; Amsalem, K.; Lewitus, G.M.; Meiri, D. Cannabinoidomics—An analytical approach to understand the effect of medical Cannabis treatment on the endocannabinoid metabolome. Talanta 2020, 219, 121336. [Google Scholar] [CrossRef] [PubMed]
- Panikashvili, D.; Simeonidou, C.; Ben-Shabat, S.; Hanus, L.; Breuer, A.; Mechoulam, R.; Shohami, E. An endogenous canna-binoid (2-AG) is neuroprotective after brain injury. Nature 2001, 413, 527–531. [Google Scholar] [CrossRef] [PubMed]
- Guindon, J.; Desroches, J.; Beaulieu, P. The antinociceptive effects of intraplantar injections of 2-arachidonoyl glycerol are mediated by cannabinoid CB2 receptors. Br. J. Pharmacol. 2007, 150, 693–701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hohmann, A.G.; Suplita, R.L.; Bolton, N.M.; Neely, M.H.; Fegley, D.; Mangieri, R.; Krey, J.F.; Walker, J.M.; Holmes, P.V.; Crystal, J.D.; et al. An endocannabinoid mechanism for stress-induced analgesia. Nat. Cell Biol. 2005, 435, 1108–1112. [Google Scholar] [CrossRef]
- Centonze, D.; Bari, M.; Rossi, S.; Prosperetti, C.; Furlan, R.; Fezza, F.; De Chiara, V.; Battistini, L.; Bernardi, G.; Bernardini, S.; et al. The endocannabinoid system is dysregulated in multiple sclerosis and in experimental autoimmune encephalomyelitis. Brain 2007, 130, 2543–2553. [Google Scholar] [CrossRef] [Green Version]
- Muthian, S.; Rademacher, D.; Roelke, C.; Gross, G.; Hillard, C. Anandamide content is increased and CB1 cannabinoid receptor blockade is protective during transient, focal cerebral ischemia. Neuroscience 2004, 129, 743–750. [Google Scholar] [CrossRef]
- Lawton, S.K.; Xu, F.; Tran, A.; Wong, E.; Prakash, A.; Schumacher, M.; Hellman, J.; Wilhelmsen, K. N-Arachidonoyl Do-pamine Modulates Acute Systemic Inflammation via Nonhematopoietic TRPV1. J. Immunol. 2017, 199, 1465–1475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bisogno, T.; Martire, A.; Petrosino, S.; Popoli, P.; Di Marzo, V. Symptom-related changes of endocannabinoid and pal-mitoylethanolamide levels in brain areas of R6/2 mice, a transgenic model of Huntington’s disease. Neurochem. Int. 2008, 52, 307–313. [Google Scholar] [CrossRef] [PubMed]
- De Filippis, D.; Luongo, L.; Cipriano, M.; Palazzo, E.; Cinelli, M.P.; de Novellis, V.; Maione, S.; Iuvone, T. Palmitoylethano-lamide reduces granuloma-induced hyperalgesia by modulation of mast cell activation in rats. Mol. Pain 2011, 7, 3. [Google Scholar] [CrossRef] [Green Version]
- Costa, B.; Comelli, F.; Bettoni, I.; Colleoni, M.; Giagnoni, G. The endogenous fatty acid amide, palmitoylethanolamide, has anti-allodynic and anti-hyperalgesic effects in a murine model of neuropathic pain: Involvement of CB(1), TRPV1 and PPARgamma receptors and neurotrophic factors. Pain 2008, 139, 541–550. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, A.; Crupi, R.; Impellizzeri, D.; Campolo, M.; Marino, A.; Esposito, E.; Cuzzocrea, S. Administration of pal-mitoylethanolamide (PEA) protects the neurovascular unit and reduces secondary injury after traumatic brain injury in mice. Brain Behav. Immun. 2012, 26, 1310–1321. [Google Scholar] [CrossRef] [PubMed]
- Zerangue, N.; Kavanaugh, M.P. Flux coupling in a neuronal glutamate transporter. Nat. Cell Biol. 1996, 383, 634–637. [Google Scholar] [CrossRef]
- Rossi, D.J.; Oshima, T.; Attwell, D. Glutamate release in severe brain ischaemia is mainly by reversed uptake. Nat. Cell Biol. 2000, 403, 316–321. [Google Scholar] [CrossRef]
- Riveros, N.; Fiedler, J.; Lagos, N.; Munoz, C.; Orrego, F. Glutamate in rat brain cortex synaptic vesicles: Influence of the vesicle isolation procedure. Brain Res. 1986, 386, 405–408. [Google Scholar] [CrossRef]
- Clements, J.D.; Lester, R.A.; Tong, G.; Jahr, C.E.; Westbrook, G.L. The time course of glutamate in the synaptic cleft. Science 1992, 258, 1498–1501. [Google Scholar] [CrossRef]
- Zhou, X.; Hollern, D.; Liao, J.; Andrechek, E.; Wang, H. NMDA receptor-mediated excitotoxicity depends on the coactivation of synaptic and extrasynaptic receptors. Cell Death Dis. 2013, 4, e560. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, J.W.; Kotermanski, S.E. Mechanism of action of memantine. Curr. Opin. Pharmacol. 2006, 6, 61–67. [Google Scholar] [CrossRef]
- Xia, P.; Chen, H.-S.V.; Zhang, D.; Lipton, S.A. Memantine Preferentially Blocks Extrasynaptic over Synaptic NMDA Receptor Currents in Hippocampal Autapses. J. Neurosci. 2010, 30, 11246–11250. [Google Scholar] [CrossRef] [Green Version]
- Doyle, S.; Hansen, D.B.; Vella, J.; Bond, P.; Harper, G.; Zammit, C.; Valentino, M.; Fern, R. Vesicular glutamate release from central axons contributes to myelin damage. Nat. Commun. 2018, 9, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Brickley, S.G.; Misra, C.; Mok, M.H.S.; Mishina, M.; Cull-Candy, S.G. NR2B and NR2D Subunits Coassemble in Cerebellar Golgi Cells to Form a Distinct NMDA Receptor Subtype Restricted to Extrasynaptic Sites. J. Neurosci. 2003, 23, 4958–4966. [Google Scholar] [CrossRef] [PubMed]
- Walter, L.; Franklin, A.; Witting, A.; Moller, T.; Stella, N. Astrocytes in culture produce anandamide and other acylethano-lamides. J. Biol. Chem. 2002, 277, 20869–20876. [Google Scholar] [CrossRef] [Green Version]
- Walter, L.; Franklin, A.; Witting, A.; Wade, C.; Xie, Y.; Kunos, G.; Mackie, K.; Stella, N. Nonpsychotropic Cannabinoid Receptors Regulate Microglial Cell Migration. J. Neurosci. 2003, 23, 1398–1405. [Google Scholar] [CrossRef] [Green Version]
- Witting, A.; Walter, L.; Wacker, J.; Moller, T.; Stella, N. P2X7 receptors control 2-arachidonoylglycerol production by microglial cells. Proc. Natl. Acad. Sci. USA 2004, 101, 3214–3219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stella, N.; Piomelli, D. Receptor-dependent formation of endogenous cannabinoids in cortical neurons. Eur. J. Pharmacol. 2001, 425, 189–196. [Google Scholar] [CrossRef] [Green Version]
- Franklin, A.; Parmentier-Batteur, S.; Walter, L.; Greenberg, D.A.; Stella, N. Palmitoylethanolamide increases after focal cerebral ischemia and potentiates microglial cell motility. J. Neurosci. 2003, 23, 7767–7775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Franklin, A.; Stella, N. Arachidonylcyclopropylamide increases microglial cell migration through cannabinoid CB2 and ab-normal-cannabidiol-sensitive receptors. Eur. J. Pharmacol. 2003, 474, 195–198. [Google Scholar] [CrossRef]
- Kreutz, S.; Koch, M.; Böttger, C.; Ghadban, C.; Korf, H.-W.; Dehghani, F. 2-Arachidonoylglycerol elicits neuroprotective effects on excitotoxically lesioned dentate gyrus granule cells via abnormal-cannabidiol-sensitive receptors on microglial cells. Glia 2009, 57, 286–294. [Google Scholar] [CrossRef] [PubMed]
- Wake, H.; Moorhouse, A.J.; Jinno, S.; Kohsaka, S.; Nabekura, J. Resting Microglia Directly Monitor the Functional State of Synapses In Vivo and Determine the Fate of Ischemic Terminals. J. Neurosci. 2009, 29, 3974. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weinhard, L.; di Bartolomei, G.; Bolasco, G.; Machado, P.; Schieber, N.L.; Neniskyte, U.; Exiga, M.; Vadisiute, A.; Raggioli, A.; Schertel, A.; et al. Microglia remodel synapses by presynaptic trogocytosis and spine head filopodia in-duction. Nat. Commun. 2018, 9, 1228. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joly, E.; Hudrisier, D. What is trogocytosis and what is its purpose? Nat. Immunol. 2003, 4, 815. [Google Scholar] [CrossRef]
- Jafari, M.; Schumacher, A.-M.; Snaidero, N.; Gavilanes, E.M.U.; Neziraj, T.; Kocsis-Jutka, V.; Engels, D.; Jürgens, T.; Wagner, I.; Weidinger, J.D.F.; et al. Phagocyte-mediated synapse removal in cortical neuroinflammation is promoted by local calcium accumulation. Nat. Neurosci. 2021, 24, 355–367. [Google Scholar] [CrossRef]
- Combes, V.; Guillemin, G.J.; Chan-Ling, T.; Hunt, N.H.; Grau, G.E. The crossroads of neuroinflammation in infectious diseases: Endothelial cells and astrocytes. Trends Parasitol. 2012, 28, 311–319. [Google Scholar] [CrossRef]
- Hume, R.; Dingledine, R.; Heinemann, S.F. Identification of a site in glutamate receptor subunits that controls calcium permeability. Science 1991, 253, 1028–1031. [Google Scholar] [CrossRef]
- Malenka, R.C.; Kauer, J.A.; Perkel, D.J.; Mauk, M.D.; Kelly, P.T.; Nicoll, R.A.; Waxham, M.N. An essential role for postsynaptic calmodulin and protein kinase activity in long-term potentiation. Nat. Cell Biol. 1989, 340, 554–557. [Google Scholar] [CrossRef] [PubMed]
