Genotypes and Phenotypes: A Search for Influential Genes in Diabetic Retinopathy

 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Role of Genetic Variance in Complex Diseases
3. Natural History and Clinical Phenotypes of Diabetic Retinopathy
3.1. Nonproliferative Diabetic Retinopathy (NPDR)
3.2. Proliferative Diabetic Retinopathy (PDR)
3.3. Diabetic Macular Edema (DME)
4. Heterogeneity of DR Phenotypes
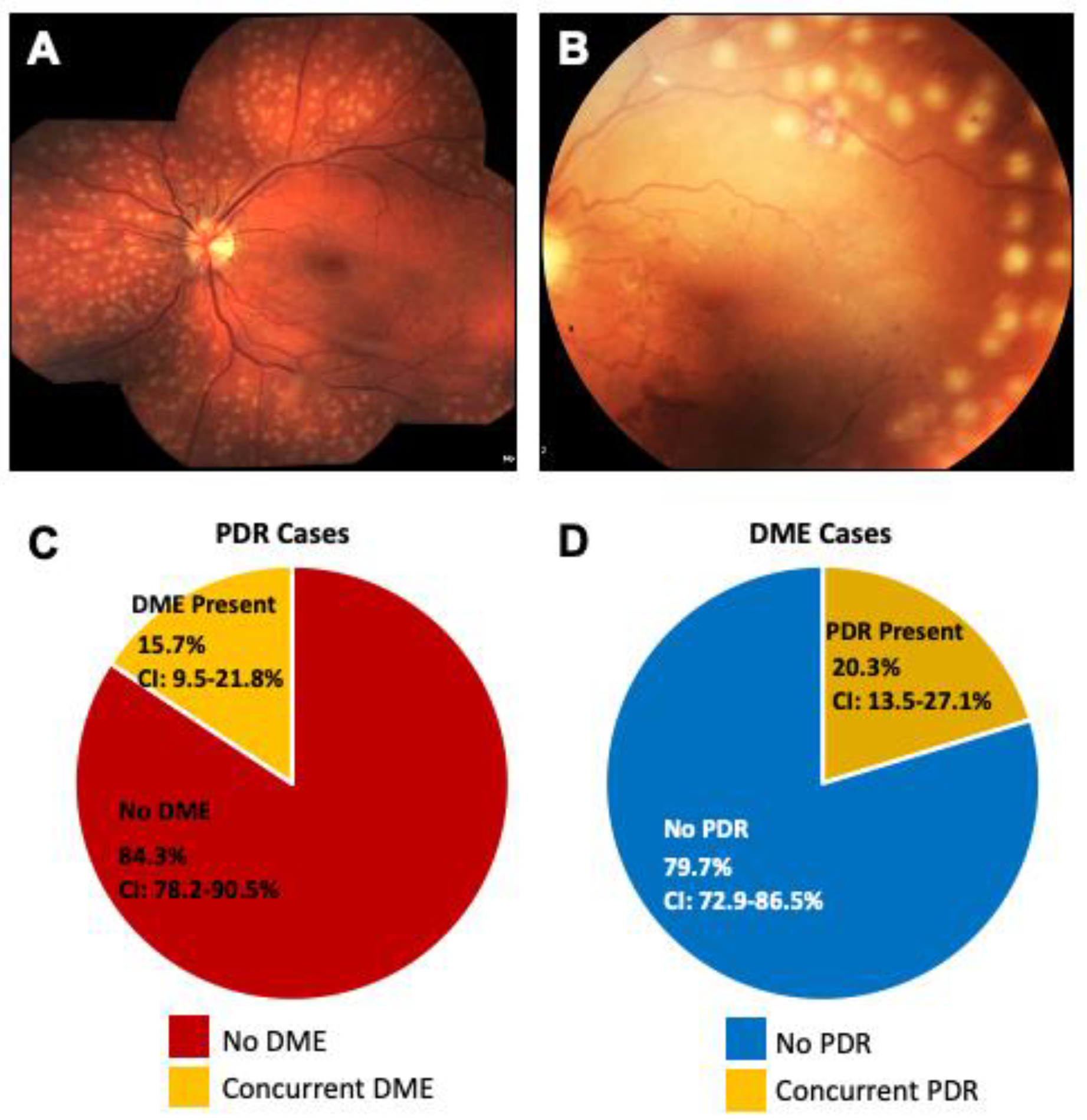
4.1. Are PDR and DME Two Distinct Diseases?
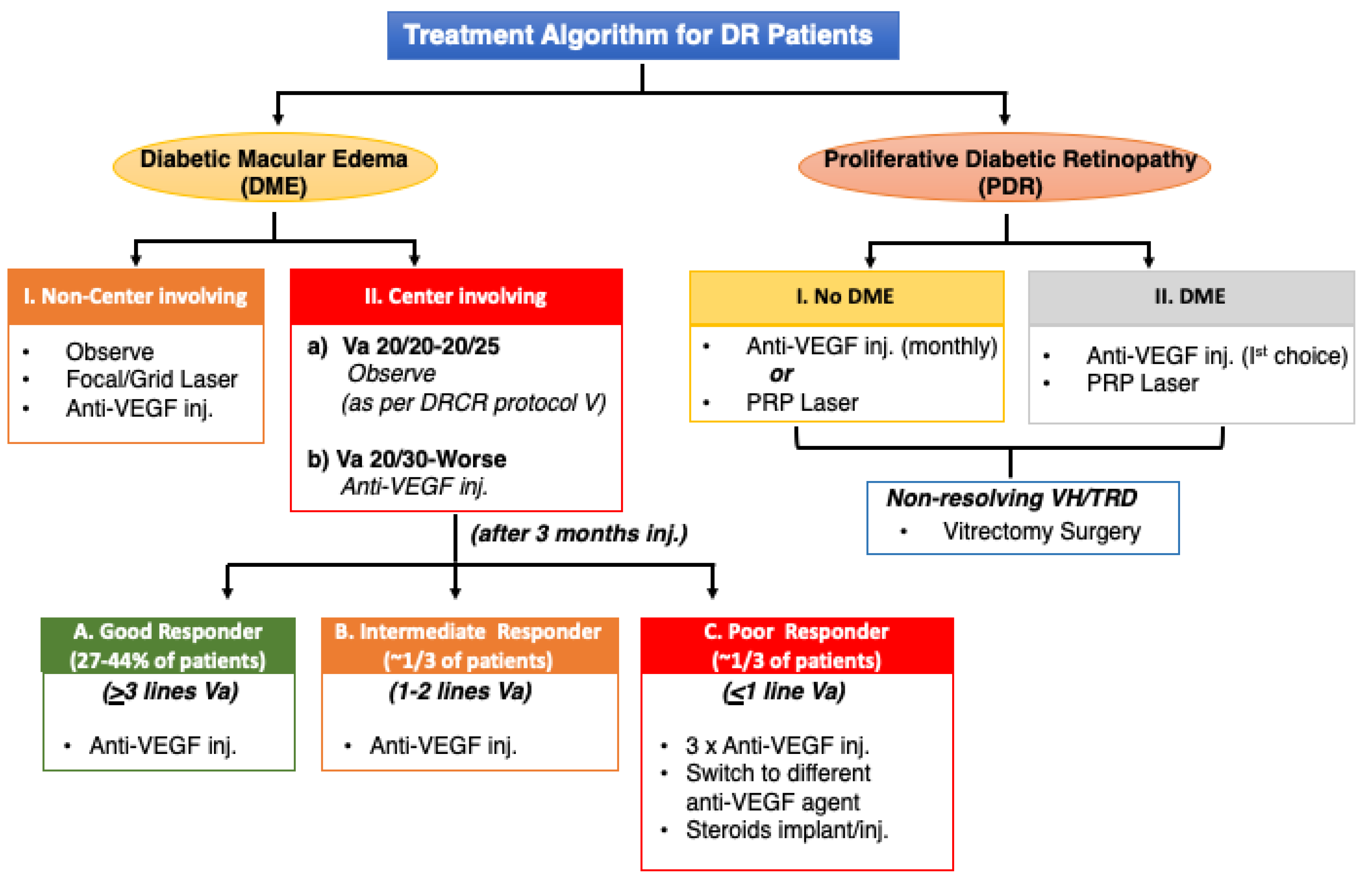
4.2. Variable Drug Response to Treatment
5. Clinical Evidence of Genetic Factors in DR Phenotypes
Ethnic Differences in Disease Manifestation
6. Are Systemic Factors Always Related to Severity of DR?
6.1. Duration of Diabetes
6.2. Hyperglycemia
6.3. Hypertension
6.4. Hyperlipidemia
7. Early Studies Point to the Role of Genetics in DR
8. Leveraging Gene Mapping and Previous Genetic Studies
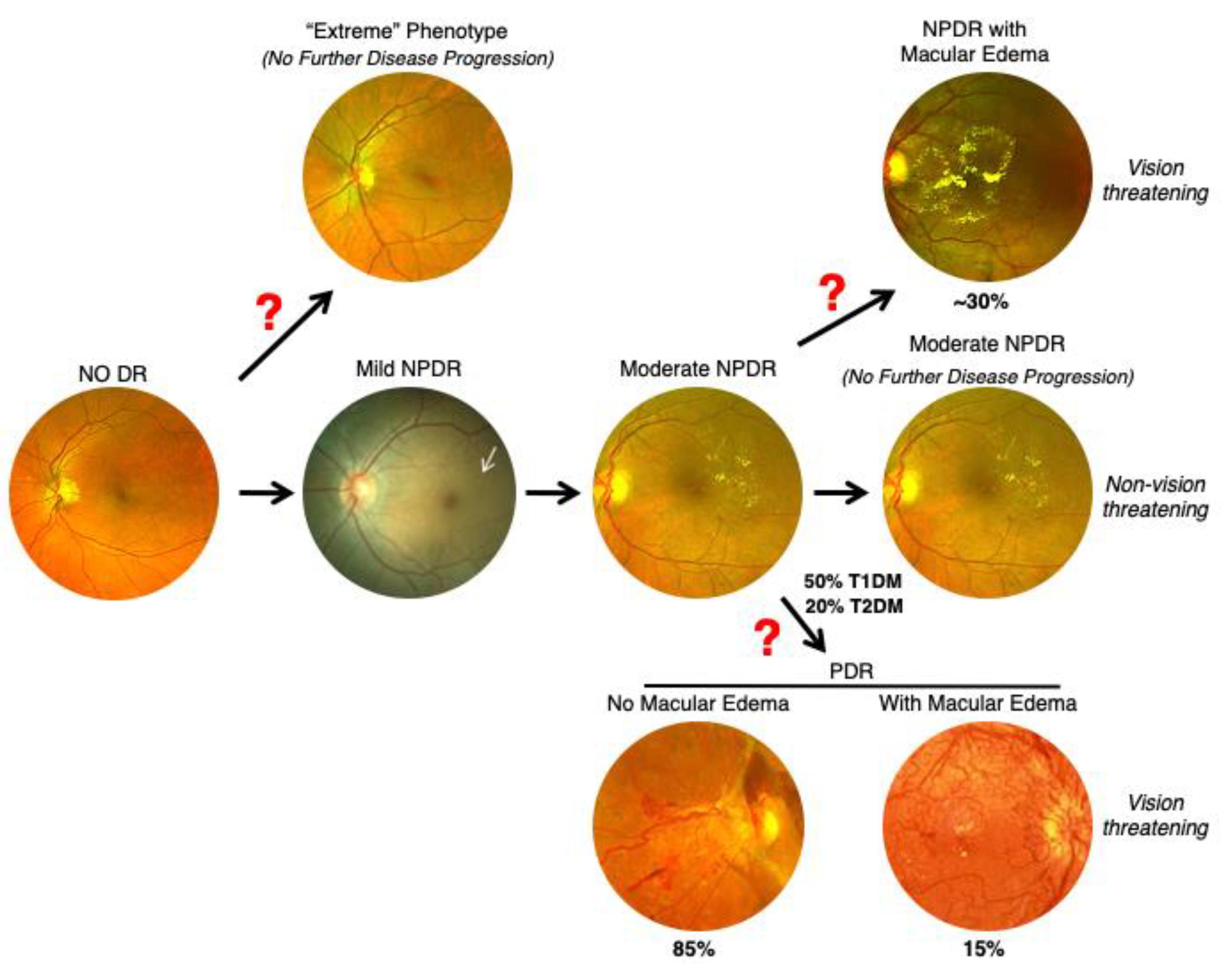
9. Nonsequential Progression of DR
10. Diabetic Nephropathy and Retinopathy
11. Diabetic Retinopathy Genomics (DRGen) Study
12. Future Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Paxton, W.A.; Martin, S.R.; Tse, D.; O’Brien, T.R.; Skurnick, J.; VanDevanter, N.L.; Padian, N.; Braun, J.F.; Kotler, D.P.; Wolinsky, S.M.; et al. Relative resistance to HIV-1 infection of CD4 lymphocytes from persons who remain uninfected despite multiple high-risk sexual exposure. Nat. Med. 1996, 2, 412–417. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Paxton, W.A.; Choe, S.; Ceradini, D.; Martin, S.R.; Horuk, R.; MacDonald, M.E.; Stuhlmann, H.; Koup, R.A.; Landau, N.R. Homozygous defect in HIV-1 coreceptor accounts for resistance of some multiply-exposed individuals to HIV-1 infection. Cell 1996, 86, 367–377. [Google Scholar] [CrossRef] [Green Version]
- Vangelista, L.; Vento, S. The Expanding Therapeutic Perspective of CCR5 Blockade. Front. Immunol. 2017, 8, 1981. [Google Scholar] [CrossRef] [PubMed]
