
Contemporary Transposon Tools: A Review and Guide through Mechanisms and Applications of Sleeping Beauty, piggyBac and Tol2 for Genome Engineering


Abstract
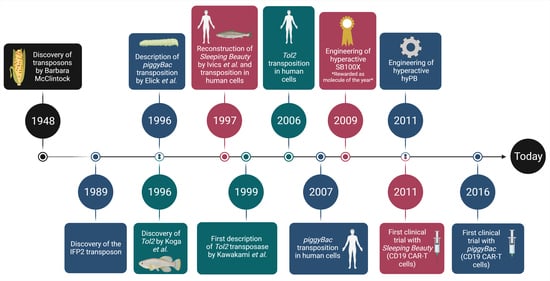
:
1. Introduction
2. The Sleeping Beauty Transposon System
3. The piggyBac Transposon System
4. The Tol2 Transposon System
5. Vectors for Enhanced Delivery of Transposon Systems
5.1. Minimized DNA Vectors
5.2. mRNA and Proteins
5.3. Hybrid Systems Relying on Non-Integrative Viral Vectors and Nanoparticles
6. Basic Research Applications
6.1. Insertional Mutagenesis Screens
6.2. Transgenic Animals
6.3. iPSC Reprogramming
7. Preclinical Applications
8. Clinical Applications
9. Conclusions and Outlook
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Kulkosky, J.; Jones, K.S.; Katz, R.A.; Mack, J.P.; Skalka, A.M. Residues critical for retroviral integrative recombination in a region that is highly conserved among retroviral/retrotransposon integrases and bacterial insertion sequence transposases. Mol. Cell. Biol. 1992, 12, 2331–2338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doak, T.G.; Doerder, F.P.; Jahn, C.L.; Herrick, G. A proposed superfamily of transposase genes: Transposon-like elements in ciliated protozoa and a common “D35E” motif. Proc. Natl. Acad. Sci. USA 1994, 91, 942–946. [Google Scholar] [CrossRef] [Green Version]
- Bujacz, G.; Alexandratos, J.; Wlodawer, A.; Merkel, G.; Andrake, M.; Katz, R.A.; Skalka, A.M. Binding of different divalent cations to the active site of avian sarcoma virus integrase and their effects on enzymatic activity. J. Biol. Chem. 1997, 272, 18161–18168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldgur, Y.; Dyda, F.; Hickman, A.B.; Jenkins, T.M.; Craigie, R.; Davies, D.R. Three new structures of the core domain of HIV-1 integrase: An active site that binds magnesium. Proc. Natl. Acad. Sci. USA 1998, 95, 9150–9154. [Google Scholar] [CrossRef] [Green Version]
- Craig, N.L. Unity in transposition reactions. Science 1995, 270, 253–254. [Google Scholar] [CrossRef]
- Hickman, A.B.; Dyda, F. Mechanisms of DNA Transposition. Microbiol. Spectr. 2015, 3, 529–553. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Mitra, R.; Atkinson, P.W.; Hickman, A.B.; Dyda, F.; Craig, N.L. Transposition of hAT elements links transposable elements and V(D)J recombination. Nature 2004, 432, 995–1001. [Google Scholar] [CrossRef]
- Hencken, C.G.; Li, X.; Craig, N.L. Functional characterization of an active Rag-like transposase. Nat. Struct. Mol. Biol. 2012, 19, 834–836. [Google Scholar] [CrossRef] [Green Version]
- Bhasin, A.; Goryshin, I.Y.; Reznikoff, W.S. Hairpin formation in Tn5 transposition. J. Biol. Chem. 1999, 274, 37021–37029. [Google Scholar] [CrossRef] [Green Version]
- Kennedy, A.K.; Guhathakurta, A.; Kleckner, N.; Haniford, D.B. Tn10 transposition via a DNA hairpin intermediate. Cell 1998, 95, 125–134. [Google Scholar] [CrossRef] [Green Version]
- Mitra, R.; Fain-Thornton, J.; Craig, N.L. piggyBac can bypass DNA synthesis during cut and paste transposition. EMBO J. 2008, 27, 1097–1109. [Google Scholar] [CrossRef] [Green Version]
- Dawson, A.; Finnegan, D.J. Excision of the Drosophila mariner transposon Mos1. Comparison with bacterial transposition and V(D)J recombination. Mol. Cell 2003, 11, 225–235. [Google Scholar] [CrossRef]
- Izsvák, Z.; Stüwe, E.E.; Fiedler, D.; Katzer, A.; Jeggo, P.A.; Ivics, Z. Healing the wounds inflicted by sleeping beauty transposition by double-strand break repair in mammalian somatic cells. Mol. Cell 2004, 13, 279–290. [Google Scholar] [CrossRef]
- Richardson, J.M.; Dawson, A.; O’Hagan, N.; Taylor, P.; Finnegan, D.J.; Walkinshaw, M.D. Mechanism of Mos1 transposition: Insights from structural analysis. EMBO J. 2006, 25, 1324–1334. [Google Scholar] [CrossRef] [Green Version]
- Bouuaert, C.C.; Chalmers, R. A single active site in the mariner transposase cleaves DNA strands of opposite polarity. Nucleic Acids Res. 2017, 45, 11467–11478. [Google Scholar] [CrossRef] [Green Version]
- Goryshin, I.Y.; Reznikoff, W.S. Tn5 in vitro transposition. J. Biol. Chem. 1998, 273, 7367–7374. [Google Scholar] [CrossRef] [Green Version]
- Van Luenen, H.G.; Colloms, S.D.; Plasterk, R.H. The mechanism of transposition of Tc3 in C. elegans. Cell 1994, 79, 293–301. [Google Scholar] [CrossRef]
- Miskey, C.; Papp, B.; Mátés, L.; Sinzelle, L.; Keller, H.; Izsvák, Z.; Ivics, Z. The ancient mariner sails again: Transposition of the human Hsmar1 element by a reconstructed transposase and activities of the SETMAR protein on transposon ends. Mol. Cell. Biol. 2007, 27, 4589–4600. [Google Scholar] [CrossRef] [Green Version]
- Luo, G.; Ivics, Z.; Izsvák, Z.; Bradley, A. Chromosomal transposition of a Tc1/mariner-like element in mouse embryonic stem cells. Proc. Natl. Acad. Sci. USA 1998, 95, 10769–10773. [Google Scholar] [CrossRef] [Green Version]
- Ivics, Z.; Izsvak, Z.; Minter, A.; Hackett, P.B. Identification of functional domains and evolution of Tc1-like transposable elements. Proc. Natl. Acad. Sci. USA 1996, 93, 5008–5013. [Google Scholar] [CrossRef] [Green Version]
- Ivics, Z.; Hackett, P.B.; Plasterk, R.H.; Izsvák, Z. Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell 1997, 91, 501–510. [Google Scholar] [CrossRef] [Green Version]
- Izsvák, Z.; Ivics, Z.; Plasterk, R.H. Sleeping Beauty, a wide host-range transposon vector for genetic transformation in vertebrates. J. Mol. Biol. 2000, 302, 93–102. [Google Scholar] [CrossRef]
- Kawakami, K.; Largaespada, D.A.; Ivics, Z. Transposons as Tools for Functional Genomics in Vertebrate Models. Trends Genet. 2017, 33, 784–801. [Google Scholar] [CrossRef]
- Amberger, M.; Ivics, Z. Latest Advances for the Sleeping Beauty Transposon System: 23 Years of Insomnia but Prettier than Ever: Refinement and Recent Innovations of the Sleeping Beauty Transposon System Enabling Novel, Nonviral Genetic Engineering Applications. Bioessays 2020, 42, e2000136. [Google Scholar] [CrossRef] [PubMed]
- Cui, Z.; Geurts, A.M.; Liu, G.; Kaufman, C.D.; Hackett, P.B. Structure-function analysis of the inverted terminal repeats of the sleeping beauty transposon. J. Mol. Biol. 2002, 318, 1221–1235. [Google Scholar] [CrossRef]
- Zayed, H.; Izsvák, Z.; Walisko, O.; Ivics, Z. Development of hyperactive sleeping beauty transposon vectors by mutational analysis. Mol. Ther. 2004, 9, 292–304. [Google Scholar] [CrossRef]
- Zayed, H.; Izsvák, Z.; Khare, D.; Heinemann, U.; Ivics, Z. The DNA-bending protein HMGB1 is a cellular cofactor of Sleeping Beauty transposition. Nucleic Acids Res. 2003, 31, 2313–2322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arensburger, P.; Hice, R.H.; Zhou, L.; Smith, R.C.; Tom, A.C.; Wright, J.A.; Knapp, J.; O’Brochta, D.A.; Craig, N.L.; Atkinson, P.W. Phylogenetic and functional characterization of the hAT transposon superfamily. Genetics 2011, 188, 45–57. [Google Scholar] [CrossRef] [Green Version]
- Koga, A.; Suzuki, M.; Inagaki, H.; Bessho, Y.; Hori, H. Transposable element in fish. Nature 1996, 383, 30. [Google Scholar] [CrossRef]
- Koga, A.; Suzuki, M.; Maruyama, Y.; Tsutsumi, M.; Hori, H. Amino acid sequence of a putative transposase protein of the medaka fish transposable element Tol2 deduced from mRNA nucleotide sequences. FEBS Lett. 1999, 461, 295–298. [Google Scholar] [CrossRef] [Green Version]
- Kawakami, K.; Shima, A. Identification of the Tol2 transposase of the medaka fish Oryzias latipes that catalyzes excision of a nonautonomous Tol2 element in zebrafish Danio rerio. Gene 1999, 240, 239–244. [Google Scholar] [CrossRef]
- Ochmann, M.T.; Ivics, Z. Jumping Ahead with Sleeping Beauty: Mechanistic Insights into Cut-and-Paste Transposition. Viruses 2021, 13, 76. [Google Scholar] [CrossRef] [PubMed]
- Izsvák, Z.; Khare, D.; Behlke, J.; Heinemann, U.; Plasterk, R.H.; Ivics, Z. Involvement of a bifunctional, paired-like DNA-binding domain and a transpositional enhancer in Sleeping Beauty transposition. J. Biol. Chem. 2002, 277, 34581–34588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carpentier, C.E.; Schreifels, J.M.; Aronovich, E.L.; Carlson, D.F.; Hackett, P.B.; Nesmelova, I.V. NMR structural analysis of Sleeping Beauty transposase binding to DNA. Protein Sci. 2014, 23, 23–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Pryputniewicz-Dobrinska, D.; Nagy, E.É.; Kaufman, C.D.; Singh, M.; Yant, S.; Wang, J.; Dalda, A.; Kay, M.A.; Ivics, Z.; et al. Regulated complex assembly safeguards the fidelity of Sleeping Beauty transposition. Nucleic Acids Res. 2017, 45, 311–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vigdal, T.J.; Kaufman, C.D.; Izsvák, Z.; Voytas, D.F.; Ivics, Z. Common physical properties of DNA affecting target site selection of sleeping beauty and other Tc1/mariner transposable elements. J. Mol. Biol. 2002, 323, 441–452. [Google Scholar] [CrossRef]