- Malinow, R.; Schulman, H.; Tsien, R.W. Inhibition of postsynaptic PKC or CaMKII blocks induction but not expression of LTP. Science 1989, 245, 862–866. [Google Scholar] [CrossRef]
- Murakoshi, H.; Wang, H.; Yasuda, R. Local, persistent activation of Rho GTPases during plasticity of single dendritic spines. Nat. Cell Biol. 2011, 472, 100–104. [Google Scholar] [CrossRef]
- Shi, S.H.; Hayashi, Y.; Petralia, R.S.; Zaman, S.H.; Wenthold, R.J.; Svoboda, K.; Malinow, R. Rapid spine delivery and redistribution of AMPA receptors after synaptic NMDA receptor activation. Science 1999, 284, 1811–1816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, M.; Penick, E.C.; Edwards, J.G.; Kauer, J.A.; Ehlers, M.D. Recycling Endosomes Supply AMPA Receptors for LTP. Science 2004, 305, 1972–1975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Derkach, V.; Barria, A.; Soderling, T.R. Ca2+/calmodulin-kinase II enhances channel conductance of al-pha-amino-3-hydroxy-5-methyl-4-isoxazolepropionate type glutamate receptors. Proc. Natl. Acad. Sci. USA 1999, 96, 3269–3274. [Google Scholar] [CrossRef] [Green Version]
- Fortin, D.A.; Davare, M.A.; Srivastava, T.; Brady, J.D.; Nygaard, S.; Derkach, V.A.; Soderling, T.R. Long-term potentiation-dependent spine enlargement requires synaptic Ca2+-permeable AMPA receptors recruited by CaM-kinase I. J. Neurosci. 2010, 30, 11565–11575. [Google Scholar] [CrossRef] [Green Version]
- Mulkey, R.M.; Endo, S.; Shenolikar, S.; Malenka, R.C. Involvement of a calcineurin/ inhibitor-1 phosphatase cascade in hippocampal long-term depression. Nat. Cell Biol. 1994, 369, 486–488. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.-K.; Kameyama, K.; Huganir, R.L.; Bear, M.F. NMDA Induces Long-Term Synaptic Depression and Dephosphorylation of the GluR1 Subunit of AMPA Receptors in Hippocampus. Neuron 1998, 21, 1151–1162. [Google Scholar] [CrossRef] [Green Version]
- Ehlers, M.D. Reinsertion or Degradation of AMPA Receptors Determined by Activity-Dependent Endocytic Sorting. Neuron 2000, 28, 511–525. [Google Scholar] [CrossRef] [Green Version]
- Mauna, J.C.; Miyamae, T.; Pulli, B.; Thiels, E. Protein phosphatases 1 and 2A are both required for long-term depression and associated dephosphorylation of cAMP response element binding protein in hippocampal area CA1 in vivo. Hippocampus 2011, 21, 1093–1104. [Google Scholar] [CrossRef] [Green Version]
- Rick, J.T.; Milgram, N.W. Frequency dependence of long-term potentiation and depression in the dentate gyrus of the freely moving rat. Hippocampus 1996, 6, 118–124. [Google Scholar] [CrossRef]
- Cui, Y.; Paillé, V.; Xu, H.; Genet, S.; Delord, B.; Fino, E.; Berry, H.; Venance, L. Endocannabinoids mediate bidirectional striatal spike-timing-dependent plasticity. J. Physiol. 2015, 593, 2833–2849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varma, N.; Carlson, G.C.; Ledent, C.; Alger, B.E. Metabotropic Glutamate Receptors Drive the Endocannabinoid System in Hippocampus. J. Neurosci. 2001, 21, RC188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasatkina, L.A.; Gumenyuk, V.P.; Sturm, E.M.; Heinemann, A.; Bernas, T.; Trikash, I.O. Modulation of neurosecretion and approaches for its multistep analysis. Biochim. Biophys. Acta BBA Gen. Subj. 2018, 1862, 2701–2713. [Google Scholar] [CrossRef]
- Singla, S.; Kreitzer, A.C.; Malenka, R.C. Mechanisms for Synapse Specificity during Striatal Long-Term Depression. J. Neurosci. 2007, 27, 5260–5264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sjöström, P.J.; Turrigiano, G.G.; Nelson, S.B. Neocortical LTD via Coincident Activation of Presynaptic NMDA and Can-nabinoid Receptors. Neuron 2003, 39, 641–654. [Google Scholar] [CrossRef] [Green Version]
- Straiker, A.; Mackie, K. Depolarization-induced suppression of excitation in murine autaptic hippocampal neurones. J. Physiol. 2005, 569, 501–517. [Google Scholar] [CrossRef]
- Colmers, P.L.; Bains, J.S. Presynaptic mGluRs Control the Duration of Endocannabinoid-Mediated DSI. J. Neurosci. 2018, 38, 10444–10453. [Google Scholar] [CrossRef] [Green Version]
- Maejima, T.; Hashimoto, K.; Yoshida, T.; Aiba, A.; Kano, M. Presynaptic Inhibition Caused by Retrograde Signal from Metabotropic Glutamate to Cannabinoid Receptors. Neuron 2001, 31, 463–475. [Google Scholar] [CrossRef] [Green Version]
- Chávez, A.E.; Chiu, C.Q.; Castillo, P.E. TRPV1 activation by endogenous anandamide triggers postsynaptic long-term depression in dentate gyrus. Nat. Neurosci. 2010, 13, 1511–1518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grueter, B.A.; Brasnjo, G.; Malenka, R.C. Postsynaptic TRPV1 triggers cell type–specific long-term depression in the nucleus accumbens. Nat. Neurosci. 2010, 13, 1519–1525. [Google Scholar] [CrossRef]
- Gibson, H.E.; Edwards, J.G.; Page, R.S.; Van Hook, M.J.; Kauer, J.A. TRPV1 Channels Mediate Long-Term Depression at Synapses on Hippocampal Interneurons. Neuron 2008, 57, 746–759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puente, N.; Cui, Y.; Lassalle, O.; Lafourcade, M.; Georges, F.; Venance, L.; Grandes, P.; Manzoni, O.J. Polymodal activation of the endocannabinoid system in the extended amygdala. Nat. Neurosci. 2011, 14, 1542–1547. [Google Scholar] [CrossRef] [PubMed]
- Mechoulam, R.; Fride, E.; Hanu, L.; Sheskin, T.; Bisogno, T.; Di Marzo, V.; Bayewitch, M.; Vogel, Z. Anandamide may mediate sleep induction. Nat. Cell Biol. 1997, 389, 25–26. [Google Scholar] [CrossRef]
- Nayak, D.; Roth, T.L.; McGavern, D.B. Microglia Development and Function. Annu. Rev. Immunol. 2014, 32, 367–402. [Google Scholar] [CrossRef] [Green Version]
- Ling, E.; Ng, Y.; Wu, C.; Kaur, C. Microglia: Its development and role as a neuropathology sensor. Progress Brain Res. 2001, 132, 61–79. [Google Scholar] [CrossRef]
- Colton, C.A.; Gilbert, D.L. Production of superoxide anions by a CNS macrophage, the microglia. FEBS Lett. 1987, 223, 284–288. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Rochford, C.D.; Neumann, H. Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells-2. J. Exp. Med. 2005, 201, 647–657. [Google Scholar] [CrossRef] [Green Version]
- Levi, G.; Minghetti, L.; Aloisi, F. Regulation of prostanoid synthesis in microglial cells and effects of prostaglandin E2 on microglial functions. Biochimie 1998, 80, 899–904. [Google Scholar] [CrossRef]
- Nakamura, Y. Regulating Factors for Microglial Activation. Biol. Pharm. Bull. 2002, 25, 945–953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, H.; Lapointe, B.M.; Clark, S.R.; Zbytnuik, L.; Kubes, P. A Requirement for Microglial TLR4 in Leukocyte Recruitment into Brain in Response to Lipopolysaccharide. J. Immunol. 2006, 177, 8103–8110. [Google Scholar] [CrossRef] [Green Version]
- Russo, M.V.; McGavern, D.B. Immune Surveillance of the CNS following Infection and Injury. Trends Immunol. 2015, 36, 637–650. [Google Scholar] [CrossRef] [Green Version]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.-B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Banati, R.B.; Gehrmann, J.; Schubert, P.; Kreutzberg, G.W. Cytotoxicity of microglia. Glia 1993, 7, 111–118. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Le, W. Differential Roles of M1 and M2 Microglia in Neurodegenerative Diseases. Mol. Neurobiol. 2016, 53, 1181–1194. [Google Scholar] [CrossRef]
- Block, M.L.; Zecca, L.; Hong, J.-S. Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nat. Rev. Neurosci. 2007, 8, 57–69. [Google Scholar] [CrossRef]
- Banati, R.B. Visualising microglial activation in vivo. Glia 2002, 40, 206–217. [Google Scholar] [CrossRef] [PubMed]
- Banati, R.B. Neuropathological imaging: In vivo detection of glial activation as a measure of disease and adaptive change in the brain. Br. Med. Bull. 2003, 65, 121–131. [Google Scholar] [CrossRef] [Green Version]
- Lehnardt, S.; Massillon, L.; Follett, P.; Jensen, F.E.; Ratan, R.; Rosenberg, P.; Volpe, J.J.; Vartanian, T. Activation of innate immunity in the CNS triggers neurodegeneration through a Toll-like receptor 4-dependent pathway. Proc. Natl. Acad. Sci. USA 2003, 100, 8514–8519. [Google Scholar] [CrossRef] [Green Version]