- Dobzhansky, T. Genetics and the Origin of Species, 1st ed.; Columbia University Press: New York, NY, USA, 1937. [Google Scholar]
- Dobzhansky, T. Genetics and the Origin of Species, 3rd ed.; Columbia University Press: New York, NY, USA, 1951. [Google Scholar]
- Szabo, C.I.; King, M.C. Population genetics of BRCA1 and BRCA2. Am. J. Hum. Genet. 1997, 60, 1013–1020. [Google Scholar] [PubMed]
- Takahashi, H.; Behbakht, K.; McGovern, P.E.; Chiu, H.C.; Couch, F.J.; Weber, B.L.; Friedman, L.S.; King, M.C.; Furusato, M.; LiVolsi, V.A.; et al. Mutation analysis of the BRCA1 gene in ovarian cancers. Cancer Res. 1995, 55, 2998–3002. [Google Scholar]
- Friedman, L.S.; Ostermeyer, E.A.; Szabo, C.I.; Dowd, P.; Lynch, E.D.; Rowell, S.E.; King, M.C. Confirmation of BRCA1 by analysis of germline mutations linked to breast and ovarian cancer in ten families. Nat. Genet. 1994, 8, 399–404. [Google Scholar] [CrossRef]
- Edwards, A.O.; Ritter, R.; Abel, K.J.; Manning, A.; Panhuysen, C.; Farrer, L.A. Complement factor H polymorphism and age-related macular degeneration. Science 2005, 308, 421–424. [Google Scholar] [CrossRef] [Green Version]
- Hageman, G.S.; Anderson, D.H.; Johnson, L.V.; Hancox, L.S.; Taiber, A.J.; Hardisty, L.I.; Hageman, J.L.; Stockman, H.A.; Borchardt, J.D.; Gehrs, K.M.; et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2005, 102, 7227–7232. [Google Scholar] [CrossRef] [Green Version]
- Haines, J.L.; Hauser, M.A.; Schmidt, S.; Scott, W.K.; Olson, L.M.; Gallins, P.; Spencer, K.L.; Kwan, S.Y.; Noureddine, M.; Gilbert, J.R.; et al. Complement factor H variant increases the risk of age-related macular degeneration. Science 2005, 308, 419–421. [Google Scholar] [CrossRef] [Green Version]
- Klein, R.J.; Zeiss, C.; Chew, E.Y.; Tsai, J.Y.; Sackler, R.S.; Haynes, C.; Henning, A.K.; SanGiovanni, J.P.; Mane, S.M.; Mayne, S.T.; et al. Complement factor H polymorphism in age-related macular degeneration. Science 2005, 308, 385–389. [Google Scholar] [CrossRef]