- Moldt, B.; Miskey, C.; Staunstrup, N.H.; Gogol-Döring, A.; Bak, R.O.; Sharma, N.; Mátés, L.; Izsvák, Z.; Chen, W.; Ivics, Z.; et al. Comparative genomic integration profiling of Sleeping Beauty transposons mobilized with high efficacy from integrase-defective lentiviral vectors in primary human cells. Mol. Ther. 2011, 19, 1499–1510. [Google Scholar] [CrossRef] [Green Version]
- Gogol-Döring, A.; Ammar, I.; Gupta, S.; Bunse, M.; Miskey, C.; Chen, W.; Uckert, W.; Schulz, T.F.; Izsvák, Z.; Ivics, Z. Genome-wide Profiling Reveals Remarkable Parallels Between Insertion Site Selection Properties of the MLV Retrovirus and the piggyBac Transposon in Primary Human CD4(+) T Cells. Mol. Ther. 2016, 24, 592–606. [Google Scholar] [CrossRef] [Green Version]
- Holstein, M.; Mesa-Nuñez, C.; Miskey, C.; Almarza, E.; Poletti, V.; Schmeer, M.; Grueso, E.; Ordóñez Flores, J.C.; Kobelt, D.; Walther, W.; et al. Efficient Non-viral Gene Delivery into Human Hematopoietic Stem Cells by Minicircle Sleeping Beauty Transposon Vectors. Mol. Ther. 2018, 26, 1137–1153. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Wang, J.; Devaraj, A.; Singh, M.; Jimenez Orgaz, A.; Chen, J.-X.; Selbach, M.; Ivics, Z.; Izsvák, Z. Suicidal autointegration of sleeping beauty and piggyBac transposons in eukaryotic cells. PLoS Genet. 2014, 10, e1004103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- English, J.J.; Harrison, K.; Jones, J. Aberrant Transpositions of Maize Double Ds-Like Elements Usually Involve Ds Ends on Sister Chromatids. Plant Cell 1995, 7, 1235–1247. [Google Scholar] [CrossRef] [Green Version]
- Yant, S.R.; Park, J.; Huang, Y.; Mikkelsen, J.G.; Kay, M.A. Mutational analysis of the N-terminal DNA-binding domain of sleeping beauty transposase: Critical residues for DNA binding and hyperactivity in mammalian cells. Mol. Cell. Biol. 2004, 24, 9239–9247. [Google Scholar] [CrossRef] [Green Version]
- Baus, J.; Liu, L.; Heggestad, A.D.; Sanz, S.; Fletcher, B.S. Hyperactive transposase mutants of the Sleeping Beauty transposon. Mol. Ther. 2005, 12, 1148–1156. [Google Scholar] [CrossRef]
- Mátés, L.; Chuah, M.K.L.; Belay, E.; Jerchow, B.; Manoj, N.; Acosta-Sanchez, A.; Grzela, D.P.; Schmitt, A.; Becker, K.; Matrai, J.; et al. Molecular evolution of a novel hyperactive Sleeping Beauty transposase enables robust stable gene transfer in vertebrates. Nat. Genet. 2009, 41, 753–761. [Google Scholar] [CrossRef]
- Voigt, F.; Wiedemann, L.; Zuliani, C.; Querques, I.; Sebe, A.; Mátés, L.; Izsvák, Z.; Ivics, Z.; Barabas, O. Sleeping Beauty transposase structure allows rational design of hyperactive variants for genetic engineering. Nat. Commun. 2016, 7, 11126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konnova, T.A.; Singer, C.M.; Nesmelova, I.V. NMR solution structure of the RED subdomain of the Sleeping Beauty transposase. Protein Sci. 2017, 26, 1171–1181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abrusán, G.; Yant, S.R.; Szilágyi, A.; Marsh, J.A.; Mátés, L.; Izsvák, Z.; Barabás, O.; Ivics, Z. Structural Determinants of Sleeping Beauty Transposase Activity. Mol. Ther. 2016, 24, 1369–1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kesselring, L.; Miskey, C.; Zuliani, C.; Querques, I.; Kapitonov, V.; Laukó, A.; Fehér, A.; Palazzo, A.; Diem, T.; Lustig, J.; et al. A single amino acid switch converts the Sleeping Beauty transposase into an efficient unidirectional excisionase with utility in stem cell reprogramming. Nucleic Acids Res. 2020, 48, 316–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Querques, I.; Mades, A.; Zuliani, C.; Miskey, C.; Alb, M.; Grueso, E.; Machwirth, M.; Rausch, T.; Einsele, H.; Ivics, Z.; et al. A highly soluble Sleeping Beauty transposase improves control of gene insertion. Nat. Biotechnol. 2019, 37, 1502–1512. [Google Scholar] [CrossRef]
- Ivics, Z.; Li, M.A.; Mátés, L.; Boeke, J.D.; Nagy, A.; Bradley, A.; Izsvák, Z. Transposon-mediated genome manipulation in vertebrates. Nat. Methods 2009, 6, 415–422. [Google Scholar] [CrossRef] [Green Version]
- Grabundzija, I.; Wang, J.; Sebe, A.; Erdei, Z.; Kajdi, R.; Devaraj, A.; Steinemann, D.; Szuhai, K.; Stein, U.; Cantz, T.; et al. Sleeping Beauty transposon-based system for cellular reprogramming and targeted gene insertion in induced pluripotent stem cells. Nucleic Acids Res. 2013, 41, 1829–1847. [Google Scholar] [CrossRef]
- Kues, W.A.; Herrmann, D.; Barg-Kues, B.; Haridoss, S.; Nowak-Imialek, M.; Buchholz, T.; Streeck, M.; Grebe, A.; Grabundzija, I.; Merkert, S.; et al. Derivation and characterization of sleeping beauty transposon-mediated porcine induced pluripotent stem cells. Stem Cells Dev. 2013, 22, 124–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talluri, T.R.; Kumar, D.; Glage, S.; Garrels, W.; Ivics, Z.; Debowski, K.; Behr, R.; Kues, W.A. Non-viral reprogramming of fibroblasts into induced pluripotent stem cells by Sleeping Beauty and piggyBac transposons. Biochem. Biophys. Res. Commun. 2014, 450, 581–587. [Google Scholar] [CrossRef] [PubMed]
- Talluri, T.R.; Kumar, D.; Glage, S.; Garrels, W.; Ivics, Z.; Debowski, K.; Behr, R.; Niemann, H.; Kues, W.A. Derivation and characterization of bovine induced pluripotent stem cells by transposon-mediated reprogramming. Cell. Reprogram. 2015, 17, 131–140. [Google Scholar] [CrossRef] [Green Version]
- Sebe, A.; Ivics, Z. Reprogramming of Human Fibroblasts to Induced Pluripotent Stem Cells with Sleeping Beauty Transposon-Based Stable Gene Delivery. Methods Mol. Biol. 2016, 1400, 419–427. [Google Scholar] [CrossRef] [PubMed]
- Molina-Estevez, F.J.; Lozano, M.L.; Navarro, S.; Torres, Y.; Grabundzija, I.; Ivics, Z.; Samper, E.; Bueren, J.A.; Guenechea, G. Brief report: Impaired cell reprogramming in nonhomologous end joining deficient cells. Stem Cells 2013, 31, 1726–1730. [Google Scholar] [CrossRef] [PubMed]
- Copeland, N.G.; Jenkins, N.A. Harnessing transposons for cancer gene discovery. Nat. Rev. Cancer 2010, 10, 696–706. [Google Scholar] [CrossRef]
- DeNicola, G.M.; Karreth, F.A.; Adams, D.J.; Wong, C.C. The utility of transposon mutagenesis for cancer studies in the era of genome editing. Genome Biol. 2015, 16, 229. [Google Scholar] [CrossRef] [Green Version]
- Ivics, Z.; Garrels, W.; Mátés, L.; Yau, T.Y.; Bashir, S.; Zidek, V.; Landa, V.; Geurts, A.; Pravenec, M.; Rülicke, T.; et al. Germline transgenesis in pigs by cytoplasmic microinjection of Sleeping Beauty transposons. Nat. Protoc. 2014, 9, 810–827. [Google Scholar] [CrossRef] [Green Version]
- Ivics, Z.; Hiripi, L.; Hoffmann, O.I.; Mátés, L.; Yau, T.Y.; Bashir, S.; Zidek, V.; Landa, V.; Geurts, A.; Pravenec, M.; et al. Germline transgenesis in rabbits by pronuclear microinjection of Sleeping Beauty transposons. Nat. Protoc. 2014, 9, 794–809. [Google Scholar] [CrossRef] [Green Version]
- Ivics, Z.; Mátés, L.; Yau, T.Y.; Landa, V.; Zidek, V.; Bashir, S.; Hoffmann, O.I.; Hiripi, L.; Garrels, W.; Kues, W.A.; et al. Germline transgenesis in rodents by pronuclear microinjection of Sleeping Beauty transposons. Nat. Protoc. 2014, 9, 773–793. [Google Scholar] [CrossRef]
- Alessio, A.P.; Fili, A.E.; Garrels, W.; Forcato, D.O.; Olmos Nicotra, M.F.; Liaudat, A.C.; Bevacqua, R.J.; Savy, V.; Hiriart, M.I.; Talluri, T.R.; et al. Establishment of cell-based transposon-mediated transgenesis in cattle. Theriogenology 2016, 85, 1297–1311.e2. [Google Scholar] [CrossRef] [Green Version]
- Garrels, W.; Mátés, L.; Holler, S.; Dalda, A.; Taylor, U.; Petersen, B.; Niemann, H.; Izsvák, Z.; Ivics, Z.; Kues, W.A. Germline transgenic pigs by Sleeping Beauty transposition in porcine zygotes and targeted integration in the pig genome. PLoS ONE 2011, 6, e23573. [Google Scholar] [CrossRef] [Green Version]
- Garrels, W.; Talluri, T.R.; Apfelbaum, R.; Carratalá, Y.P.; Bosch, P.; Pötzsch, K.; Grueso, E.; Ivics, Z.; Kues, W.A. One-step Multiplex Transgenesis via Sleeping Beauty Transposition in Cattle. Sci. Rep. 2016, 6, 21953. [Google Scholar] [CrossRef] [Green Version]
- Katter, K.; Geurts, A.M.; Hoffmann, O.; Mátés, L.; Landa, V.; Hiripi, L.; Moreno, C.; Lazar, J.; Bashir, S.; Zidek, V.; et al. Transposon-mediated transgenesis, transgenic rescue, and tissue-specific gene expression in rodents and rabbits. FASEB J. 2013, 27, 930–941. [Google Scholar] [CrossRef] [Green Version]