- Heneka, M.T.; Sastre, M.; Dumitrescu-Ozimek, L.; Dewachter, I.; Walter, J.; Klockgether, T.; Van Leuven, F. Focal glial acti-vation coincides with increased BACE1 activation and precedes amyloid plaque deposition in APP[V717I] transgenic mice. J. Neuroinflamm. 2005, 2, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, M.; Jin, J.; Lim, J.E.; Kou, J.; Pattanayak, A.; Rehman, J.A.; Kim, H.D.; Tahara, K.; Lalonde, R.; Fukuchi, K. TLR4 mutation reduces microglial activation, increases Abeta deposits and exacerbates cognitive deficits in a mouse model of Alzheimer’s disease. J. Neuroinflamm. 2011, 8, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caso, J.R.; Pradillo, J.M.; Hurtado, O.; Lorenzo, P.; Moro, M.A.; Lizasoain, I. Toll-Like Receptor 4 Is Involved in Brain Damage and Inflammation After Experimental Stroke. Circulation 2007, 115, 1599–1608. [Google Scholar] [CrossRef]
- Maresz, K.; Carrier, E.J.; Ponomarev, E.D.; Hillard, C.J.; Dittel, B.N. Modulation of the cannabinoid CB2 receptor in mi-croglial cells in response to inflammatory stimuli. J. Neurochem. 2005, 95, 437–445. [Google Scholar] [CrossRef]
- Hohmann, U.; Pelzer, M.; Kleine, J.; Hohmann, T.; Ghadban, C.; Dehghani, F. Opposite Effects of Neuroprotective Cannabinoids, Palmitoylethanolamide, and 2-Arachidonoylglycerol on Function and Morphology of Microglia. Front. Neurosci. 2019, 13, 1180. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Jia, J.; Liu, X.; Bai, F.; Wang, Q.; Xiong, L. Activation of murine microglial N9 cells is attenuated through cannabinoid receptor CB2 signaling. Biochem. Biophys. Res. Commun. 2015, 458, 92–97. [Google Scholar] [CrossRef] [PubMed]
- Carrier, E.J.; Kearn, C.S.; Barkmeier, A.J.; Breese, N.M.; Yang, W.; Nithipatikom, K.; Pfister, S.L.; Campbell, W.B.; Hillard, C.J. Cultured Rat Microglial Cells Synthesize the Endocannabinoid 2-Arachidonylglycerol, Which Increases Proliferation via a CB2Receptor-Dependent Mechanism. Mol. Pharmacol. 2004, 65, 999–1007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Araque, A.; Parpura, V.; Sanzgiri, R.P.; Haydon, P.G. Tripartite synapses: Glia, the unacknowledged partner. Trends Neurosci. 1999, 22, 208–215. [Google Scholar] [CrossRef]
- Han, J.; Kesner, P.; Metna-Laurent, M.; Duan, T.; Xu, L.; Georges, F.; Koehl, M.; Abrous, N.; Mendizabal-Zubiaga, J.; Grandes, P.; et al. Acute Cannabinoids Impair Working Memory through Astroglial CB1 Receptor Modulation of Hippocampal LTD. Cell 2012, 148, 1039–1050. [Google Scholar] [CrossRef] [Green Version]
- Rossi, D.; Volterra, A. Astrocytic dysfunction: Insights on the role in neurodegeneration. Brain Res. Bull. 2009, 80, 224–232. [Google Scholar] [CrossRef]
- Liddelow, S.A.; Barres, B.A. Reactive Astrocytes: Production, Function, and Therapeutic Potential. Immunity 2017, 46, 957–967. [Google Scholar] [CrossRef] [Green Version]
- Lian, H.; Yang, L.; Cole, A.; Sun, L.; Chiang, A.C.; Fowler, S.W.; Shim, D.J.; Rodriguez-Rivera, J.; Taglialatela, G.; Jankowsky, J.L.; et al. NFkappaB-activated astroglial release of complement C3 compromises neuronal morphology and function associated with Alzheimer’s disease. Neuron 2015, 85, 101–115. [Google Scholar] [CrossRef] [Green Version]
- Phatnani, H.P.; Guarnieri, P.; Friedman, B.A.; Carrasco, M.A.; Muratet, M.; O’Keeffe, S.; Nwakeze, C.; Behn, F.P.; Newberry, K.M.; Meadows, S.K.; et al. Intricate interplay between astrocytes and motor neurons in ALS. Proc. Natl. Acad. Sci. USA 2013, 110, E756–E765. [Google Scholar] [CrossRef] [Green Version]
- Aebischer, J.; Cassina, P.; Otsmane, B.; Moumen, A.; Seilhean, D.; Meininger, V.; Barbeito, L.; Pettmann, B.; Raoul, C. IFNgamma triggers a LIGHT-dependent selective death of motoneurons contributing to the non-cell-autonomous effects of mutant SOD1. Cell Death Differ. 2011, 18, 754–768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.-S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Okada, S.; Nakamura, M.; Katoh, H.; Miyao, T.; Shimazaki, T.; Ishii, K.; Yamane, J.; Yoshimura, A.; Iwamoto, Y.; Toyama, Y.; et al. Conditional ablation of Stat3 or Socs3 discloses a dual role for reactive astrocytes after spinal cord injury. Nat. Med. 2006, 12, 829–834. [Google Scholar] [CrossRef]
- Christopherson, K.S.; Ullian, E.M.; Stokes, C.C.A.; Mullowney, C.E.; Hell, J.W.; Agah, A.; Lawler, J.; Mosher, D.F.; Bornstein, P.; Barres, B.A. Thrombospondins Are Astrocyte-Secreted Proteins that Promote CNS Synaptogenesis. Cell 2005, 120, 421–433. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Lin, F.; Wang, J.; Wu, J.; Han, R.; Zhu, L.; Zhang, G.; DiFiglia, M.; Qin, Z. Truncated N-terminal huntingtin fragment with expanded-polyglutamine (htt552-100Q) suppresses brain-derived neurotrophic factor transcription in astrocytes. Acta Biochim. Biophys. Sin. 2012, 44, 249–258. [Google Scholar] [CrossRef] [Green Version]
- Arregui Jorge, L.; Benítez, J.A.; Razgado Paula, L.F.; Vergara, P.; Segovia, J. Adenoviral Astrocyte-Specific Expression of BDNF in the Striata of Mice Transgenic for Huntington’s Disease Delays the Onset of the Motor Phenotype. Cell. Mol. Neurobiol. 2011, 31, 1229–1243. [Google Scholar] [CrossRef] [PubMed]
- Walter, L.; Dinh, T.; Stella, N. ATP induces a rapid and pronounced increase in 2-arachidonoylglycerol production by astro-cytes, a response limited by monoacylglycerol lipase. J. Neurosci. 2004, 24, 8068–8074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, L.; Stella, N. Endothelin-1 increases 2-arachidonoyl glycerol (2-AG) production in astrocytes. Glia 2003, 44, 85–90. [Google Scholar] [CrossRef]
- Hegyi, Z.; Olah, T.; Koszeghy, A.; Piscitelli, F.; Hollo, K.; Pal, B.; Csernoch, L.; Di Marzo, V.; Antal, M. CB1 receptor activation induces intracellular Ca(2+) mobilization and 2-arachidonoylglycerol release in rodent spinal cord astrocytes. Sci. Rep. 2018, 8, 10562. [Google Scholar] [CrossRef] [PubMed]
- Del Pulgar, T.G.; de Ceballos, M.L.; Guzmán, M.; Velasco, G. Cannabinoids Protect Astrocytes from Ceramide-induced Apoptosis through the Phosphatidylinositol 3-Kinase/Protein Kinase B Pathway. J. Biol. Chem. 2002, 277, 36527–36533. [Google Scholar] [CrossRef] [Green Version]
- Carracedo, A.; Geelen, M.J.H.; Diez, M.; Hanada, K.; Guzman, M.; Velasco, G. Ceramide sensitizes astrocytes to oxidative stress: Protective role of cannabinoids. Biochem. J. 2004, 380, 435–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Molina-Holgado, F.; Molina-Holgado, E.; Guaza, C.; Rothwell, N.J. Role of CB1 and CB2 receptors in the inhibitory effects of cannabinoids on lipopolysaccharide-induced nitric oxide release in astrocyte cultures. J. Neurosci. Res. 2002, 67, 829–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Zhang, H.; Geng, B.; Xie, Q.; Li, W.; Deng, Y.; Shi, W.; Pan, Y.; Kang, X.; Wang, J. 2-arachidonyl glycerol modulates astrocytic glutamine synthetase via p38 and ERK1/2 pathways. J. Neuroinflamm. 2018, 15, 220. [Google Scholar] [CrossRef]
- Feliu, A.; Mestre, L.; Carrillo-Salinas, F.J.; Yong, V.W.; Mecha, M.; Guaza, C. 2-arachidonoylglycerol reduces chondroitin sulphate proteoglycan production by astrocytes and enhances oligodendrocyte differentiation under inhibitory conditions. Glia 2020, 68, 1255–1273. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.; Li, M.; Li, X.; Kinden, R.; Zhou, H.; Guo, F.; Wang, Q.; Xiong, L. 2-Arachidonylglycerol Protects Primary Astrocytes Exposed to Oxygen-Glucose Deprivation Through a Blockade of NDRG2 Signaling and STAT3 Phosphorylation. Rejuvenation Res. 2016, 19, 215–222. [Google Scholar] [CrossRef]
- Scuderi, C.; Esposito, G.; Blasio, A.; Valenza, M.; Arietti, P.; Steardo, L., Jr.; Carnuccio, R.; De Filippis, D.; Petrosino, S.; Iuvone, T.; et al. Palmitoylethanolamide counteracts reactive astrogliosis induced by beta-amyloid peptide. J. Cell Mol. Med. 2011, 15, 2664–2674. [Google Scholar] [CrossRef] [Green Version]
- Gajardo-Gomez, R.; Labra, V.C.; Maturana, C.J.; Shoji, K.F.; Santibanez, C.A.; Saez, J.C.; Giaume, C.; Orellana, J.A. Cannabinoids prevent the amyloid beta-induced activation of astroglial hemichannels: A neuroprotective mechanism. Glia 2017, 65, 122–137. [Google Scholar] [CrossRef]
- Beggiato, S.; Borelli, A.C.; Ferraro, L.; Tanganelli, S.; Antonelli, T.; Tomasini, M.C. Palmitoylethanolamide Blunts Amy-loid-beta42-Induced Astrocyte Activation and Improves Neuronal Survival in Primary Mouse Cortical Astrocyte-Neuron Co-Cultures. J. Alzheimers Dis. 2018, 61, 389–399. [Google Scholar] [CrossRef]