- Russell, S.; Bennett, J.; Wellman, J.A.; Chung, D.C.; Yu, Z.F.; Tillman, A.; Wittes, J.; Pappas, J.; Elci, O.; McCague, S.; et al. Efficacy and safety of voretigene neparvovec (AAV2-hRPE65v2) in patients with RPE65-mediated inherited retinal dystrophy: A randomised, controlled, open-label, phase 3 trial. Lancet 2017, 390, 849–860. [Google Scholar] [CrossRef]
- Awata, T.; Yamashita, H.; Kurihara, S.; Morita-Ohkubo, T.; Miyashita, Y.; Katayama, S.; Mori, K.; Yoneya, S.; Kohda, M.; Okazaki, Y.; et al. Correction: A Genome-Wide Association Study for Diabetic Retinopathy in a Japanese Population: Potential Association with a Long Intergenic Non-Coding RNA. PLoS ONE 2015, 10, e0126789. [Google Scholar] [CrossRef] [PubMed]
- Burdon, K.P.; Fogarty, R.D.; Shen, W.; Abhary, S.; Kaidonis, G.; Appukuttan, B.; Hewitt, A.W.; Sharma, S.; Daniell, M.; Essex, R.W.; et al. Genome-wide association study for sight-threatening diabetic retinopathy reveals association with genetic variation near the GRB2 gene. Diabetologia 2015, 58, 2288–2297. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.P.; Hallman, D.M.; Gonzalez, V.H.; Klein, B.E.; Klein, R.; Hayes, M.G.; Cox, N.J.; Bell, G.I.; Hanis, C.L. Identification of Diabetic Retinopathy Genes through a Genome-Wide Association Study among Mexican-Americans from Starr County, Texas. J. Ophthalmol. 2010, 2010. [Google Scholar] [CrossRef] [PubMed]
- Grassi, M.A.; Tikhomirov, A.; Ramalingam, S.; Below, J.E.; Cox, N.J.; Nicolae, D.L. Genome-wide meta-analysis for severe diabetic retinopathy. Hum. Mol. Genet. 2011, 20, 2472–2481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, Y.C.; Lin, J.M.; Lin, H.J.; Chen, C.C.; Chen, S.Y.; Tsai, C.H.; Tsai, F.J. Genome-wide association study of diabetic retinopathy in a Taiwanese population. Ophthalmology 2011, 118, 642–648. [Google Scholar] [CrossRef]
- Lin, H.J.; Huang, Y.C.; Lin, J.M.; Wu, J.Y.; Chen, L.A.; Tsai, F.J. Association of genes on chromosome 6, GRIK2, TMEM217 and TMEM63B (linked to MRPL14 ) with diabetic retinopathy. Ophthalmologica 2013, 229, 54–60. [Google Scholar] [CrossRef]