- Narayanavari, S.A.; Chilkunda, S.S.; Ivics, Z.; Izsvák, Z. Sleeping Beauty transposition: From biology to applications. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 18–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hackett, P.B.; Largaespada, D.A.; Cooper, L.J.N. A transposon and transposase system for human application. Mol. Ther. 2010, 18, 674–683. [Google Scholar] [CrossRef]
- Boehme, P.; Doerner, J.; Solanki, M.; Jing, L.; Zhang, W.; Ehrhardt, A. The sleeping beauty transposon vector system for treatment of rare genetic diseases: An unrealized hope? Curr. Gene Ther. 2015, 15, 255–265. [Google Scholar] [CrossRef] [PubMed]
- Hudecek, M.; Ivics, Z. Non-viral therapeutic cell engineering with the Sleeping Beauty transposon system. Curr. Opin. Genet. Dev. 2018, 52, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Hudecek, M.; Izsvák, Z.; Johnen, S.; Renner, M.; Thumann, G.; Ivics, Z. Going non-viral: The Sleeping Beauty transposon system breaks on through to the clinical side. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 355–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Matteo, M.; Belay, E.; Chuah, M.K.; VandenDriessche, T. Recent developments in transposon-mediated gene therapy. Expert Opin. Biol. Ther. 2012, 12, 841–858. [Google Scholar] [CrossRef]
- Potter, K.N.; Faulkner, P.; MacKinnon, E.A. Strain Selection During Serial Passage of Trichoplusia ni Nuclear Polyhedrosis Virus. J. Virol. 1976, 18, 1040–1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraser, M.J.; Smith, G.E.; Summers, M.D. Acquisition of Host Cell DNA Sequences by Baculoviruses: Relationship Between Host DNA Insertions and FP Mutants of Autographa californica and Galleria mellonella Nuclear Polyhedrosis Viruses. J. Virol. 1983, 47, 287–300. [Google Scholar] [CrossRef] [Green Version]
- Cary, L.C.; Goebel, M.; Corsaro, B.G.; Wang, H.-G.; Rosen, E.; Fraser, M.J. Transposon mutagenesis of baculoviruses: Analysis of Trichoplusia ni transposon IFP2 insertions within the FP-locus of nuclear polyhedrosis viruses. Virology 1989, 172, 156–169. [Google Scholar] [CrossRef]
- Elick, T.A.; Bauser, C.A.; Fraser, M.J. Excision of the piggyBac transposable element in vitro is a precise event that is enhanced by the expression of its encoded transposase. Genetica 1996, 98, 33–41. [Google Scholar] [CrossRef]
- Chen, Q.; Luo, W.; Veach, R.A.; Hickman, A.B.; Wilson, M.H.; Dyda, F. Structural basis of seamless excision and specific targeting by piggyBac transposase. Nat. Commun. 2020, 11, 3446. [Google Scholar] [CrossRef]
- Keith, J.H.; Fraser, T.S.; Fraser, M.J. Analysis of the piggyBac transposase reveals a functional nuclear targeting signal in the 94 c-terminal residues. BMC Mol. Biol. 2008, 9, 72. [Google Scholar] [CrossRef] [Green Version]
- Morellet, N.; Li, X.; Wieninger, S.A.; Taylor, J.L.; Bischerour, J.; Moriau, S.; Lescop, E.; Bardiaux, B.; Mathy, N.; Assrir, N.; et al. Sequence-specific DNA binding activity of the cross-brace zinc finger motif of the piggyBac transposase. Nucleic Acids Res. 2018, 46, 2660–2677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johnson, E.T.; Dowd, P.F. A non-autonomous insect piggyBac transposable element is mobile in tobacco. Mol. Genet. Genom. 2014, 289, 895–902. [Google Scholar] [CrossRef]
- Ding, S.; Wu, X.; Li, G.; Han, M.; Zhuang, Y.; Xu, T. Efficient transposition of the piggyBac (PB) transposon in mammalian cells and mice. Cell 2005, 122, 473–483. [Google Scholar] [CrossRef] [Green Version]
- Wilson, M.H.; Coates, C.J.; George, A.L. PiggyBac transposon-mediated gene transfer in human cells. Mol. Ther. 2007, 15, 139–145. [Google Scholar] [CrossRef]
- Li, M.A.; Turner, D.J.; Ning, Z.; Yusa, K.; Liang, Q.; Eckert, S.; Rad, L.; Fitzgerald, T.W.; Craig, N.L.; Bradley, A. Mobilization of giant piggyBac transposons in the mouse genome. Nucleic Acids Res. 2011, 39, e148. [Google Scholar] [CrossRef] [Green Version]
- Rostovskaya, M.; Fu, J.; Obst, M.; Baer, I.; Weidlich, S.; Wang, H.; Smith, A.J.H.; Anastassiadis, K.; Stewart, A.F. Transposon-mediated BAC transgenesis in human ES cells. Nucleic Acids Res. 2012, 40, e150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fraser, M.J.; Cary, L.; Boonvisudhi, K.; Wang, H.H. Assay for Movement of Lepidopteran Transposon IFP2 in Insect Cells Using a Baculovis Genome as Target DNA. Virology 1995, 211, 397–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yusa, K.; Zhou, L.; Li, M.A.; Bradley, A.; Craig, N.L. A hyperactive piggyBac transposase for mammalian applications. Proc. Natl. Acad. Sci. USA 2011, 108, 1531–1536. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Burnight, E.R.; Cooney, A.L.; Malani, N.; Brady, T.; Sander, J.D.; Staber, J.; Wheelan, S.J.; Joung, J.K.; McCray, P.B.; et al. piggyBac transposase tools for genome engineering. Proc. Natl. Acad. Sci. USA 2013, 110, E2279–E2287. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Li, Q.; Helfer, C.M.; Jiao, J.; You, J. Bromodomain protein Brd4 associated with acetylated chromatin is important for maintenance of higher-order chromatin structure. J. Biol. Chem. 2012, 287, 10738–10752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; Larue, R.C.; Plumb, M.R.; Malani, N.; Male, F.; Slaughter, A.; Kessl, J.J.; Shkriabai, N.; Coward, E.; Aiyer, S.S.; et al. BET proteins promote efficient murine leukemia virus integration at transcription start sites. Proc. Natl. Acad. Sci. USA 2013, 110, 12036–12041. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.S.; Maetzig, T.; Maertens, G.N.; Sharif, A.; Rothe, M.; Weidner-Glunde, M.; Galla, M.; Schambach, A.; Cherepanov, P.; Schulz, T.F. Bromo- and extraterminal domain chromatin regulators serve as cofactors for murine leukemia virus integration. J. Virol. 2013, 87, 12721–12736. [Google Scholar] [CrossRef] [Green Version]
- Hamada, M.; Nishio, N.; Okuno, Y.; Suzuki, S.; Kawashima, N.; Muramatsu, H.; Tsubota, S.; Wilson, M.H.; Morita, D.; Kataoka, S.; et al. Integration Mapping of piggyBac-Mediated CD19 Chimeric Antigen Receptor T Cells Analyzed by Novel Tagmentation-Assisted PCR. EBioMedicine 2018, 34, 18–26. [Google Scholar] [CrossRef] [Green Version]
- Pavelitz, T.; Gray, L.T.; Padilla, S.L.; Bailey, A.D.; Weiner, A.M. PGBD5: A neural-specific intron-containing piggyBac transposase domesticated over 500 million years ago and conserved from cephalochordates to humans. Mobile DNA 2013, 4, 23. [Google Scholar] [CrossRef] [Green Version]
- Henssen, A.G.; Henaff, E.; Jiang, E.; Eisenberg, A.R.; Carson, J.R.; Villasante, C.M.; Ray, M.; Still, E.; Burns, M.; Gandara, J.; et al. Genomic DNA transposition induced by human PGBD5. eLife 2015, 4. [Google Scholar] [CrossRef]
- Li, X.; Harrell, R.A.; Handler, A.M.; Beam, T.; Hennessy, K.; Fraser, M.J. piggyBac internal sequences are necessary for efficient transformation of target genomes. Insect Mol. Biol. 2005, 14, 17–30. [Google Scholar] [CrossRef]
- Lacoste, A.; Berenshteyn, F.; Brivanlou, A.H. An efficient and reversible transposable system for gene delivery and lineage-specific differentiation in human embryonic stem cells. Cell Stem Cell 2009, 5, 332–342. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cadiñanos, J.; Bradley, A. Generation of an inducible and optimized piggyBac transposon system. Nucleic Acids Res. 2007, 35, e87. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, W.; Song, S.; Han, Y.; Chen, H.; Liu, X.; Qian, Q. An efficient Screening System in Yeast to Select a Hyperactive piggyBac Transposase for Mammalian Applications. Int. J. Mol. Sci. 2020, 21, 3064. [Google Scholar] [CrossRef]
- Calvi, B.R.; Hong, T.J.; Findley, S.D.; Gelbart, W.M. Evidence for a common evolutionary origin of inverted repeat transposons in Drosophila and plants: Hobo, Activator, and Tam3. Cell 1991, 66, 465–471. [Google Scholar] [CrossRef]
- McGinnis, W.; Shermoen, A.W.; Beckendorf, S.K. A transposable element inserted just 5’ to a Drosophila glue protein gene alters gene expression and chromatin structure. Cell 1983, 34, 75–84. [Google Scholar] [CrossRef]
- McClintock, B. Mutable Loci in Maize; Carnegie Institution of Washington Yearbook: Washington, DC, USA, 1948; pp. 155–169. [Google Scholar]
- Sommer, H.; Carpenter, R.; Harrison, B.J.; Saedler, H. The transposable element Tam3 of Antirrhinum majus generates a novel type of sequence alterations upon excision. Mol. Gen. Genet. MGG 1985, 199, 225–231. [Google Scholar] [CrossRef]