- Beggiato, S.; Cassano, T.; Ferraro, L.; Tomasini, M.C. Astrocytic palmitoylethanolamide pre-exposure exerts neuroprotective effects in astrocyte-neuron co-cultures from a triple transgenic mouse model of Alzheimer’s disease. Life Sci. 2020, 257, 118037. [Google Scholar] [CrossRef]
- Bronzuoli, M.R.; Facchinetti, R.; Steardo, L.; Romano, A.; Stecca, C.; Passarella, S.; Cassano, T.; Scuderi, C. Palmitoylethanolamide Dampens Reactive Astrogliosis and Improves Neuronal Trophic Support in a Triple Transgenic Model of Alzheimer’s Disease: In Vitro and In Vivo Evidence. Oxid. Med. Cell. Longev. 2018, 2018, 4720532. [Google Scholar] [CrossRef] [Green Version]
- Luo, D.; Zhang, Y.; Yuan, X.; Pan, Y.; Yang, L.; Zhao, Y.; Zhuo, R.; Chen, C.; Peng, L.; Li, W.; et al. Oleoylethan-olamide inhibits glial activation via moudulating PPARalpha and promotes motor function recovery after brain ischemia. Pharmacol. Res. 2019, 141, 530–540. [Google Scholar] [CrossRef]
- Scuderi, C.; Valenza, M.; Stecca, C.; Esposito, G.; Carratù, M.R.; Steardo, L. Palmitoylethanolamide exerts neuroprotective effects in mixed neuroglial cultures and organotypic hippocampal slices via peroxisome proliferator-activated receptor-α. J. Neuroinflamm. 2012, 9, 49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.Y.; Zhou, Y.Q.; Yu, Z.P.; Zhang, X.Q.; Shi, J.; Shen, H.W. Restoring glutamate homeostasis in the nucleus accumbens via endocannabinoid-mimetic drug prevents relapse to cocaine seeking behavior in rats. Neuropsychopharmacology 2021, 46, 970–981. [Google Scholar] [CrossRef] [PubMed]
- Moreno-García, Álvaro; Bernal-Chico, A.; Colomer, T.; Rodríguez-Antigüedad, A.; Matute, C.; Mato, S. Gene Expression Analysis of Astrocyte and Microglia Endocannabinoid Signaling during Autoimmune Demyelination. Biomolecules 2020, 10, 1228. [Google Scholar] [CrossRef] [PubMed]
- Viader, A.; Blankman, J.L.; Zhong, P.; Liu, X.; Schlosburg, J.E.; Joslyn, C.M.; Liu, Q.-S.; Tomarchio, A.J.; Lichtman, A.H.; Selley, D.E.; et al. Metabolic Interplay between Astrocytes and Neurons Regulates Endocannabinoid Action. Cell Rep. 2015, 12, 798–808. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Hu, Y.; Cao, Z.; Liu, Q.; Cheng, Y. Cerebrospinal Fluid Inflammatory Cytokine Aberrations in Alzheimer’s Disease, Parkinson’s Disease and Amyotrophic Lateral Sclerosis: A Systematic Review and Meta-Analysis. Front. Immunol. 2018, 9, 2122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almutairi, M.M.A.; Gong, C.; Xu, Y.G.; Chang, Y.; Shi, H. Factors controlling permeability of the blood–brain barrier. Cell. Mol. Life Sci. 2016, 73, 57–77. [Google Scholar] [CrossRef]
- Rothwell, N.J.; Luheshi, G.N. Interleukin 1 in the brain: Biology, pathology and therapeutic target. Trends Neurosci. 2000, 23, 618–625. [Google Scholar] [CrossRef]
- Garcia, E.; Aguilar-Cevallos, J.; Silva-Garcia, R.; Ibarra, A. Cytokine and Growth Factor Activation In Vivo and In Vitro after Spinal Cord Injury. Mediat. Inflamm. 2016, 2016, 1–21. [Google Scholar] [CrossRef] [Green Version]
- Barnes, C.A. Long-term potentiation and the ageing brain. Philos. Trans. R. Soc. B Biol. Sci. 2003, 358, 765–772. [Google Scholar] [CrossRef] [Green Version]
- Zhu, H.; Wang, Z.; Yu, J.; Yang, X.; He, F.; Liu, Z.; Che, F.; Chen, X.; Ren, H.; Hong, M.; et al. Role and mechanisms of cytokines in the secondary brain injury after intracerebral hemorrhage. Prog. Neurobiol. 2019, 178, 101610. [Google Scholar] [CrossRef]
- Li, B.; Chen, M.; Guo, L.; Yun, Y.; Li, G.; Sang, N. Endocannabinoid 2-arachidonoylglycerol protects inflammatory insults from sulfur dioxide inhalation via cannabinoid receptors in the brain. J. Environ. Sci. 2017, 51, 265–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabner, G.F.; Eichmann, T.O.; Wagner, B.; Gao, Y.; Farzi, A.; Taschler, U.; Radner, F.P.; Schweiger, M.; Lass, A.; Holzer, P.; et al. Deletion of Monoglyceride Lipase in Astrocytes Attenuates Lipopolysaccharide-induced Neuroinflammation. J. Biol. Chem. 2016, 291, 913–923. [Google Scholar] [CrossRef] [Green Version]
- Wen, J.; Jones, M.; Tanaka, M.; Selvaraj, P.; Symes, A.J.; Cox, B.; Zhang, Y. WWL70 protects against chronic constriction injury-induced neuropathic pain in mice by cannabinoid receptor-independent mechanisms. J. Neuroinflamm. 2018, 15, 9. [Google Scholar] [CrossRef] [PubMed]
- Piro, J.R.; Suidan, G.L.; Quan, J.; Pi, Y.; O’Neill, S.M.; Ilardi, M.; Pozdnyakov, N.; Lanz, T.A.; Xi, H.; Bell, R.D.; et al. Inhibition of 2-AG hydrolysis differentially regulates blood brain barrier permeability after injury. J. Neuroinflamm. 2018, 15, 142. [Google Scholar] [CrossRef]
- Murphy, N.; Cowley, T.R.; Blau, C.W.; Dempsey, C.N.; Noonan, J.; Gowran, A.; Tanveer, R.; Olango, W.M.; Finn, D.P.; Campbell, V.A.; et al. The fatty acid amide hydrolase inhibitor URB597 exerts anti-inflammatory effects in hippo-campus of aged rats and restores an age-related deficit in long-term potentiation. J. Neuroinflamm. 2012, 9, 79. [Google Scholar] [CrossRef] [Green Version]
- Rivera, P.; Fernández-Arjona, M.D.M.; Silva-Peña, D.; Blanco, E.; Vargas, A.; López-Ávalos, M.D.; Grondona, J.M.; Serrano, A.; Pavón, F.J.; De Fonseca, F.R.; et al. Pharmacological blockade of fatty acid amide hydrolase (FAAH) by URB597 improves memory and changes the phenotype of hippocampal microglia despite ethanol exposure. Biochem. Pharmacol. 2018, 157, 244–257. [Google Scholar] [CrossRef] [PubMed]
- Henry, R.J.; Kerr, D.M.; Flannery, L.E.; Killilea, M.; Hughes, E.M.; Corcoran, L.; Finn, D.P.; Roche, M. Pharmacological inhibition of FAAH modulates TLR-induced neuroinflammation, but not sickness behaviour: An effect partially mediated by central TRPV1. Brain Behav. Immun. 2017, 62, 318–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paterniti, I.; Impellizzeri, D.; Crupi, R.; Morabito, R.; Campolo, M.; Esposito, E.; Cuzzocrea, S. Molecular evidence for the involvement of PPAR-delta and PPAR-gamma in anti-inflammatory and neuroprotective activities of palmitoylethanolamide after spinal cord trauma. J. Neuroinflamm. 2013, 10, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Impellizzeri, D.; Cordaro, M.; Bruschetta, G.; Siracusa, R.; Crupi, R.; Esposito, E.; Cuzzocrea, S. N-Palmitoylethanolamine-Oxazoline as a New Therapeutic Strategy to Control Neuroinflammation: Neuroprotective Effects in Experimental Models of Spinal Cord and Brain Injury. J. Neurotrauma 2017, 34, 2609–2623. [Google Scholar] [CrossRef]
- Cordaro, M.; Impellizzeri, D.; Paterniti, I.; Bruschetta, G.; Siracusa, R.; De Stefano, D.; Cuzzocrea, S.; Esposito, E. Neuropro-tective Effects of Co-UltraPEALut on Secondary Inflammatory Process and Autophagy Involved in Traumatic Brain Injury. J. Neurotrauma 2016, 33, 132–146. [Google Scholar] [CrossRef]
- Gugliandolo, E.; D’Amico, R.; Cordaro, M.; Fusco, R.; Siracusa, R.; Crupi, R.; Impellizzeri, D.; Cuzzocrea, S.; Di Paola, R. Effect of PEA-OXA on neuropathic pain and functional recovery after sciatic nerve crush. J. Neuroinflamm. 2018, 15, 1–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siracusa, R.; Paterniti, I.; Impellizzeri, D.; Cordaro, M.; Crupi, R.; Navarra, M.; Cuzzocrea, S.; Esposito, E. The Association of Palmitoylethanolamide with Luteolin Decreases Neuroinflammation and Stimulates Autophagy in Parkinson’s Disease Model. CNS Neurol. Disord. Drug Targets 2015, 14, 1350–1366. [Google Scholar] [CrossRef]
- Crupi, R.; Impellizzeri, D.; Cordaro, M.; Siracusa, R.; Casili, G.; Evangelista, M.; Cuzzocrea, S. N-palmitoylethanolamide Prevents Parkinsonian Phenotypes in Aged Mice. Mol. Neurobiol. 2018, 55, 8455–8472. [Google Scholar] [CrossRef] [PubMed]
- Orefice, N.S.; Alhouayek, M.; Carotenuto, A.; Montella, S.; Barbato, F.; Comelli, A.; Calignano, A.; Muccioli, G.G.; Orefice, G. Oral Palmitoylethanolamide Treatment Is Associated with Reduced Cutaneous Adverse Effects of Interferon-beta1a and Circulating Proinflammatory Cytokines in Relapsing-Remitting Multiple Sclerosis. Neurotherapeutics 2016, 13, 428–438. [Google Scholar] [CrossRef]
- Sayd, A.; Antón, M.; Alén, F.; Caso, J.R.; Pavón, J.; Leza, J.C.; De Fonseca, F.R.; García-Bueno, B.; Orio, L. Systemic Administration of Oleoylethanolamide Protects from Neuroinflammation and Anhedonia Induced by LPS in Rats. Int. J. Neuropsychopharmacol. 2015, 18, pyu111. [Google Scholar] [CrossRef] [Green Version]