- Sheu, W.H.; Kuo, J.Z.; Lee, I.T.; Hung, Y.J.; Lee, W.J.; Tsai, H.Y.; Wang, J.S.; Goodarzi, M.O.; Klein, R.; Klein, B.E.; et al. Genome-wide association study in a Chinese population with diabetic retinopathy. Hum. Mol. Genet. 2013, 22, 3165–3173. [Google Scholar] [CrossRef] [Green Version]
- Altmuller, J.; Palmer, L.J.; Fischer, G.; Scherb, H.; Wjst, M. Genomewide scans of complex human diseases: True linkage is hard to find. Am. J. Hum. Genet. 2001, 69, 936–950. [Google Scholar] [CrossRef] [Green Version]
- Newton-Cheh, C.; Hirschhorn, J.N. Genetic association studies of complex traits: Design and analysis issues. Mutat. Res. 2005, 573, 54–69. [Google Scholar] [CrossRef]
- Ananthakrishnan, A.N.; Xavier, R.J. How does genotype influence disease phenotype in inflammatory bowel disease? Inflamm. Bowel Dis. 2013, 19, 2021–2030. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, C.J.; Crock, P.A.; King, B.R.; Meldrum, C.J.; Scott, R.J. Phenotype-genotype correlations in a series of wolfram syndrome families. Diabetes Care 2004, 27, 2003–2009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stessman, H.A.; Bernier, R.; Eichler, E.E. A genotype-first approach to defining the subtypes of a complex disease. Cell 2014, 156, 872–877. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, R.; Klein, B.E.; Moss, S.E.; Davis, M.D.; DeMets, D.L. The Wisconsin epidemiologic study of diabetic retinopathy. II. Prevalence and risk of diabetic retinopathy when age at diagnosis is less than 30 years. Arch. Ophthalmol. 1984, 102, 520–526. [Google Scholar] [CrossRef] [PubMed]
- Klein, R.; Klein, B.E.; Moss, S.E.; Davis, M.D.; DeMets, D.L. The Wisconsin epidemiologic study of diabetic retinopathy. III. Prevalence and risk of diabetic retinopathy when age at diagnosis is 30 or more years. Arch. Ophthalmol. 1984, 102, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Diabetes Control and Complications Trial Research Group. The relationship of glycemic exposure (HbA1c) to the risk of development and progression of retinopathy in the diabetes control and complications trial. Diabetes 1995, 44, 968–983. [CrossRef]