- Ni, J.; Wangensteen, K.J.; Nelsen, D.; Balciunas, D.; Skuster, K.J.; Urban, M.D.; Ekker, S.C. Active recombinant Tol2 transposase for gene transfer and gene discovery applications. Mobile DNA 2016, 7, 6. [Google Scholar] [CrossRef] [Green Version]
- Hickman, A.B.; Perez, Z.N.; Zhou, L.; Musingarimi, P.; Ghirlando, R.; Hinshaw, J.E.; Craig, N.L.; Dyda, F. Molecular architecture of a eukaryotic DNA transposase. Nat. Struct. Mol. Biol. 2005, 12, 715–721. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.-W.; Wessler, S.R. The catalytic domain of all eukaryotic cut-and-paste transposase superfamilies. Proc. Natl. Acad. Sci. USA 2011, 108, 7884. [Google Scholar] [CrossRef] [Green Version]
- Tsutsumi, M.; Koga, A.; Hori, H. Long and short mRnas transcribed from the medaka fish transposon Tol2 respectively exert positive and negative effects on excision. Genet. Res. 2003, 82, 33–40. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, K.; Koga, A.; Hori, H.; Shima, A. Excision of the tol2 transposable element of the medaka fish, Oryzias latipes, in zebrafish, Danio rerio. Gene 1998, 225, 17–22. [Google Scholar] [CrossRef]
- Huang, P.; Xu, L.; Liang, W.; Tam, C.I.; Zhang, Y.; Qi, F.; Zhu, Z.; Lin, S.; Zhang, B. Genomic deletion induced by Tol2 transposon excision in zebrafish. Nucleic Acids Res. 2013, 41, e36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grabundzija, I.; Irgang, M.; Mátés, L.; Belay, E.; Matrai, J.; Gogol-Döring, A.; Kawakami, K.; Chen, W.; Ruiz, P.; Chuah, M.K.L.; et al. Comparative analysis of transposable element vector systems in human cells. Mol. Ther. 2010, 18, 1200–1209. [Google Scholar] [CrossRef]
- Kondrychyn, I.; Garcia-Lecea, M.; Emelyanov, A.; Parinov, S.; Korzh, V. Genome-wide analysis of Tol2 transposon reintegration in zebrafish. BMC Genom. 2009, 10, 418. [Google Scholar] [CrossRef] [Green Version]
- Huang, X.; Guo, H.; Tammana, S.; Jung, Y.-C.; Mellgren, E.; Bassi, P.; Cao, Q.; Tu, Z.J.; Kim, Y.C.; Ekker, S.C.; et al. Gene transfer efficiency and genome-wide integration profiling of Sleeping Beauty, Tol2, and piggyBac transposons in human primary T cells. Mol. Ther. 2010, 18, 1803–1813. [Google Scholar] [CrossRef]
- Vrljicak, P.; Tao, S.; Varshney, G.K.; Quach, H.N.B.; Joshi, A.; LaFave, M.C.; Burgess, S.M.; Sampath, K. Genome-Wide Analysis of Transposon and Retroviral Insertions Reveals Preferential Integrations in Regions of DNA Flexibility. G3 Genes Genomes Genet. 2016, 6, 805. [Google Scholar] [CrossRef] [Green Version]
- Urasaki, A.; Morvan, G.; Kawakami, K. Functional dissection of the Tol2 transposable element identified the minimal cis-sequence and a highly repetitive sequence in the subterminal region essential for transposition. Genetics 2006, 174, 639–649. [Google Scholar] [CrossRef] [Green Version]
- Balciunas, D.; Wangensteen, K.J.; Wilber, A.; Bell, J.; Geurts, A.; Sivasubbu, S.; Wang, X.; Hackett, P.B.; Largaespada, D.A.; McIvor, R.S.; et al. Harnessing a high cargo-capacity transposon for genetic applications in vertebrates. PLoS Genet. 2006, 2, e169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsukahara, T.; Iwase, N.; Kawakami, K.; Iwasaki, M.; Yamamoto, C.; Ohmine, K.; Uchibori, R.; Teruya, T.; Ido, H.; Saga, Y.; et al. The Tol2 transposon system mediates the genetic engineering of T-cells with CD19-specific chimeric antigen receptors for B-cell malignancies. Gene Ther. 2015, 22, 209–215. [Google Scholar] [CrossRef] [PubMed]
- Suster, M.L.; Sumiyama, K.; Kawakami, K. Transposon-mediated BAC transgenesis in zebrafish and mice. BMC Genom. 2009, 10, 477. [Google Scholar] [CrossRef] [Green Version]
- Kawakami, K. Tol2: A versatile gene transfer vector in vertebrates. Genome Biol. 2007, 8, S7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keng, V.W.; Ryan, B.J.; Wangensteen, K.J.; Balciunas, D.; Schmedt, C.; Ekker, S.C.; Largaespada, D.A. Efficient transposition of Tol2 in the mouse germline. Genetics 2009, 183, 1565–1573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackey, A.S.; Redd, P.S.; DeLaurier, A.; Hancock, C.N. Codon optimized Tol2 transposase results in increased transient expression of a crystallin-GFP transgene in zebrafish. MicroPubl. Biol. 2020, 2020. [Google Scholar] [CrossRef]
- Yagita, K.; Yamanaka, I.; Emoto, N.; Kawakami, K.; Shimada, S. Real-time monitoring of circadian clock oscillations in primary cultures of mammalian cells using Tol2 transposon-mediated gene transfer strategy. BMC Biotechnol. 2010, 10, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paludan, S.R.; Bowie, A.G. Immune sensing of DNA. Immunity 2013, 38, 870–880. [Google Scholar] [CrossRef] [Green Version]
- Hemmi, H.; Takeuchi, O.; Kawai, T.; Kaisho, T.; Sato, S.; Sanjo, H.; Matsumoto, M.; Hoshino, K.; Wagner, H.; Takeda, K.; et al. A Toll-like receptor recognizes bacterial DNA. Nature 2000, 408, 740–745. [Google Scholar] [CrossRef]
- Murphy, D.B.; Epstein, S.L. Guidance for human somatic cell therapy and gene therapy. March 1998. Center for Biologics Evaluation and Research, Food and Drug Administration. Hum. Gene Ther. 1998, 9, 1513–1524. [Google Scholar] [CrossRef]
- Huang, X.; Haley, K.; Wong, M.; Guo, H.; Lu, C.; Wilber, A.; Zhou, X. Unexpectedly high copy number of random integration but low frequency of persistent expression of the Sleeping Beauty transposase after trans delivery in primary human T cells. Hum. Gene Ther. 2010, 21, 1577–1590. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.; Troilo, P.J.; Wang, X.; Griffiths, T.G.; Pacchione, S.J.; Barnum, A.B.; Harper, L.B.; Pauley, C.J.; Niu, Z.; Denisova, L.; et al. Detection of integration of plasmid DNA into host genomic DNA following intramuscular injection and electroporation. Gene Ther. 2004, 11, 711–721. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Kong, J.; Stalker, J.; Bradley, A. Chromosomal mobilization and reintegration of Sleeping Beauty and PiggyBac transposons. Genesis 2009, 47, 404–408. [Google Scholar] [CrossRef]
- Bishop, D.C.; Caproni, L.; Gowrishankar, K.; Legiewicz, M.; Karbowniczek, K.; Tite, J.; Gottlieb, D.J.; Micklethwaite, K.P. CAR T Cell Generation by piggyBac Transposition from Linear Doggybone DNA Vectors Requires Transposon DNA-Flanking Regions. Mol. Ther. Methods Clin. Dev. 2020, 17, 359–368. [Google Scholar] [CrossRef] [Green Version]
- Prommersberger, S.; Reiser, M.; Beckmann, J.; Danhof, S.; Amberger, M.; Quade-Lyssy, P.; Einsele, H.; Hudecek, M.; Bonig, H.; Ivics, Z. CARAMBA: A first-in-human clinical trial with SLAMF7 CAR-T cells prepared by virus-free Sleeping Beauty gene transfer to treat multiple myeloma. Gene Ther. 2021. [Google Scholar] [CrossRef]
- Marie, C.; Vandermeulen, G.; Quiviger, M.; Richard, M.; Préat, V.; Scherman, D. pFARs, plasmids free of antibiotic resistance markers, display high-level transgene expression in muscle, skin and tumour cells. J. Gene Med. 2010, 12, 323–332. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Garcia, L.; Recalde, S.; Hernandez, M.; Bezunartea, J.; Rodriguez-Madoz, J.R.; Johnen, S.; Diarra, S.; Marie, C.; Izsvák, Z.; Ivics, Z.; et al. Long-Term PEDF Release in Rat Iris and Retinal Epithelial Cells after Sleeping Beauty Transposon-Mediated Gene Delivery. Mol. Ther. Nucleic Acids 2017, 9, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hernandez, M.; Recalde, S.; Garcia-Garcia, L.; Bezunartea, J.; Miskey, C.; Johnen, S.; Diarra, S.; Sebe, A.; Rodriguez-Madoz, J.R.; Pouillot, S.; et al. Preclinical Evaluation of a Cell-Based Gene Therapy Using the Sleeping Beauty Transposon System in Choroidal Neovascularization. Mol. Ther. Methods Clin. Dev. 2019, 15, 403–417. [Google Scholar] [CrossRef] [Green Version]
- Darquet, A.M.; Cameron, B.; Wils, P.; Scherman, D.; Crouzet, J. A new DNA vehicle for nonviral gene delivery: Supercoiled minicircle. Gene Ther. 1997, 4, 1341–1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monjezi, R.; Miskey, C.; Gogishvili, T.; Schleef, M.; Schmeer, M.; Einsele, H.; Ivics, Z.; Hudecek, M. Enhanced CAR T-cell engineering using non-viral Sleeping Beauty transposition from minicircle vectors. Leukemia 2017, 31, 186–194. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Chen, Y.; Zhao, S.; Guan, K.-L.; Zhuang, Y.; Zhou, W.; Wu, X.; Xu, T. DNA-PK facilitates piggyBac transposition by promoting paired-end complex formation. Proc. Natl. Acad. Sci. USA 2017, 114, 7408–7413. [Google Scholar] [CrossRef] [Green Version]