- Sahu, P.; Mudgal, J.; Arora, D.; Kinra, M.; Mallik, S.B.; Rao, C.M.; Pai, K.S.R.; Nampoothiri, M. Cannabinoid receptor 2 activation mitigates lipopolysaccharide-induced neuroinflammation and sickness behavior in mice. Psychopharmacology 2019, 236, 1829–1838. [Google Scholar] [CrossRef]
- Li, L.; Yun, D.; Zhang, Y.; Tao, Y.; Tan, Q.; Qiao, F.; Luo, B.; Liu, Y.; Fan, R.; Xian, J.; et al. A cannabinoid receptor 2 agonist reduces blood–brain barrier damage via induction of MKP-1 after intracerebral hemorrhage in rats. Brain Res. 2018, 1697, 113–123. [Google Scholar] [CrossRef]
- Sun, L.; Dong, R.; Xu, X.; Yang, X.; Peng, M. Activation of cannabinoid receptor type 2 attenuates surgery-induced cognitive impairment in mice through anti-inflammatory activity. J. Neuroinflamm. 2017, 14, 138. [Google Scholar] [CrossRef]
- Javed, H.; Azimullah, S.; Haque, M.E.; Ojha, S.K. Cannabinoid Type 2 (CB2) Receptors Activation Protects against Oxidative Stress and Neuroinflammation Associated Dopaminergic Neurodegeneration in Rotenone Model of Parkinson’s Disease. Front. Neurosci. 2016, 10, 321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roche, M.; Diamond, M.; Kelly, J.P.; Finn, D.P. In vivo modulation of LPS-induced alterations in brain and peripheral cyto-kines and HPA axis activity by cannabinoids. J. Neuroimmunol. 2006, 181, 57–67. [Google Scholar] [CrossRef] [PubMed]
- Lou, Z.-Y.; Zhao, C.-B.; Xiao, B.-G. Immunoregulation of experimental autoimmune encephalomyelitis by the selective CB1 receptor antagonist. J. Neurosci. Res. 2012, 90, 84–95. [Google Scholar] [CrossRef]
- Chen, Y.; Luo, X.; Liu, S.; Shen, Y. Neuroprotective effect of cannabinoid receptor 1 antagonist in the MNU-induced retinal degeneration model. Exp. Eye Res. 2018, 167, 145–151. [Google Scholar] [CrossRef]
- Reichenbach, Z.W.; Li, H.; Ward, S.J.; Tuma, R.F. The CB1 antagonist, SR141716A, is protective in permanent photo-thrombotic cerebral ischemia. Neurosci. Lett. 2016, 630, 9–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmes, C.; Cunningham, C.; Zotova, E.; Woolford, J.; Dean, C.; Kerr, S.; Culliford, D.; Perry, V.H. Systemic inflammation and disease progression in Alzheimer disease. Neurology 2009, 73, 768–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez-Oroz, M.C.; Jahanshahi, M.; Krack, P.; Litvan, I.; Macias, R.; Bezard, E.; Obeso, J.A. Initial clinical manifestations of Parkinson’s disease: Features and pathophysiological mechanisms. Lancet Neurol. 2009, 8, 1128–1139. [Google Scholar] [CrossRef] [Green Version]
- Raymond, L.A.; Andre, V.M.; Cepeda, C.; Gladding, C.M.; Milnerwood, A.J.; Levine, M.S. Pathophysiology of Huntington’s disease: Time-dependent alterations in synaptic and receptor function. Neuroscience 2011, 198, 252–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crotti, A.; Glass, C.K. The choreography of neuroinflammation in Huntington’s disease. Trends Immunol. 2015, 36, 364–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robberecht, W.; Philips, T. The changing scene of amyotrophic lateral sclerosis. Nat. Rev. Neurosci. 2013, 14, 248–264. [Google Scholar] [CrossRef] [PubMed]
- Philips, T.; Robberecht, W. Neuroinflammation in amyotrophic lateral sclerosis: Role of glial activation in motor neuron disease. Lancet Neurol. 2011, 10, 253–263. [Google Scholar] [CrossRef]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef]
- Wang, Q.; Peng, Y.; Chen, S.; Gou, X.; Hu, B.; Du, J.; Lu, Y.; Xiong, L. Pretreatment with Electroacupuncture Induces Rapid Tolerance to Focal Cerebral Ischemia Through Regulation of Endocannabinoid System. Stroke 2009, 40, 2157–2164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez-Vargas, M.; Morales-Gomez, J.; Gonzalez-Rivera, R.; Hernandez-Enriquez, C.; Perez-Arredondo, A.; Estrada-Rojo, F.; Navarro, L. Does the Neuroprotective Role of Anandamide Display Diurnal Variations? Int. J. Mol. Sci. 2013, 14, 23341–23355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boccella, S.; Marabese, I.; Iannotta, M.; Belardo, C.; Neugebauer, V.; Mazzitelli, M.; Pieretti, G.; Maione, S.; Palazzo, E. Metabotropic Glutamate Receptor 5 and 8 Modulate the Ameliorative Effect of Ultramicronized Palmitoylethanolamide on Cognitive Decline Associated with Neuropathic Pain. Int. J. Mol. Sci. 2019, 20, 1757. [Google Scholar] [CrossRef] [Green Version]
- Scuderi, C.; Bronzuoli, M.R.; Facchinetti, R.; Pace, L.; Ferraro, L.; Broad, K.D.; Serviddio, G.; Bellanti, F.; Palombelli, G.; Carpinelli, G.; et al. Ultramicronized palmitoylethanolamide rescues learning and memory impairments in a triple transgenic mouse model of Alzheimer’s disease by exerting anti-inflammatory and neuroprotective effects. Transl. Psychiatry 2018, 8, 32. [Google Scholar] [CrossRef]
- Fusco, R.; Gugliandolo, E.; Campolo, M.; Evangelista, M.; Di Paola, R.; Cuzzocrea, S. Effect of a new formulation of micronized and ultramicronized N-palmitoylethanolamine in a tibia fracture mouse model of complex regional pain syndrome. PLoS ONE 2017, 12, e0178553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schweiger, V.; Martini, A.; Bellamoli, P.; Donadello, K.; Schievano, C.; Balzo, G.D.; Sarzi-Puttini, P.; Parolini, M.; Polati, E. Ultramicronized Palmitoylethanolamide (um-PEA) as Add-on Treatment in Fibromyalgia Syndrome (FMS): Retrospective Observational Study on 407 Patients. CNS Neurol. Disord. Drug Targets 2019, 18, 326–333. [Google Scholar] [CrossRef]
- Caltagirone, C.; Cisari, C.; Schievano, C.; Di Paola, R.; Cordaro, M.; Bruschetta, G.; Esposito, E.; Cuzzocrea, S.; Stroke Study Group. Co-ultramicronized Palmitoylethanolamide/Luteolin in the Treatment of Cerebral Ischemia: From Rodent to Man. Transl. Stroke Res. 2016, 7, 54–69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aso, E.; Ferrer, I. CB2 Cannabinoid Receptor as Potential Target against Alzheimer’s Disease. Front. Neurosci. 2016, 10, 243. [Google Scholar] [CrossRef] [Green Version]
- Han, Q.W.; Yuan, Y.H.; Chen, N.H. The therapeutic role of cannabinoid receptors and its agonists or antagonists in Parkinson’s disease. Prog. Neuropsychopharmacol. Biol. Psychiatry 2020, 96, 109745. [Google Scholar] [CrossRef]
- González, S.; Scorticati, C.; García-Arencibia, M.; de Miguel, R.; Ramos, J.A.; Fernández-Ruiz, J. Effects of rimonabant, a selective cannabinoid CB1 receptor antagonist, in a rat model of Parkinson’s disease. Brain Res. 2006, 1073–1074, 209–219. [Google Scholar] [CrossRef]
- Di Marzo, V.; Hill, M.P.; Bisogno, T.; Crossman, A.R.; Brotchie, J.M. Enhanced levels of endogenous cannabinoids in the globus pallidus are associated with a reduction in movement in an animal model of Parkinson’s disease. FASEB J. 2000, 14, 1432–1438. [Google Scholar] [CrossRef]
- Mesnage, V.; Houeto, J.L.; Bonnet, A.M.; Clavier, I.; Arnulf, I.; Cattelin, F.; Le Fur, G.; Damier, P.; Welter, M.L.; Agid, Y. Neurokinin B, Neurotensin, and Cannabinoid Receptor Antagonists and Parkinson Disease. Clin. Neuropharmacol. 2004, 27, 108–110. [Google Scholar] [CrossRef] [PubMed]
- Benito, C.; Nunez, E.; Tolon, R.M.; Carrier, E.J.; Rabano, A.; Hillard, C.J.; Romero, J. Cannabinoid CB2 receptors and fatty acid amide hydrolase are selectively overexpressed in neuritic plaque-associated glia in Alzheimer’s disease brains. J. Neurosci. 2003, 23, 11136–11141. [Google Scholar] [CrossRef] [Green Version]
- Núñez, E.; Benito, C.; Pazos, M.R.; Barbachano, A.; Fajardo, O.; González, S.; Tolón, R.M.; Romero, J. Cannabinoid CB2receptors are expressed by perivascular microglial cells in the human brain: An immunohistochemical study. Synapse 2004, 53, 208–213. [Google Scholar] [CrossRef]
- Ren, S.-Y.; Wang, Z.-Z.; Zhang, Y.; Chen, N.-H. Potential application of endocannabinoid system agents in neuropsychiatric and neurodegenerative diseases—focusing on FAAH/MAGL inhibitors. Acta Pharmacol. Sin. 2020, 41, 1263–1271. [Google Scholar] [CrossRef]
- Deng, H.; Li, W. Monoacylglycerol lipase inhibitors: Modulators for lipid metabolism in cancer malignancy, neurological and metabolic disorders. Acta Pharm. Sin. B 2020, 10, 582–602. [Google Scholar] [CrossRef] [PubMed]
- Montanari, S.; Scalvini, L.; Bartolini, M.; Belluti, F.; Gobbi, S.; Andrisano, V.; Ligresti, A.; Di Marzo, V.; Rivara, S.; Mor, M.; et al. Fatty Acid Amide Hydrolase (FAAH), Acetylcholinesterase (AChE), and Butyrylcholinesterase (BuChE): Networked Targets for the Development of Carbamates as Potential Anti-Alzheimer’s Disease Agents. J. Med. Chem. 2016, 59, 6387–6406. [Google Scholar] [CrossRef]