- Maa, A.Y.; Sullivan, B.R. Relationship of hemoglobin A1C with the presence and severity of retinopathy upon initial screening of Type II diabetes mellitus. Am. J. Ophthalmol. 2007, 144, 456–457. [Google Scholar] [CrossRef]
- Nittala, M.G.; Keane, P.A.; Zhang, K.; Sadda, S.R. Risk factors for proliferative diabetic retinopathy in a Latino American population. Retina 2014, 34, 1594–1599. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.K.; Keenan, H.A.; Cavallerano, J.D.; Asztalos, B.F.; Schaefer, E.J.; Sell, D.R.; Strauch, C.M.; Monnier, V.M.; Doria, A.; Aiello, L.P.; et al. Protection from retinopathy and other complications in patients with type 1 diabetes of extreme duration: The joslin 50-year medalist study. Diabetes Care 2011, 34, 968–974. [Google Scholar] [CrossRef] [Green Version]
- Sartore, G.; Chilelli, N.C.; Burlina, S.; Lapolla, A. Association between glucose variability as assessed by continuous glucose monitoring (CGM) and diabetic retinopathy in type 1 and type 2 diabetes. Acta Diabetol. 2013, 50, 437–442. [Google Scholar] [CrossRef]
- Von Graefe, A. Ueber die mit Diabetes mellitus vorkommenden Sehstörungen. Dies Arch IV 1858, 10, 230–234. [Google Scholar]
- Nettleship, G. On oedema or cystic disease of the retina. Roy. Ophth. Lond. Hosp. Rep. 1872, VII, 343–351. [Google Scholar]
- Polonsky, K.S. The past 200 years in diabetes. N. Engl. J. Med. 2012, 367, 1332–1340. [Google Scholar] [CrossRef]
- Goldberg, M.F.; Fine, S.L. Symposium on the Treatment of Diabetic Retinopathy (No. 1890); Airlie House: Warrenton, VA, USA, 1969; US Neurological and Sensory Disease Control Program. [Google Scholar]
- Harris Nwanyanwu, K.; Talwar, N.; Gardner, T.W.; Wrobel, J.S.; Herman, W.H.; Stein, J.D. Predicting development of proliferative diabetic retinopathy. Diabetes Care 2013, 36, 1562–1568. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yun, W.L.; Acharya, A.U.; Venkatesh, Y.V.; Chee, C.; Min, L.C.; Ng, E.Y.K. Identification of different stages of diabetic retinopathy using retinal optical images. Inf. Sci. 2008, 178, 106–121. [Google Scholar] [CrossRef]