- Walters, A.A.; Kinnear, E.; Shattock, R.J.; McDonald, J.U.; Caproni, L.J.; Porter, N.; Tregoning, J.S. Comparative analysis of enzymatically produced novel linear DNA constructs with plasmids for use as DNA vaccines. Gene Ther. 2014, 21, 645–652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinmyo, Y.; Mito, T.; Matsushita, T.; Sarashina, I.; Miyawaki, K.; Ohuchi, H.; Noji, S. piggyBac-mediated somatic transformation of the two-spotted cricket, Gryllus bimaculatus. Dev. Growth Differ. 2004, 46, 343–349. [Google Scholar] [CrossRef] [PubMed]
- Wilber, A.; Frandsen, J.L.; Geurts, J.L.; Largaespada, D.A.; Hackett, P.B.; McIvor, R.S. RNA as a source of transposase for Sleeping Beauty-mediated gene insertion and expression in somatic cells and tissues. Mol. Ther. 2006, 13, 625–630. [Google Scholar] [CrossRef]
- Wilber, A.; Wangensteen, K.J.; Chen, Y.; Zhuo, L.; Frandsen, J.L.; Bell, J.B.; Chen, Z.J.; Ekker, S.C.; McIvor, R.S.; Wang, X. Messenger RNA as a source of transposase for sleeping beauty transposon-mediated correction of hereditary tyrosinemia type I. Mol. Ther. 2007, 15, 1280–1287. [Google Scholar] [CrossRef]
- Wiehe, J.M.; Ponsaerts, P.; Rojewski, M.T.; Homann, J.M.; Greiner, J.; Kronawitter, D.; Schrezenmeier, H.; Hombach, V.; Wiesneth, M.; Zimmermann, O.; et al. mRNA-mediated gene delivery into human progenitor cells promotes highly efficient protein expression. J. Cell. Mol. Med. 2007, 11, 521–530. [Google Scholar] [CrossRef]
- Galla, M.; Schambach, A.; Falk, C.S.; Maetzig, T.; Kuehle, J.; Lange, K.; Zychlinski, D.; Heinz, N.; Brugman, M.H.; Göhring, G.; et al. Avoiding cytotoxicity of transposases by dose-controlled mRNA delivery. Nucleic Acids Res. 2011, 39, 7147–7160. [Google Scholar] [CrossRef]
- Kormann, M.S.D.; Hasenpusch, G.; Aneja, M.K.; Nica, G.; Flemmer, A.W.; Herber-Jonat, S.; Huppmann, M.; Mays, L.E.; Illenyi, M.; Schams, A.; et al. Expression of therapeutic proteins after delivery of chemically modified mRNA in mice. Nat. Biotechnol. 2011, 29, 154–157. [Google Scholar] [CrossRef] [PubMed]
- Järver, P.; Fernaeus, S.; EL-Andaloussi, S.; Tjörnhammar, M.-L.; Langel, Ü. Co-transduction of Sleeping Beauty Transposase and Donor Plasmid via a Cell-penetrating Peptide: A simple one step Method. Int. J. Pept. Res. Ther. 2008, 14, 58–63. [Google Scholar] [CrossRef]
- Lee, C.-Y.; Li, J.-F.; Liou, J.-S.; Charng, Y.-C.; Huang, Y.-W.; Lee, H.-J. A gene delivery system for human cells mediated by both a cell-penetrating peptide and a piggyBac transposase. Biomaterials 2011, 32, 6264–6276. [Google Scholar] [CrossRef] [PubMed]
- La, Q.T.; Ren, B.; Logan, G.J.; Cunningham, S.C.; Khandekar, N.; Nassif, N.T.; O’Brien, B.A.; Alexander, I.E.; Simpson, A.M. Use of a Hybrid Adeno-Associated Viral Vector Transposon System to Deliver the Insulin Gene to Diabetic NOD Mice. Cells 2020, 9, 2227. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, S.C.; Siew, S.M.; Hallwirth, C.V.; Bolitho, C.; Sasaki, N.; Garg, G.; Michael, I.P.; Hetherington, N.A.; Carpenter, K.; de Alencastro, G.; et al. Modeling correction of severe urea cycle defects in the growing murine liver using a hybrid recombinant adeno-associated virus/piggyBac transposase gene delivery system. Hepatology 2015, 62, 417–428. [Google Scholar] [CrossRef]
- Siew, S.M.; Cunningham, S.C.; Zhu, E.; Tay, S.S.; Venuti, E.; Bolitho, C.; Alexander, I.E. Prevention of Cholestatic Liver Disease and Reduced Tumorigenicity in a Murine Model of PFIC Type 3 Using Hybrid AAV-piggyBac Gene Therapy. Hepatology 2019, 70, 2047–2061. [Google Scholar] [CrossRef]
- Cooney, A.L.; Thornell, I.M.; Singh, B.K.; Shah, V.S.; Stoltz, D.A.; McCray, P.B.; Zabner, J.; Sinn, P.L. A Novel AAV-mediated Gene Delivery System Corrects CFTR Function in Pigs. Am. J. Respir. Cell Mol. Biol. 2019, 61, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Tipanee, J.; VandenDriessche, T.; Chuah, M.K. Transposons: Moving Forward from Preclinical Studies to Clinical Trials. Hum. Gene Ther. 2017, 28, 1087–1104. [Google Scholar] [CrossRef] [Green Version]
- Kebriaei, P.; Singh, H.; Huls, M.H.; Figliola, M.J.; Bassett, R.; Olivares, S.; Jena, B.; Dawson, M.J.; Kumaresan, P.R.; Su, S.; et al. Phase I trials using Sleeping Beauty to generate CD19-specific CAR T cells. J. Clin. Investig. 2016, 126, 3363–3376. [Google Scholar] [CrossRef]
- Smith, R.P.; Riordan, J.D.; Feddersen, C.R.; Dupuy, A.J. A Hybrid Adenoviral Vector System Achieves Efficient Long-Term Gene Expression in the Liver via piggyBac Transposition. Hum. Gene Ther. 2015, 26, 377–385. [Google Scholar] [CrossRef] [Green Version]
- Pan, Y.; Lv, J.; Pan, D.; Yang, M.; Ju, H.; Zhou, J.; Zhu, L.; Zhang, Y. Retrofitting baculoviral vector with Sleeping Beauty transposon system: Competent for long-term reporter gene imaging in vivo. Appl. Microbiol. Biotechnol. 2018, 102, 1933–1943. [Google Scholar] [CrossRef]
- Chen, C.-L.; Tseng, Y.-W.; Wu, J.-C.; Chen, G.-Y.; Lin, K.-C.; Hwang, S.-M.; Hu, Y.-C. Suppression of hepatocellular carcinoma by baculovirus-mediated expression of long non-coding RNA PTENP1 and MicroRNA regulation. Biomaterials 2015, 44, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.-L.; Wu, J.-C.; Chen, G.-Y.; Yuan, P.-H.; Tseng, Y.-W.; Li, K.-C.; Hwang, S.-M.; Hu, Y.-C. Baculovirus-mediated miRNA regulation to suppress hepatocellular carcinoma tumorigenicity and metastasis. Mol. Ther. 2015, 23, 79–88. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.-L.; Luo, W.-Y.; Lo, W.-H.; Lin, K.-J.; Sung, L.-Y.; Shih, Y.-S.; Chang, Y.-H.; Hu, Y.-C. Development of hybrid baculovirus vectors for artificial MicroRNA delivery and prolonged gene suppression. Biotechnol. Bioeng. 2011, 108, 2958–2967. [Google Scholar] [CrossRef]
- Boehme, P.; Zhang, W.; Solanki, M.; Ehrke-Schulz, E.; Ehrhardt, A. A High-Capacity Adenoviral Hybrid Vector System Utilizing the Hyperactive Sleeping Beauty Transposase SB100X for Enhanced Integration. Mol. Ther. Nucleic Acids 2016, 5, e337. [Google Scholar] [CrossRef] [Green Version]
- Richter, M.; Saydaminova, K.; Yumul, R.; Krishnan, R.; Liu, J.; Nagy, E.-E.; Singh, M.; Izsvák, Z.; Cattaneo, R.; Uckert, W.; et al. In vivo transduction of primitive mobilized hematopoietic stem cells after intravenous injection of integrating adenovirus vectors. Blood 2016, 128, 2206–2217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skipper, K.A.; Nielsen, M.G.; Andersen, S.; Ryø, L.B.; Bak, R.O.; Mikkelsen, J.G. Time-Restricted PiggyBac DNA Transposition by Transposase Protein Delivery Using Lentivirus-Derived Nanoparticles. Mol. Ther. Nucleic Acids 2018, 11, 253–262. [Google Scholar] [CrossRef] [Green Version]
- Kren, B.T.; Unger, G.M.; Sjeklocha, L.; Trossen, A.A.; Korman, V.; Diethelm-Okita, B.M.; Reding, M.T.; Steer, C.J. Nanocapsule-delivered Sleeping Beauty mediates therapeutic Factor VIII expression in liver sinusoidal endothelial cells of hemophilia A mice. J. Clin. Investig. 2009, 119, 2086–2099. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, S.; Munder, A.; Hedtfeld, S.; Braubach, P.; Glage, S.; Zhang, L.; Lienenklaus, S.; Schultze, A.; Hasenpusch, G.; Garrels, W.; et al. Self-assembled peptide–poloxamine nanoparticles enable in vitro and in vivo genome restoration for cystic fibrosis. Nat. Nanotechnol. 2019, 14, 287–297. [Google Scholar] [CrossRef] [PubMed]
- Smith, T.T.; Stephan, S.B.; Moffett, H.F.; McKnight, L.E.; Ji, W.; Reiman, D.; Bonagofski, E.; Wohlfahrt, M.E.; Pillai, S.P.S.; Stephan, M.T. In situ programming of leukaemia-specific T cells using synthetic DNA nanocarriers. Nat. Nanotechnol. 2017, 12, 813–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parinov, S.; Kondrichin, I.; Korzh, V.; Emelyanov, A. Tol2 transposon-mediated enhancer trap to identify developmentally regulated zebrafish genes in vivo. Dev. Dyn. 2004, 231, 449–459. [Google Scholar] [CrossRef]
- Beckmann, P.J.; Largaespada, D.A. Transposon Insertion Mutagenesis in Mice for Modeling Human Cancers: Critical Insights Gained and New Opportunities. Int. J. Mol. Sci. 2020, 21, 1172. [Google Scholar] [CrossRef] [Green Version]
- Chang, H.; Pan, Y.; Landrette, S.; Ding, S.; Yang, D.; Liu, L.; Tian, L.; Chai, H.; Li, P.; Li, D.-M.; et al. Efficient genome-wide first-generation phenotypic screening system in mice using the piggyBac transposon. Proc. Natl. Acad. Sci. USA 2019, 116, 18507–18516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levitan, A.; Gale, A.N.; Dallon, E.K.; Kozan, D.W.; Cunningham, K.W.; Sharan, R.; Berman, J. Comparing the utility of in vivo transposon mutagenesis approaches in yeast species to infer gene essentiality. Curr. Genet. 2020, 66, 1117–1134. [Google Scholar] [CrossRef] [PubMed]