- Bottemanne, P.; Muccioli, G.G.; Alhouayek, M. N-acylethanolamine hydrolyzing acid amidase inhibition: Tools and potential therapeutic opportunities. Drug Discov. Today 2018, 23, 1520–1529. [Google Scholar] [CrossRef] [PubMed]
- Piomelli, D.; Scalvini, L.; Fotio, Y.; Lodola, A.; Spadoni, G.; Tarzia, G.; Mor, M. N-Acylethanolamine Acid Amidase (NAAA): Structure, Function, and Inhibition. J. Med. Chem. 2020, 63, 7475–7490. [Google Scholar] [CrossRef] [PubMed]
- Malamas, M.S.; Farah, S.I.; Lamani, M.; Pelekoudas, D.N.; Perry, N.T.; Rajarshi, G.; Miyabe, C.Y.; Chandrashekhar, H.; West, J.; Pavlopoulos, S.; et al. Design and synthesis of cyanamides as potent and selective N-acylethanolamine acid amidase inhibitors. Bioorg. Med. Chem. 2020, 28, 115195. [Google Scholar] [CrossRef] [PubMed]
- Kerbrat, A.; Ferré, J.-C.; Fillatre, P.; Ronzière, T.; Vannier, S.; Carsin-Nicol, B.; Lavoué, S.; Vérin, M.; Gauvrit, J.-Y.; Le Tulzo, Y.; et al. Acute Neurologic Disorder from an Inhibitor of Fatty Acid Amide Hydrolase. N. Engl. J. Med. 2016, 375, 1717–1725. [Google Scholar] [CrossRef] [Green Version]
- Pasquarelli, N.; Porazik, C.; Bayer, H.; Buck, E.; Schildknecht, S.; Weydt, P.; Witting, A.; Ferger, B. Contrasting effects of selective MAGL and FAAH inhibition on dopamine depletion and GDNF expression in a chronic MPTP mouse model of Parkinson’s disease. Neurochem. Int. 2017, 110, 14–24. [Google Scholar] [CrossRef]
- Tchantchou, F.; Tucker, L.B.; Fu, A.H.; Bluett, R.J.; McCabe, J.T.; Patel, S.; Zhang, Y. The fatty acid amide hydrolase inhibitor PF-3845 promotes neuronal survival, attenuates inflammation and improves functional recovery in mice with traumatic brain injury. Neuropharmacology 2014, 85, 427–439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnston, T.H.; Huot, P.; Fox, S.H.; Wakefield, J.D.; Sykes, K.A.; Bartolini, W.P.; Milne, G.T.; Pearson, J.P.; Brotchie, J.M. Fatty acid amide hydrolase (FAAH) inhibition reduces L-3,4-dihydroxyphenylalanine-induced hyperactivity in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-lesioned non-human primate model of Parkinson’s disease. J. Pharmacol. Exp. Ther. 2011, 336, 423–430. [Google Scholar] [CrossRef] [Green Version]
- Viveros-Paredes, J.; Gonzalez-Castañeda, R.; Escalante-Castañeda, A.; Tejeda-Martínez, A.; Castañeda-Achutiguí, F.; Flores-Soto, M. Effect of inhibition of fatty acid amide hydrolase on MPTP-induced dopaminergic neuronal damage. Neurología 2019, 34, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Celorrio, M.; Fernández-Suárez, D.; Rojo-Bustamante, E.; Echeverry-Alzate, V.; Ramírez, M.J.; Hillard, C.J.; López-Moreno, J.A.; Maldonado, R.; Oyarzábal, J.; Franco, R.; et al. Fatty acid amide hydrolase inhibition for the symptomatic relief of Parkinson’s disease. Brain Behav. Immun. 2016, 57, 94–105. [Google Scholar] [CrossRef] [Green Version]
- Pryce, G.; Cabranes, A.; Fernández-Ruiz, J.; Bisogno, T.; Di Marzo, V.; Long, J.Z.; Cravatt, B.F.; Giovannoni, G.; Baker, D. Control of experimental spasticity by targeting the degradation of endocannabinoids using selective fatty acid amide hydrolase inhibitors. Mult. Scler. J. 2013, 19, 1896–1904. [Google Scholar] [CrossRef]
- Kerr, D.M.; Harhen, B.; Okine, B.N.; Egan, L.J.; Finn, D.P.; Roche, M. The monoacylglycerol lipase inhibitor JZL184 atten-uates LPS-induced increases in cytokine expression in the rat frontal cortex and plasma: Differential mechanisms of action. Br. J. Pharmacol. 2013, 169, 808–819. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Chen, C. Alleviation of Neuropathology by Inhibition of Monoacylglycerol Lipase in APP Transgenic Mice Lacking CB2 Receptors. Mol. Neurobiol. 2017, 55, 4802–4810. [Google Scholar] [CrossRef]
- Pihlaja, R.; Takkinen, J.; Eskola, O.; Vasara, J.; López-Picón, F.R.; Haaparanta-Solin, M.; Rinne, J.O. Monoacylglycerol lipase inhibitor JZL184 reduces neuroinflammatory response in APdE9 mice and in adult mouse glial cells. J. Neuroinflamm. 2015, 12, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayeux, J.; Katz, P.; Edwards, S.; Middleton, J.W.; Molina, P.E. Inhibition of Endocannabinoid Degradation Improves Outcomes from Mild Traumatic Brain Injury: A Mechanistic Role for Synaptic Hyperexcitability. J. Neurotrauma 2017, 34, 436–443. [Google Scholar] [CrossRef] [PubMed]
- Darmani, N.A.; Izzo, A.; Degenhardt, B.; Valenti, M.; Scaglione, G.; Capasso, R.; Sorrentini, I.; Di Marzo, V. Involvement of the cannabimimetic compound, N-palmitoyl-ethanolamine, in inflammatory and neuropathic conditions: Review of the available pre-clinical data, and first human studies. Neuropharmacology 2005, 48, 1154–1163. [Google Scholar] [CrossRef]
- Cisar, J.S.; Weber, O.D.; Clapper, J.R.; Blankman, J.L.; Henry, C.L.; Simon, G.M.; Alexander, J.P.; Jones, T.K.; Ezekowitz, R.A.B.; O’Neill, G.P.; et al. Identification of ABX-1431, a Selective Inhibitor of Monoacylglycerol Lipase and Clinical Candidate for Treatment of Neurological Disorders. J. Med. Chem. 2018, 61, 9062–9084. [Google Scholar] [CrossRef] [Green Version]
- Fucich, E.A.; Stielper, Z.F.; Cancienne, H.L.; Edwards, S.; Gilpin, N.W.; Molina, P.E.; Middleton, J.W. Endocannabinoid degradation inhibitors ameliorate neuronal and synaptic alterations following traumatic brain injury. J. Neurophysiol. 2020, 123, 707–717. [Google Scholar] [CrossRef] [PubMed]
- Toma, W.; Caillaud, M.; Patel, N.H.; Tran, T.H.; Donvito, G.; Roberts, J.; Bagdas, D.; Jackson, A.; Lichtman, A.; Gewirtz, D.A.; et al. N-acylethanolamine-hydrolysing acid amidase: A new potential target to treat paclitaxel-induced neuropathy. Eur. J. Pain 2021. [Google Scholar] [CrossRef]
- Migliore, M.; Pontis, S.; Fuentes de Arriba, A.L.; Realini, N.; Torrente, E.; Armirotti, A.; Romeo, E.; Di Martino, S.; Russo, D.; Pizzirani, D.; et al. Second-Generation Non-Covalent NAAA Inhibitors are Protective in a Model of Multiple Sclerosis. Angew. Chem. Int. Ed. 2016, 55, 11193–11197. [Google Scholar] [CrossRef]
- Yang, L.; Li, L.; Chen, L.; Li, Y.; Chen, H.; Li, Y.; Ji, G.; Lin, D.; Liu, Z.; Qiu, Y. Potential analgesic effects of a novel N-acylethanolamine acid amidase inhibitor F96 through PPAR-α. Sci. Rep. 2015, 5, srep13565. [Google Scholar] [CrossRef] [Green Version]
- Petrosino, S.; Ahmad, A.; Marcolongo, G.; Esposito, E.; Allarà, M.; Verde, R.; Cuzzocrea, S.; Di Marzo, V. Diacerein is a potent and selective inhibitor of palmitoylethanolamide inactivation with analgesic activity in a rat model of acute inflammatory pain. Pharmacol. Res. 2015, 91, 9–14. [Google Scholar] [CrossRef]
- Sasso, O.; Moreno-Sanz, G.; Martucci, C.; Realini, N.; Dionisi, M.; Mengatto, L.; Duranti, A.; Tarozzo, G.; Tarzia, G.; Mor, M.; et al. Antinociceptive effects of the N-acylethanolamine acid amidase inhibitor ARN077 in rodent pain models. Pain 2013, 154, 350–360. [Google Scholar] [CrossRef] [Green Version]
- Banks, W. Blood-Brain Barrier Transport of Cytokines: A Mechanism for Neuropathology. Curr. Pharm. Des. 2005, 11, 973–984. [Google Scholar] [CrossRef]
- Varatharaj, A.; Galea, I. The blood-brain barrier in systemic inflammation. Brain Behav. Immun. 2017, 60, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Panikashvili, D.; Shein, N.A.; Mechoulam, R.; Trembovler, V.; Kohen, R.; Alexandrovich, A.; Shohami, E. The endocannabinoid 2-AG protects the blood–brain barrier after closed head injury and inhibits mRNA expression of proinflammatory cytokines. Neurobiol. Dis. 2006, 22, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Katz, P.S.; Sulzer, J.K.; Impastato, R.A.; Teng, S.X.; Rogers, E.K.; Molina, P.E. Endocannabinoid Degradation Inhibition Improves Neurobehavioral Function, Blood–Brain Barrier Integrity, and Neuroinflammation following Mild Traumatic Brain Injury. J. Neurotrauma 2015, 32, 297–306. [Google Scholar] [CrossRef] [Green Version]