- Jones, C.D.; Greenwood, R.H.; Misra, A.; Bachmann, M.O. Incidence and progression of diabetic retinopathy during 17 years of a population-based screening program in England. Diabetes Care 2012, 35, 592–596. [Google Scholar] [CrossRef] [Green Version]
- Wong, T.Y.; Mwamburi, M.; Klein, R.; Larsen, M.; Flynn, H.; Hernandez-Medina, M.; Ranganathan, G.; Wirostko, B.; Pleil, A.; Mitchell, P. Rates of progression in diabetic retinopathy during different time periods: A systematic review and meta-analysis. Diabetes Care 2009, 32, 2307–2313. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Y.; He, M.; Congdon, N. The worldwide epidemic of diabetic retinopathy. Indian J. Ophthalmol. 2012, 60, 428–431. [Google Scholar] [CrossRef]
- Das, A.; McGuire, P.G.; Rangasamy, S. Diabetic Macular Edema: Pathophysiology and Novel Therapeutic Targets. Ophthalmology 2015, 122, 1375–1394. [Google Scholar] [CrossRef]
- Joslin, E.P.; Kahn, C.R. (Eds.) Joslin’s Diabetes Mellitus; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2005. [Google Scholar]
- The Diabetic Retinopathy Study Research Group. Preliminary report on effects of photocoagulation therapy. Am. J. Ophthalmol. 1976, 81, 383–396. [Google Scholar] [CrossRef]
- The Diabetic Retinopathy Study Research Group. Four risk factors for severe visual loss in diabetic retinopathy. The third report from the Diabetic Retinopathy Study. Arch. Ophthalmol. 1979, 97, 654–655. [Google Scholar] [CrossRef] [PubMed]
- Beetham, W.P.; Aiello, L.M.; Balodimos, M.C.; Koncz, L. Ruby laser photocoagulation of early diabetic neovascular retinopathy. Preliminary report of a long-term controlled study. Arch. Ophthalmol. 1970, 83, 261–272. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, S.; Genovesi-Ebert, F.; Di Bartolo, E.; Vento, A.; Miniaci, S.; Williams, G. Injection of intravitreal bevacizumab (Avastin) as a preoperative adjunct before vitrectomy surgery in the treatment of severe proliferative diabetic retinopathy (PDR). Graefe’s Arch. Clin. Exp. Ophthalmol. 2008, 246, 837–842. [Google Scholar] [CrossRef] [PubMed]