- Kawakami, K.; Takeda, H.; Kawakami, N.; Kobayashi, M.; Matsuda, N.; Mishina, M. A transposon-mediated gene trap approach identifies developmentally regulated genes in zebrafish. Dev. Cell 2004, 7, 133–144. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dooley, K. Zebrafish: A model system for the study of human disease. Curr. Opin. Genet. Dev. 2000, 10, 252–256. [Google Scholar] [CrossRef]
- Rafferty, S.A.; Quinn, T.A. A beginner’s guide to understanding and implementing the genetic modification of zebrafish. Prog. Biophys. Mol. Biol. 2018, 138, 3–19. [Google Scholar] [CrossRef] [PubMed]
- Gordon, J.W.; Scangos, G.A.; Plotkin, D.J.; Barbosa, J.A.; Ruddle, F.H. Genetic transformation of mouse embryos by microinjection of purified DNA. Proc. Natl. Acad. Sci. USA 1980, 77, 7380–7384. [Google Scholar] [CrossRef] [Green Version]
- Brinster, R.L.; Chen, H.Y.; Trumbauer, M.E.; Yagle, M.K.; Palmiter, R.D. Factors affecting the efficiency of introducing foreign DNA into mice by microinjecting eggs. Proc. Natl. Acad. Sci. USA 1985, 82, 4438–4442. [Google Scholar] [CrossRef] [Green Version]
- Bishop, J.O.; Smith, P. Mechanism of chromosomal integration of microinjected DNA. Mol. Biol. Med. 1989, 6, 283–298. [Google Scholar]
- Cousens, C.; Carver, A.S.; Wilmut, I.; Colman, A.; Garner, I.; O’Neill, G.T. Use of PCR-based methods for selection of integrated transgenes in preimplantation embryos. Mol. Reprod. Dev. 1994, 39, 384–391. [Google Scholar] [CrossRef]
- Garrick, D.; Fiering, S.; Martin, D.I.; Whitelaw, E. Repeat-induced gene silencing in mammals. Nat. Genet. 1998, 18, 56–59. [Google Scholar] [CrossRef]
- Whitelaw, C.B.; Springbett, A.J.; Webster, J.; Clark, J. The majority of G0 transgenic mice are derived from mosaic embryos. Transgenic Res. 1993, 2, 29–32. [Google Scholar] [CrossRef]
- Covarrubias, L.; Nishida, Y.; Mintz, B. Early postimplantation embryo lethality due to DNA rearrangements in a transgenic mouse strain. Proc. Natl. Acad. Sci. USA 1986, 83, 6020–6024. [Google Scholar] [CrossRef] [Green Version]
- Kohrman, D.C.; Plummer, N.W.; Schuster, T.; Jones, J.M.; Jang, W.; Burgess, D.L.; Galt, J.; Spear, B.T.; Meisler, M.H. Insertional mutation of the motor endplate disease (med) locus on mouse chromosome 15. Genomics 1995, 26, 171–177. [Google Scholar] [CrossRef]
- Wilkie, T.M.; Palmiter, R.D. Analysis of the integrant in MyK-103 transgenic mice in which males fail to transmit the integrant. Mol. Cell. Biol. 1987, 7, 1646–1655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davidson, A.E.; Balciunas, D.; Mohn, D.; Shaffer, J.; Hermanson, S.; Sivasubbu, S.; Cliff, M.P.; Hackett, P.B.; Ekker, S.C. Efficient gene delivery and gene expression in zebrafish using the Sleeping Beauty transposon. Dev. Biol. 2003, 263, 191–202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, D.; Xue, S.; Chan, S.; Sang, Y.; Wang, S.; Wang, Y.; Chen, C.; Gao, B.; Mueller, F.; Song, C. Enhancer Trapping and Annotation in Zebrafish Mediated with Sleeping Beauty, piggyBac and Tol2 Transposons. Genes (Basel) 2018, 9, 630. [Google Scholar] [CrossRef] [Green Version]
- Dupuy, A.J.; Clark, K.; Carlson, C.M.; Fritz, S.; Davidson, A.E.; Markley, K.M.; Finley, K.; Fletcher, C.F.; Ekker, S.C.; Hackett, P.B.; et al. Mammalian germ-line transgenesis by transposition. Proc. Natl. Acad. Sci. USA 2002, 99, 4495. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sumiyama, K.; Kawakami, K.; Yagita, K. A simple and highly efficient transgenesis method in mice with the Tol2 transposon system and cytoplasmic microinjection. Genomics 2010, 95, 306–311. [Google Scholar] [CrossRef]
- Li, T.; Shuai, L.; Mao, J.; Wang, X.; Wang, M.; Zhang, X.; Wang, L.; Li, Y.; Li, W.; Zhou, Q. Efficient Production of Fluorescent Transgenic Rats using the piggyBac Transposon. Sci. Rep. 2016, 6, 33225. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Zeng, F.; Meng, F.; Xu, Z.; Zhang, X.; Huang, X.; Tang, F.; Gao, W.; Shi, J.; He, X.; et al. Generation of transgenic pigs by cytoplasmic injection of piggyBac transposase-based pmGENIE-3 plasmids. Biol. Reprod. 2014, 90, 93. [Google Scholar] [CrossRef] [Green Version]
- Yum, S.-Y.; Lee, S.-J.; Kim, H.-M.; Choi, W.-J.; Park, J.-H.; Lee, W.-W.; Kim, H.-S.; Kim, H.-J.; Bae, S.-H.; Lee, J.-H.; et al. Efficient generation of transgenic cattle using the DNA transposon and their analysis by next-generation sequencing. Sci. Rep. 2016, 6, 27185. [Google Scholar] [CrossRef]
- Zakrzewski, W.; Dobrzyński, M.; Szymonowicz, M.; Rybak, Z. Stem cells: Past, present, and future. Stem Cell Res. Ther. 2019, 10, 68. [Google Scholar] [CrossRef]
- Wu, S.C.-Y.; Meir, Y.-J.J.; Coates, C.J.; Handler, A.M.; Pelczar, P.; Moisyadi, S.; Kaminski, J.M. piggyBac is a flexible and highly active transposon as compared to sleeping beauty, Tol2, and Mos1 in mammalian cells. Proc. Natl. Acad. Sci. USA 2006, 103, 15008–15013. [Google Scholar] [CrossRef] [Green Version]
- Davis, R.P.; Nemes, C.; Varga, E.; Freund, C.; Kosmidis, G.; Gkatzis, K.; de Jong, D.; Szuhai, K.; Dinnyés, A.; Mummery, C.L. Generation of induced pluripotent stem cells from human foetal fibroblasts using the Sleeping Beauty transposon gene delivery system. Differentiation 2013, 86, 30–37. [Google Scholar] [CrossRef]
- Yu, J.; Hu, K.; Smuga-Otto, K.; Tian, S.; Stewart, R.; Slukvin, I.I.; Thomson, J.A. Human induced pluripotent stem cells free of vector and transgene sequences. Science 2009, 324, 797–801. [Google Scholar] [CrossRef] [Green Version]
- Jia, F.; Wilson, K.D.; Sun, N.; Gupta, D.M.; Huang, M.; Li, Z.; Panetta, N.J.; Chen, Z.Y.; Robbins, R.C.; Kay, M.A.; et al. A nonviral minicircle vector for deriving human iPS cells. Nat. Methods 2010, 7, 197–199. [Google Scholar] [CrossRef] [Green Version]
- Kumar, D.; Anand, T.; Talluri, T.R.; Kues, W.A. Potential of transposon-mediated cellular reprogramming towards cell-based therapies. World J. Stem Cells 2020, 12, 527–544. [Google Scholar] [CrossRef]
- Li, M.A.; Pettitt, S.J.; Eckert, S.; Ning, Z.; Rice, S.; Cadiñanos, J.; Yusa, K.; Conte, N.; Bradley, A. The piggyBac transposon displays local and distant reintegration preferences and can cause mutations at noncanonical integration sites. Mol. Cell. Biol. 2013, 33, 1317–1330. [Google Scholar] [CrossRef] [Green Version]
- Woltjen, K.; Michael, I.P.; Mohseni, P.; Desai, R.; Mileikovsky, M.; Hämäläinen, R.; Cowling, R.; Wang, W.; Liu, P.; Gertsenstein, M.; et al. piggyBac transposition reprograms fibroblasts to induced pluripotent stem cells. Nature 2009, 458, 766–770. [Google Scholar] [CrossRef]
- Itoh, M.; Kawagoe, S.; Tamai, K.; Nakagawa, H.; Asahina, A.; Okano, H.J. Footprint-free gene mutation correction in induced pluripotent stem cell (iPSC) derived from recessive dystrophic epidermolysis bullosa (RDEB) using the CRISPR/Cas9 and piggyBac transposon system. J. Dermatol. Sci. 2020, 98, 163–172. [Google Scholar] [CrossRef]
- Teque, F.; Ye, L.; Xie, F.; Wang, J.; Morvan, M.G.; Kan, Y.W.; Levy, J.A. Genetically-edited induced pluripotent stem cells derived from HIV-1-infected patients on therapy can give rise to immune cells resistant to HIV-1 infection. AIDS 2020, 34, 1141–1149. [Google Scholar] [CrossRef] [PubMed]
- Hütter, G.; Nowak, D.; Mossner, M.; Ganepola, S.; Müssig, A.; Allers, K.; Schneider, T.; Hofmann, J.; Kücherer, C.; Blau, O.; et al. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N. Engl. J. Med. 2009, 360, 692–698. [Google Scholar] [CrossRef] [Green Version]
- Neumeyer, J.; Lin, R.-Z.; Wang, K.; Hong, X.; Hua, T.; Croteau, S.E.; Neufeld, E.J.; Melero-Martin, J.M. Bioengineering hemophilia A-specific microvascular grafts for delivery of full-length factor VIII into the bloodstream. Blood Adv. 2019, 3, 4166–4176. [Google Scholar] [CrossRef]
- Iyer, P.S.; Mavoungou, L.O.; Ronzoni, F.; Zemla, J.; Schmid-Siegert, E.; Antonini, S.; Neff, L.A.; Dorchies, O.M.; Jaconi, M.; Lekka, M.; et al. Autologous Cell Therapy Approach for Duchenne Muscular Dystrophy using PiggyBac Transposons and Mesoangioblasts. Mol. Ther. 2018, 26, 1093–1108. [Google Scholar] [CrossRef] [Green Version]