- Tchantchou, F.; Zhang, Y. Selective Inhibition of Alpha/Beta-Hydrolase Domain 6 Attenuates Neurodegeneration, Alleviates Blood Brain Barrier Breakdown, and Improves Functional Recovery in a Mouse Model of Traumatic Brain Injury. J. Neurotrauma 2013, 30, 565–579. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Li, Y.; Cai, S.; Li, R.; Cao, G. Cannabinoid receptor 2 agonist attenuates blood-brain barrier damage in a rat model of intracerebral hemorrhage by activating the Rac1 pathway. Int. J. Mol. Med. 2018, 42, 2914–2922. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chung, Y.C.; Shin, W.-H.; Baek, J.Y.; Cho, E.J.; Baik, H.H.; Kim, S.R.; Won, S.-Y.; Jin, B.K. CB2 receptor activation prevents glial-derived neurotoxic mediator production, BBB leakage and peripheral immune cell infiltration and rescues dopamine neurons in the MPTP model of Parkinson’s disease. Exp. Mol. Med. 2016, 48, e205. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.-C.; Zhang, H.-Z.; Wang, Z.; You, F.-L.; Wang, Y.-F. The molecular mechanism and effect of cannabinoid-2 receptor agonist on the blood–spinal cord barrier permeability induced by ischemia-reperfusion injury. Brain Res. 2016, 1636, 81–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, L.; Tao, Y.; Tang, J.; Chen, Q.; Yang, Y.; Feng, Z.; Chen, Y.; Yang, L.; Yang, Y.; Zhu, G.; et al. A Cannabinoid Receptor 2 Agonist Prevents Thrombin-Induced Blood–Brain Barrier Damage via the Inhibition of Microglial Activation and Matrix Metalloproteinase Expression in Rats. Transl. Stroke Res. 2015, 6, 467–477. [Google Scholar] [CrossRef] [PubMed]
- Amenta, P.S.; Jallo, J.I.; Tuma, R.F.; Hooper, D.C.; Elliott, M.B. Cannabinoid receptor type-2 stimulation, blockade, and deletion alter the vascular inflammatory responses to traumatic brain injury. J. Neuroinflamm. 2014, 11, 191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vendel, E.; de Lange, E.C. Functions of the CB1 and CB 2 receptors in neuroprotection at the level of the blood-brain barrier. Neuromol. Med. 2014, 16, 620–642. [Google Scholar] [CrossRef] [PubMed]
- Fujii, M.; Sherchan, P.; Krafft, P.R.; Rolland, W.B.; Soejima, Y.; Zhang, J.H. Cannabinoid type 2 receptor stimulation attenuates brain edema by reducing cerebral leukocyte infiltration following subarachnoid hemorrhage in rats. J. Neurol. Sci. 2014, 342, 101–106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kong, W.; Li, H.; Tuma, R.F.; Ganea, D. Selective CB2 receptor activation ameliorates EAE by reducing Th17 differentiation and immune cell accumulation in the CNS. Cell. Immunol. 2014, 287, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Rom, S.; Zuluaga-Ramirez, V.; Dykstra, H.; Reichenbach, N.L.; Pacher, P.; Persidsky, Y. Selective Activation of Cannabinoid Receptor 2 in Leukocytes Suppresses Their Engagement of the Brain Endothelium and Protects the Blood-Brain Barrier. Am. J. Pathol. 2013, 183, 1548–1558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amenta, P.S.; Jallo, J.I.; Tuma, R.F.; Elliott, M.B. A cannabinoid type 2 receptor agonist attenuates blood-brain barrier damage and neurodegeneration in a murine model of traumatic brain injury. J. Neurosci. Res. 2012, 90, 2293–2305. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, S.H.; Haskó, J.; Skuba, A.; Fan, S.; Dykstra, H.; McCormick, R.; Reichenbach, N.; Krizbai, I.; Mahadevan, A.; Zhang, M.; et al. Activation of Cannabinoid Receptor 2 Attenuates Leukocyte-Endothelial Cell Interactions and Blood-Brain Barrier Dysfunction under Inflammatory Conditions. J. Neurosci. 2012, 32, 4004–4016. [Google Scholar] [CrossRef]
- Zhang, M.; Adler, M.W.; Abood, M.E.; Ganea, D.; Jallo, J.; Tuma, R.F. CB2 receptor activation attenuates microcirculatory dysfunction during cerebral ischemic/reperfusion injury. Microvasc. Res. 2009, 78, 86–94. [Google Scholar] [CrossRef] [Green Version]
- Hind, W.H.; Tufarelli, C.; Neophytou, M.; Anderson, S.I.; England, T.J.; O’Sullivan, S.E. Endocannabinoids modulate human blood-brain barrier permeability in vitro. Br. J. Pharmacol. 2015, 172, 3015–3027. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Yang, L.; Ma, A.; Zhang, X.; Li, W.; Yang, W.; Chen, C.; Jin, X. Orally administered oleoylethanolamide protects mice from focal cerebral ischemic injury by activating peroxisome proliferator-activated receptor α. Neuropharmacology 2012, 63, 242–249. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, L.; Zeng, K.; Xu, Z.; Suo, D.; Peng, L.; Ren, T.; Sun, Z.; Yang, W.; Jin, X.; et al. Propane-2-sulfonic acid octadec-9-enyl-amide, a novel PPARα/γ dual agonist, protects against ischemia-induced brain damage in mice by inhibiting inflammatory responses. Brain Behav. Immun. 2017, 66, 289–301. [Google Scholar] [CrossRef]
- Mestre, L.; Inigo, P.M.; Mecha, M.; Correa, F.G.; Hernangomez-Herrero, M.; Loria, F.; Docagne, F.; Borrell, J.; Guaza, C. Anandamide inhibits Theiler’s virus induced VCAM-1 in brain endothelial cells and reduces leukocyte transmigration in a model of blood brain barrier by activation of CB(1) receptors. J. Neuroinflamm. 2011, 8, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kivisäkk, P.; Mahad, D.J.; Callahan, M.K.; Trebst, C.; Tucky, B.; Wei, T.; Wu, L.; Baekkevold, E.S.; Lassmann, H.; Staugaitis, S.M.; et al. Human cerebrospinal fluid central memory CD4+T cells: Evidence for trafficking through choroid plexus and meninges via P-selectin. Proc. Natl. Acad. Sci. USA 2003, 100, 8389–8394. [Google Scholar] [CrossRef] [Green Version]
- Wilson, E.H.; Weninger, W.; Hunter, C.A. Trafficking of immune cells in the central nervous system. J. Clin. Investig. 2010, 120, 1368–1379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abadier, M.; Boscacci, R.; Vestweber, D.; Barnum, S.; Deutsch, U.; Engelhardt, B.; Lyck, R.; Cardoso-Alves, L.; Jahromi, N.H. Cell surface levels of endothelial ICAM-1 influence the transcellular or paracellular T-cell diapedesis across the blood-brain barrier. Eur. J. Immunol. 2015, 45, 1043–1058. [Google Scholar] [CrossRef]
- Banks, W.A.; Niehoff, M.L.; Ponzio, N.M.; Erickson, M.A.; Zalcman, S.S. Pharmacokinetics and modeling of immune cell trafficking: Quantifying differential influences of target tissues versus lymphocytes in SJL and lipopolysaccharide-treated mice. J. Neuroinflamm. 2012, 9, 231. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bohatschek, M.; Werner, A.; Raivich, G. Systemic LPS Injection Leads to Granulocyte Influx into Normal and Injured Brain: Effects of ICAM-1 Deficiency. Exp. Neurol. 2001, 172, 137–152. [Google Scholar] [CrossRef]
- Wang, H.; Sun, J.; Goldstein, H. Human Immunodeficiency Virus Type 1 Infection Increases the In Vivo Capacity of Peripheral Monocytes to Cross the Blood-Brain Barrier into the Brain and the In Vivo Sensitivity of the Blood-Brain Barrier to Disruption by Lipopolysaccharide. J. Virol. 2008, 82, 7591–7600. [Google Scholar] [CrossRef] [Green Version]
- Graham, E.; Angel, C.; Schwarcz, L.; Dunbar, P.; Glass, M. Detailed Characterisation of CB2 Receptor Protein Expression in Peripheral Blood Immune Cells from Healthy Human Volunteers Using Flow Cytometry. Int. J. Immunopathol. Pharmacol. 2010, 23, 25–34. [Google Scholar] [CrossRef] [Green Version]
- Jean-Gilles, L.; Braitch, M.; Latif, M.L.; Aram, J.; Fahey, A.J.; Edwards, L.J.; Robins, R.A.; Tanasescu, R.; Tighe, P.J.; Gran, B.; et al. Effects of pro-inflammatory cytokines on cannabinoid CB1and CB2receptors in immune cells. Acta Physiol. 2015, 214, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Carlisle, S.; Marciano-Cabral, F.; Staab, A.; Ludwick, C.; Cabral, G. Differential expression of the CB2 cannabinoid receptor by rodent macrophages and macrophage-like cells in relation to cell activation. Int. Immunopharmacol. 2002, 2, 69–82. [Google Scholar] [CrossRef]
- Han, K.H.; Lim, S.; Ryu, J.; Lee, C.-W.; Kim, Y.; Kang, J.-H.; Kang, S.-S.; Ahn, Y.K.; Park, C.-S.; Kim, J.J. CB1 and CB2 cannabinoid receptors differentially regulate the production of reactive oxygen species by macrophages. Cardiovasc. Res. 2009, 84, 378–386. [Google Scholar] [CrossRef] [PubMed]
- Kurihara, R.; Tohyama, Y.; Matsusaka, S.; Naruse, H.; Kinoshita, E.; Tsujioka, T.; Katsumata, Y.; Yamamura, H. Effects of Peripheral Cannabinoid Receptor Ligands on Motility and Polarization in Neutrophil-like HL60 Cells and Human Neutrophils. J. Biol. Chem. 2006, 281, 12908–12918. [Google Scholar] [CrossRef] [Green Version]
- Balenga, N.A.; Aflaki, E.; Kargl, J.; Platzer, W.; Schroder, R.; Blattermann, S.; Kostenis, E.; Brown, A.J.; Heinemann, A.; Waldhoer, M. GPR55 regulates cannabinoid 2 receptor-mediated responses in human neutrophils. Cell Res. 2011, 21, 1452–1469. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murikinati, S.; Jüttler, E.; Keinert, T.; Ridder, D.A.; Muhammad, S.; Waibler, Z.; Ledent, C.; Zimmer, A.; Kalinke, U.; Schwaninger, M. Activation of cannabinoid 2 receptors protects against cerebral ischemia by inhibiting neutrophil recruitment. FASEB J. 2009, 24, 788–798. [Google Scholar] [CrossRef] [Green Version]