- Xiao, J.; Meng, X.-M.; Huang, X.R.; Chung, A.C.; Feng, Y.-L.; Hui, D.S.; Yu, C.-M.; Sung, J.J.; Lan, H.Y. miR-29 inhibits bleomycin-induced pulmonary fibrosis in mice. Mol. Ther. 2012, 20, 1251–1260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.J.; Kren, B.T.; Wong, P.Y.-P.; Low, W.C.; Steer, C.J. Sleeping Beauty-mediated down-regulation of huntingtin expression by RNA interference. Biochem. Biophys. Res. Commun. 2005, 329, 646–652. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Liu, H.; Visner, G.; Fletcher, B.S. Sleeping Beauty-mediated eNOS gene therapy attenuates monocrotaline-induced pulmonary hypertension in rats. FASEB J. 2006, 20, 2594–2596. [Google Scholar] [CrossRef] [PubMed]
- Thumann, G.; Harmening, N.; Prat-Souteyrand, C.; Marie, C.; Pastor, M.; Sebe, A.; Miskey, C.; Hurst, L.D.; Diarra, S.; Kropp, M.; et al. Engineering of PEDF-Expressing Primary Pigment Epithelial Cells by the SB Transposon System Delivered by pFAR4 Plasmids. Mol. Ther. Nucleic Acids 2017, 6, 302–314. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bascuas, T.; Kropp, M.; Harmening, N.; Wong, B.M.; Johnen, S.; Izsvák, Z.; Thumann, G. Isolation, Culture, and Genetic Engineering of Mammalian Primary Pigment Epithelial Cells for Non-Viral Gene Therapy. J. Vis. Exp. 2021. [Google Scholar] [CrossRef]
- Belur, L.R.; Podetz-Pedersen, K.M.; Sorenson, B.S.; Hsu, A.H.; Parker, J.B.; Carlson, C.S.; Saltzman, D.A.; Ramakrishnan, S.; McIvor, R.S. Inhibition of angiogenesis and suppression of colorectal cancer metastatic to the liver using the Sleeping Beauty Transposon System. Mol. Cancer 2011, 10, 14. [Google Scholar] [CrossRef] [Green Version]
- Ohlfest, J.R.; Demorest, Z.L.; Motooka, Y.; Vengco, I.; Oh, S.; Chen, E.; Scappaticci, F.A.; Saplis, R.J.; Ekker, S.C.; Low, W.C.; et al. Combinatorial antiangiogenic gene therapy by nonviral gene transfer using the sleeping beauty transposon causes tumor regression and improves survival in mice bearing intracranial human glioblastoma. Mol. Ther. 2005, 12, 778–788. [Google Scholar] [CrossRef]
- Bagheri-Mohammadi, S.; Moradian-Tehrani, R.; Noureddini, M.; Alani, B. Novel application of adipose-derived mesenchymal stem cells via producing antiangiogenic factor TSP-1 in lung metastatic melanoma animal model. Biologicals 2020, 68, 9–18. [Google Scholar] [CrossRef]
- Song, J.S.; Kim, C.W.; Ochoa, E.R. Sleeping Beauty-mediated suicide gene therapy of hepatocellular carcinoma. Biosci. Biotechnol. Biochem. 2009, 73, 165–168. [Google Scholar] [CrossRef] [Green Version]
- Gross, G.; Waks, T.; Eshhar, Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc. Natl. Acad. Sci. USA 1989, 86, 10024–10028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feins, S.; Kong, W.; Williams, E.F.; Milone, M.C.; Fraietta, J.A. An introduction to chimeric antigen receptor (CAR) T-cell immunotherapy for human cancer. Am. J. Hematol. 2019, 94, S3–S9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magnani, C.F.; Tettamanti, S.; Alberti, G.; Pisani, I.; Biondi, A.; Serafini, M.; Gaipa, G. Transposon-Based CAR T Cells in Acute Leukemias: Where Are We Going? Cells 2020, 9, 1337. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, K.; Kato, I.; Tanaka, M.; Morita, D.; Matsuda, K.; Takahashi, Y.; Nakahata, T.; Umeda, K.; Hiramatsu, H.; Adachi, S.; et al. Direct Delivery of piggyBac CD19 CAR T Cells Has Potent Anti-tumor Activity against ALL Cells in CNS in a Xenograft Mouse Model. Mol. Ther. Oncolytics 2020, 18, 37–46. [Google Scholar] [CrossRef]
- Štach, M.; Ptáčková, P.; Mucha, M.; Musil, J.; Klener, P.; Otáhal, P. Inducible secretion of IL-21 augments anti-tumor activity of piggyBac-manufactured chimeric antigen receptor T cells. Cytotherapy 2020, 22, 744–754. [Google Scholar] [CrossRef]
- Morokawa, H.; Yagyu, S.; Hasegawa, A.; Tanaka, M.; Saito, S.; Mochizuki, H.; Sakamoto, K.; Shimoi, A.; Nakazawa, Y. Autologous non-human primate model for safety assessment of piggyBac transposon-mediated chimeric antigen receptor T cells on granulocyte-macrophage colony-stimulating factor receptor. Clin. Transl. Immunol. 2020, 9, e1207. [Google Scholar] [CrossRef]
- Li, H.; Huang, Y.; Jiang, D.-Q.; Cui, L.-Z.; He, Z.; Wang, C.; Zhang, Z.-W.; Zhu, H.-L.; Ding, Y.-M.; Li, L.-F.; et al. Antitumor activity of EGFR-specific CAR T cells against non-small-cell lung cancer cells in vitro and in mice. Cell Death Dis. 2018, 9, 177. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.; Qin, W.; Liu, T.; Jiang, D.; Cui, L.; Liu, X.; Fang, Y.; Tang, X.; Jin, H.; Qian, Q. PiggyBac-engineered T cells expressing a glypican-3-specific chimeric antigen receptor show potent activities against hepatocellular carcinoma. Immunobiology 2020, 225, 151850. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Chen, Y.; Yan, L.; Cao, H.-X.; Han, S.-Y.; Cui, J.-J.; Wen, J.G.; Zheng, Y. EGFRvIII-specific CAR-T cells produced by piggyBac transposon exhibit efficient growth suppression against hepatocellular carcinoma. Int. J. Med. Sci. 2020, 17, 1406–1414. [Google Scholar] [CrossRef]
- Zhang, Z.; Jiang, D.; Yang, H.; He, Z.; Liu, X.; Qin, W.; Li, L.; Wang, C.; Li, Y.; Li, H.; et al. Modified CAR T cells targeting membrane-proximal epitope of mesothelin enhances the antitumor function against large solid tumor. Cell Death Dis. 2019, 10, 476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Batchu, R.B.; Gruzdyn, O.V.; Tavva, P.S.; Kolli, B.K.; Dachepalli, R.; Weaver, D.W.; Gruber, S.A. Engraftment of mesothelin chimeric antigen receptor using a hybrid Sleeping Beauty/minicircle vector into NK-92MI cells for treatment of pancreatic cancer. Surgery 2019, 166, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Lupo, K.B.; Chambers, A.M.; Matosevic, S. Purinergic targeting enhances immunotherapy of CD73+ solid tumors with piggyBac-engineered chimeric antigen receptor natural killer cells. J. Immunother. Cancer 2018, 6, 136. [Google Scholar] [CrossRef]
- Magnani, C.F.; Turazzi, N.; Benedicenti, F.; Calabria, A.; Tenderini, E.; Tettamanti, S.; Giordano Attianese, G.M.P.; Cooper, L.J.N.; Aiuti, A.; Montini, E.; et al. Immunotherapy of acute leukemia by chimeric antigen receptor-modified lymphocytes using an improved Sleeping Beauty transposon platform. Oncotarget 2016, 7, 51581–51597. [Google Scholar] [CrossRef] [PubMed]
- Rotiroti, M.C.; Buracchi, C.; Arcangeli, S.; Galimberti, S.; Valsecchi, M.G.; Perriello, V.M.; Rasko, T.; Alberti, G.; Magnani, C.F.; Cappuzzello, C.; et al. Targeting CD33 in Chemoresistant AML Patient-Derived Xenografts by CAR-CIK Cells Modified with an Improved SB Transposon System. Mol. Ther. 2020, 28, 1974–1986. [Google Scholar] [CrossRef]
- Clauss, J.; Obenaus, M.; Miskey, C.; Ivics, Z.; Izsvák, Z.; Uckert, W.; Bunse, M. Efficient Non-Viral T-Cell Engineering by Sleeping Beauty Minicircles Diminishing DNA Toxicity and miRNAs Silencing the Endogenous T-Cell Receptors. Hum. Gene Ther. 2018, 29, 569–584. [Google Scholar] [CrossRef]
- Singh, H.; Manuri, P.R.; Olivares, S.; Dara, N.; Dawson, M.J.; Huls, H.; Hackett, P.B.; Kohn, D.B.; Shpall, E.J.; Champlin, R.E.; et al. Redirecting specificity of T-cell populations for CD19 using the Sleeping Beauty system. Cancer Res. 2008, 68, 2961–2971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, T.; Shi, T.; Zhang, H.; Hu, J.; Song, Y.; Wei, J.; Ren, S.; Zhou, C. Tumor neoantigens: From basic research to clinical applications. J. Hematol. Oncol. 2019, 12, 93. [Google Scholar] [CrossRef] [Green Version]
- Deniger, D.C.; Pasetto, A.; Tran, E.; Parkhurst, M.R.; Cohen, C.J.; Robbins, P.F.; Cooper, L.J.; Rosenberg, S.A. Stable, Nonviral Expression of Mutated Tumor Neoantigen-specific T-cell Receptors Using the Sleeping Beauty Transposon/Transposase System. Mol. Ther. 2016, 24, 1078–1089. [Google Scholar] [CrossRef] [Green Version]
- Cuello, A.C.; Bruno, M.A.; Allard, S.; Leon, W.; Iulita, M.F. Cholinergic Involvement in Alzheimer’s Disease. A Link with NGF Maturation and Degradation. J. Mol. Neurosci. 2010, 40, 230–235. [Google Scholar] [CrossRef]
- Eyjolfsdottir, H.; Eriksdotter, M.; Linderoth, B.; Lind, G.; Juliusson, B.; Kusk, P.; Almkvist, O.; Andreasen, N.; Blennow, K.; Ferreira, D.; et al. Targeted delivery of nerve growth factor to the cholinergic basal forebrain of Alzheimer’s disease patients: Application of a second-generation encapsulated cell biodelivery device. Alzheimer’s Res. Ther. 2016, 8, 30. [Google Scholar] [CrossRef] [Green Version]
- Bishop, D.C.; Clancy, L.E.; Burgess, J.; Mathew, G.; Atkins, E.; Advic, S.; Maddock, K.; Street, J.; Moezzi, L.; Simms, R.; et al. Matched sibling donor-derived piggybac CAR19 T cells induce remission of relapsed/refractory CD19+ malignancy following haematopoietic stem cell transplant. Cytotherapy 2019, 21, S9. [Google Scholar] [CrossRef]
- Gregory, T.; Cohen, A.D.; Costello, C.L.; Ali, S.A.; Berdeja, J.G.; Ostertag, E.M.; Martin, C.; Shedlock, D.J.; Resler, M.L.; Spear, M.A.; et al. Efficacy and Safety of P-Bcma-101 CAR-T Cells in Patients with Relapsed/Refractory (r/r) Multiple Myeloma (MM). Blood 2018, 132, 1012. [Google Scholar] [CrossRef]
- Costello, C.L.; Gregory, T.K.; Ali, S.A.; Berdeja, J.G.; Patel, K.K.; Shah, N.D.; Ostertag, E.; Martin, C.; Ghoddusi, M.; Shedlock, D.J.; et al. Phase 2 Study of the Response and Safety of P-Bcma-101 CAR-T Cells in Patients with Relapsed/Refractory (r/r) Multiple Myeloma (MM) (PRIME). Blood 2019, 134, 3184. [Google Scholar] [CrossRef]
- De Wit, T.; Dekker, S.; Maas, A.; Breedveld, G.; Knoch, T.A.; Langeveld, A.; Szumska, D.; Craig, R.; Bhattacharya, S.; Grosveld, F.; et al. Tagged mutagenesis by efficient Minos-based germ line transposition. Mol. Cell. Biol. 2010, 30, 68–77. [Google Scholar] [CrossRef] [Green Version]
- Zagoraiou, L.; Drabek, D.; Alexaki, S.; Guy, J.A.; Klinakis, A.G.; Langeveld, A.; Skavdis, G.; Mamalaki, C.; Grosveld, F.; Savakis, C. In vivo transposition of Minos, a Drosophila mobile element, in mammalian tissues. Proc. Natl. Acad. Sci. USA 2001, 98, 11474–11478. [Google Scholar] [CrossRef] [Green Version]
- Sasakura, Y.; Awazu, S.; Chiba, S.; Satoh, N. Germ-line transgenesis of the Tc1/mariner superfamily transposon Minos in Ciona intestinalis. Proc. Natl. Acad. Sci. USA 2003, 100, 7726–7730. [Google Scholar] [CrossRef] [Green Version]
- Miskey, C.; Izsvák, Z.; Plasterk, R.H.; Ivics, Z. The Frog Prince: A reconstructed transposon from Rana pipiens with high transpositional activity in vertebrate cells. Nucleic Acids Res. 2003, 31, 6873–6881. [Google Scholar] [CrossRef] [Green Version]
- Sinzelle, L.; Kapitonov, V.V.; Grzela, D.P.; Jursch, T.; Jurka, J.; Izsvák, Z.; Ivics, Z. Transposition of a reconstructed Harbinger element in human cells and functional homology with two transposon-derived cellular genes. Proc. Natl. Acad. Sci. USA 2008, 105, 4715–4720. [Google Scholar] [CrossRef] [Green Version]
- Koga, A.; Cheah, F.S.H.; Hamaguchi, S.; Yeo, G.H.; Chong, S.S. Germline transgenesis of zebrafish using the medaka Tol1 transposon system. Dev. Dyn. 2008, 237, 2466–2474. [Google Scholar] [CrossRef]
- Grabundzija, I.; Messing, S.A.; Thomas, J.; Cosby, R.L.; Bilic, I.; Miskey, C.; Gogol-Döring, A.; Kapitonov, V.; Diem, T.; Dalda, A.; et al. A Helitron transposon reconstructed from bats reveals a novel mechanism of genome shuffling in eukaryotes. Nat. Commun. 2016, 7, 10716. [Google Scholar] [CrossRef] [Green Version]
- Shen, D.; Song, C.; Miskey, C.; Chan, S.; Guan, Z.; Sang, Y.; Wang, Y.; Chen, C.; Wang, X.; Müller, F.; et al. A native, highly active Tc1/mariner transposon from zebrafish (ZB) offers an efficient genetic manipulation tool for vertebrates. Nucleic Acids Res. 2021, 49, 2126–2140. [Google Scholar] [CrossRef]
- Lundstrom, K. Viral Vectors in Gene Therapy. Diseases 2018, 6, 42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moleirinho, M.G.; Silva, R.J.S.; Alves, P.M.; Carrondo, M.J.T.; Peixoto, C. Current challenges in biotherapeutic particles manufacturing. Expert Opin. Biol. Ther. 2020, 20, 451–465. [Google Scholar] [CrossRef] [Green Version]
- Shirley, J.L.; de Jong, Y.P.; Terhorst, C.; Herzog, R.W. Immune Responses to Viral Gene Therapy Vectors. Mol. Ther. 2020, 28, 709–722. [Google Scholar] [CrossRef]
- Bushman, F.D. Retroviral Insertional Mutagenesis in Humans: Evidence for Four Genetic Mechanisms Promoting Expansion of Cell Clones. Mol. Ther. 2020, 28, 352–356. [Google Scholar] [CrossRef] [PubMed]
- Cemazar, M.; Grosel, A.; Glavac, D.; Kotnik, V.; Skobrne, M.; Kranjc, S.; Mir, L.M.; Andre, F.; Opolon, P.; Sersa, G. Effects of electrogenetherapy with p53wt combined with cisplatin on curvival of human tumor cell lines with different p53 status. DNA Cell Biol. 2003, 22, 765–775. [Google Scholar] [CrossRef] [PubMed]
- Liu, F.; Song, Y.; Liu, D. Hydrodynamics-based transfection in animals by systemic administration of plasmid DNA. Gene Ther. 1999, 6, 1258–1266. [Google Scholar] [CrossRef] [Green Version]
- Riley, M.K.; Vermerris, W. Recent Advances in Nanomaterials for Gene Delivery-A Review. Nanomaterials 2017, 7, 94. [Google Scholar] [CrossRef] [Green Version]
- Kovač, A.; Ivics, Z. Specifically integrating vectors for targeted gene delivery: Progress and prospects. Cell Gene Ther. Insights 2017, 3, 103–123. [Google Scholar] [CrossRef]
- Ma, W.; Xu, Y.-S.; Sun, X.-M.; Huang, H. Transposon-Associated CRISPR-Cas System: A Powerful DNA Insertion Tool. Trends Microbiol. 2021. [Google Scholar] [CrossRef] [PubMed]
- Ma, S.; Wang, X.; Hu, Y.; Lv, J.; Liu, C.; Liao, K.; Guo, X.; Wang, D.; Lin, Y.; Rong, Z. Enhancing site-specific DNA integration by a Cas9 nuclease fused with a DNA donor-binding domain. Nucleic Acids Res. 2020, 48, 10590–10601. [Google Scholar] [CrossRef]
- Kosicki, M.; Tomberg, K.; Bradley, A. Repair of double-strand breaks induced by CRISPR-Cas9 leads to large deletions and complex rearrangements. Nat. Biotechnol. 2018, 36, 765–771. [Google Scholar] [CrossRef] [PubMed]
- Hew, B.E.; Sato, R.; Mauro, D.; Stoytchev, I.; Owens, J.B. RNA-guided piggyBac transposition in human cells. Synth. Biol. 2019, 4, ysz018. [Google Scholar] [CrossRef] [Green Version]
- Kovač, A.; Miskey, C.; Menzel, M.; Grueso, E.; Gogol-Döring, A.; Ivics, Z. RNA-guided retargeting of Sleeping Beauty transposition in human cells. eLife 2020, 9, e53868. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sleeping Beauty (SB) | piggyBac (PB) | Tol2 | |
|---|---|---|---|
| Species of origin | Salmonid fish [21] | Cabbage looper moth | Medaka fish [29] |
| Classification | Tc1/mariner superfamily [21] | PB superfamily | hAT superfamily [30] |
| Transposable element | ~1.6 kb long | ~2.5 kb long | ~4.7 kb long |
| Terminal regions | IR/DRs of ~ 230 bp | 35–63 bp with outer TIRs and inner subterminal IRs | 150–200 bp containing the TIRs and subterminal regions |
| Transposase | 340 aa | 594 aa | 649 aa (most active isoform) |
| Footprint | CAG [13] | None [84] | Variable [29] |
| Target site preference | TA [36] | TTAA [84] | Weak consensus sequence TNA(C/G)TTATAA(G/C)TNA [101] |
| Target site duplication | TA [13] | TTAA [84] | 8 bp [101] |
| Activity in species | Various vertebrates | Vertebrates, insects, plants, yeast | Various vertebrates |
| Efficiency in human cells | Comparable to retroviral vectors [44] | Comparable to retroviral vectors [85] | Lower than PB and SB [107] |
| Cargo capacity | >100 kb [83] | >100 kb [83] | >100 kb [114] |
| Overproduction inhibition | Yes [107] | To some extent [107] | Lower than PB and SB [107] |
| Integration profile | Close-to-random [107] | Biased towards TSSs, CpG islands and DNaseI hypersensitivity sites [107] | Biased towards TSSs, CpG islands and DNaseI hypersensitivity sites [107] |
| Most common parental plasmid | pT2 | pXL-BacII | pTol2, miniTol2 |
| Most hyperactive transposase | hySB100X [45] | hyPB [85] | hTol2-M [101] |
| Vectors for transposon delivery | Plasmid DNA, pFAR, MC, non-integrative viral vectors, nanoparticles | Plasmid DNA, dbDNA, non-integrative viral vectors, nanoparticles | Plasmid DNA |
| Vectors for transposase delivery | Plasmid DNA, mRNA, SNIM RNA, recombinant protein (hsSB), non-integrative viral vectors, nanoparticles | Plasmid DNA, mRNA, non-integrative viral vectors, nanoparticles | Plasmid DNA, mRNA, recombinant protein (His-Tol2) |
| Clinical trials | Yes | Yes | No |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sandoval-Villegas, N.; Nurieva, W.; Amberger, M.; Ivics, Z. Contemporary Transposon Tools: A Review and Guide through Mechanisms and Applications of Sleeping Beauty, piggyBac and Tol2 for Genome Engineering. Int. J. Mol. Sci. 2021, 22, 5084. https://doi.org/10.3390/ijms22105084
Sandoval-Villegas N, Nurieva W, Amberger M, Ivics Z. Contemporary Transposon Tools: A Review and Guide through Mechanisms and Applications of Sleeping Beauty, piggyBac and Tol2 for Genome Engineering. International Journal of Molecular Sciences. 2021; 22(10):5084. https://doi.org/10.3390/ijms22105084
Chicago/Turabian StyleSandoval-Villegas, Nicolás, Wasifa Nurieva, Maximilian Amberger, and Zoltán Ivics. 2021. "Contemporary Transposon Tools: A Review and Guide through Mechanisms and Applications of Sleeping Beauty, piggyBac and Tol2 for Genome Engineering" International Journal of Molecular Sciences 22, no. 10: 5084. https://doi.org/10.3390/ijms22105084