- Joseph, J.; Niggemann, B.; Zaenker, K.S.; Entschladen, F. Anandamide is an endogenous inhibitor for the migration of tumor cells and T lymphocytes. Cancer Immunol. Immunother. 2004, 53, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Coopman, K.; Smith, L.D.; Wright, K.L.; Ward, S.G. Temporal variation in CB2R levels following T lymphocyte activation: Evidence that cannabinoids modulate CXCL12-induced chemotaxis. Int. Immunopharmacol. 2007, 7, 360–371. [Google Scholar] [CrossRef] [PubMed]
- Chouinard, F.; Lefebvre, J.S.; Navarro, P.; Bouchard, L.; Ferland, C.; Lalancette-Hébert, M.; Marsolais, D.; LaViolette, M.; Flamand, N. The Endocannabinoid 2-Arachidonoyl-Glycerol Activates Human Neutrophils: Critical Role of Its Hydrolysis and De Novo Leukotriene B4 Biosynthesis. J. Immunol. 2011, 186, 3188–3196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chouinard, F.; Turcotte, C.; Guan, X.; LaRose, M.-C.; Poirier, S.; Bouchard, L.; Provost, V.; Flamand, L.; Grandvaux, N.; Flamand, N. 2-Arachidonoyl-glycerol- and arachidonic acid-stimulated neutrophils release antimicrobial effectors against E. coli, S. aureus, HSV-1, and RSV. J. Leukoc. Biol. 2012, 93, 267–276. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Receptor | Location/ Cell Type | Process Regulated | Intracellular Pathway Involved | Physiological Relevance |
|---|---|---|---|---|
| CB1 | Neurons | Presynaptic vesicular neurotransmitter release | Multiple, including inhibition of AC and VGCC; resulting in regulation of [Ca2+]i | Synaptic plasticity |
| [Ca2+] influx | Neuronal survival | |||
| Astrocytes | Glutamate–glutamine cycle | Upregulation of glutamine-synthase | Perisynaptic glutamate scavenging, prevention of excitoxicity | |
| Gliotransmission | Ca2+ influx | Modulation of synaptic strength; role in synaptic plasticity | ||
| Endothelial cells | Growth and proliferation | Coupled to the MAP kinase cascade | Maintenance of BBB integrity and selectivity | |
| CB2 | Ventral tegmental area dopamine neurons | Neuronal excitability | Modulation of K+ channels [30] | Long-term neuronal hyperpolarization; synaptic plasticity |
| Hippocampal CA3/CA2 pyramidal neurons | Activation of the Na+/Bicarbonate co-transporter [34] | |||
| Microglia | Migration | Extracellular signal-regulated kinase (ERK) 1/2 signal transduction pathway | Shift between the pro-inflammatory and pro-resolving phenotype |
| Compound | Organism | Disease/Model | Finding | References |
|---|---|---|---|---|
| 2-AG | Mouse | Closed head injury | Increased 2-AG brain levels; neuroprotection | [56] |
| Rat | Inflammatory model of pain | Antinociceptive effect | [57] | |
| Stress-induced analgesia | Increased 2-AG and AEA mid-brain levels; analgesia | [58] | ||
| AEA | Mouse | Experimental autoimmune encephalomyelitis | Increased AEA brain levels | [59] |
| Human | Multiple sclerosis | Increased AEA levels in cerebrospinal fluid | [59] | |
| Rat | Focal cerebral ischemia model | Increased AEA brain levels | [60] | |
| NADA | Mouse | Acute systemic inflammation | Reduces inflammation in vivo; increases survival in endotoxemic mice | [61] |
| 2-AG, AEA, PEA | Mouse | Transgenic model of Huntington’s disease | Changed levels of 2-AG, AEA, and PEA in a disease phase- and brain region-specific way | [62] |
| PEA | Rat | Granulomatous inflammation | Reduced granuloma-induced hyperalgesia | [63] |
| Mouse | Paw model of hyperalgesia | Reduced mechanical hyperalgesia | [39] | |
| Model of neuropathic pain | Anti-allodynic and anti-hyperalgesic effects | [64] | ||
| Alzheimer’s disease | Rescued cognitive deficit and reduced neuroinflammation and oxidative stress | [40] | ||
| Traumatic brain injury model | Reduced edema and brain infractions; blocked infiltration of astrocytes | [65] | ||
| SEA | Mouse | LPS-induced neuroinflammation | Decreased activation of resident microglia and leukocyte trafficking into the brain; increased CB1/2 expression and 2-AG brain levels | [47] |
| Inhibitor | Compound | Disease/Model | Finding | References |
|---|---|---|---|---|
| FAAH inhibitors | PF-3845 | Chronic mouse model of PD | decreased CB2 expression | [223] |
| TBI mouse model | Enhanced AEA brain levels; reduced neurodegeneration in the dentate gyrus; suppressed production of amyloid precursor protein; suppressed expression of iNOS and COX-2 | [224] | ||
| URB597 | Mouse model of PD | Increased AEA, PEA, and OEA levels; reduced L-DOPA-induced side effects Inhibition of dopaminergic neuronal death; decreased microglial immunoreactivity; improved motor capacity | [225] [226] | |
| URB597 JNJ1661010 TCF2 | Chronic mouse model of PD | Anti-cataleptic effects | [227] | |
| URB597 CAY100400 CAY100402 | Experimental autoimmune encephalomyelitis (EAE) in mice | Anti-spastic effects | [228] | |
| MAGL inhibitors | JZL184 | LPS-induced neuroinflammation in rats | Decreased peripheral and central cytokine production | [229] |
| EAE in mice | Anti-spastic effects | [228] | ||
| Mouse model of AD | Improvements in spatial learning and memory decreased proinflammatory reactions of microglia and reduced amyloid β burden | [230] [231] | ||
| TBI rat model | Improved neurobehavioral recovery; reduce astrocyte activation; less glutamate dyshomeostasis | [232] | ||
| CPD-4645 | LPS-induced neuroinflammation and focal photothrombotic ischemic insult in mice | Restored functional homeostasis of the brain vasculature | [175] | |
| KML29 | chronic MPTP/probenecid mouse model of PD | Attenuated striatal dopamine depletion; increase in Gdnf expression | [223] | |
| Ulcerative colitis in humans back pain in humans | Increased PEA levels in colon enhanced PEA blood levels after therapeutic manipulation | [233] | ||
| ABX-1431 | Formalin pain model in rats | Increased brain 2-AG concentrations; suppressed pain behavior | [234] | |
| dual MAGL FAAH inhibitors | JZL195 | Traumatic brain injury in rats | Attenuated neuronal dysfunction | [235] |
| NAAA inhibitors | AM9053 | Neuropathy in mice | Reversed and prevented peripheral neuropathy via PPAR-α | [236] |
| Compound 8 | EAE in mice | Delayed disease onset; attenuated symptom intensity; normalized body weight; reduced leukocyte infiltration and microglia activation | [237] | |
| oxazolidin-2-one (F96) | Ear edema model in mice | Prevented allodynia via PPAR-α | [238] | |
| EPT4900 | Carrageenan-induced pain in rats | Inhibited inflammation as well as hyperalgesia | [239] | |
| ARN077 | Rodent models of hyperalgesia and allodynia | Prevented heat hyperalgesia and mechanical allodynia via PPAR-α | [240] |
| CB receptor | Compound | Cell Type | Effect | References |
|---|---|---|---|---|
| CB1 | AEA ACEA | Monocytes Macrophages | Promotes ROS and cytokine production | [271] |
| CB2 | JWH-015 | Macrophages | Inhibits ROS production | [271] |
| 2-AG | Neutrophils | Inhibits migration | [272] | |
| Promotes migration | [273] | |||
| JWH-133 | Neutrophils | Inhibits adhesion to cerebral endothelial cells | [274] | |
| T cells | Inhibits migration | [275,276] | ||
| 2-AG | T cells | Inhibits migration | [276] | |
| CB1/CB2 independent | 2-AG | Neutrophils | Stimulates myeloperoxidase and leukotriene release, kinase activation, and calcium mobilization | [277] |
| Release antimicrobial effectors | [278] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kasatkina, L.A.; Rittchen, S.; Sturm, E.M. Neuroprotective and Immunomodulatory Action of the Endocannabinoid System under Neuroinflammation. Int. J. Mol. Sci. 2021, 22, 5431. https://doi.org/10.3390/ijms22115431
Kasatkina LA, Rittchen S, Sturm EM. Neuroprotective and Immunomodulatory Action of the Endocannabinoid System under Neuroinflammation. International Journal of Molecular Sciences. 2021; 22(11):5431. https://doi.org/10.3390/ijms22115431
Chicago/Turabian StyleKasatkina, Ludmila A., Sonja Rittchen, and Eva M. Sturm. 2021. "Neuroprotective and Immunomodulatory Action of the Endocannabinoid System under Neuroinflammation" International Journal of Molecular Sciences 22, no. 11: 5431. https://doi.org/10.3390/ijms22115431