- Smiddy, W.E.; Flynn, H.W., Jr. Vitrectomy in the management of diabetic retinopathy. Surv. Ophthalmol. 1999, 43, 491–507. [Google Scholar] [CrossRef]
- Pacella, F.; Romano, M.R.; Turchetti, P.; Tarquini, G.; Carnovale, A.; Mollicone, A.; Mastromatteo, A.; Pacella, E. An eighteen-month follow-up study on the effects of Intravitreal Dexamethasone Implant in diabetic macular edema refractory to anti-VEGF therapy. Int. J. Ophthalmol. 2016, 9, 1427–1432. [Google Scholar] [CrossRef]
- Ciulla, T.A.; Amador, A.G.; Zinman, B. Diabetic retinopathy and diabetic macular edema: Pathophysiology, screening, and novel therapies. Diabetes Care 2003, 26, 2653–2664. [Google Scholar] [CrossRef] [Green Version]
- Ding, J.; Wong, T.Y. Current epidemiology of diabetic retinopathy and diabetic macular edema. Curr. Diab. Rep. 2012, 12, 346–354. [Google Scholar] [CrossRef]
- Lee, R.; Wong, T.Y.; Sabanayagam, C. Epidemiology of diabetic retinopathy, diabetic macular edema and related vision loss. Eye Vis. (Lond.) 2015, 2, 17. [Google Scholar] [CrossRef] [Green Version]
- Frank, R.N. Diabetic retinopathy. N. Engl. J. Med. 2004, 350, 48–58. [Google Scholar] [CrossRef]
- Romero-Aroca, P.; Baget-Bernaldiz, M.; Pareja-Rios, A.; Lopez-Galvez, M.; Navarro-Gil, R.; Verges, R. Diabetic Macular Edema Pathophysiology: Vasogenic versus Inflammatory. J. Diabetes Res. 2016, 2016, 2156273. [Google Scholar] [CrossRef] [Green Version]
- Wilkinson, C.P.; Ferris, F.L.; Klein, R.E.; Lee, P.P.; Agardh, C.D.; Davis, M.; Dills, D.; Kampik, A.; Pararajasegaram, R.; Verdaguer, J.T.; et al. Proposed international clinical diabetic retinopathy and diabetic macular edema disease severity scales. Ophthalmology 2003, 110, 1677–1682. [Google Scholar] [CrossRef]
- Elman, M.J.; Aiello, L.P.; Beck, R.W.; Bressler, N.M.; Bressler, S.B.; Edwards, A.R.; Ferris, F.L., III; Friedman, S.M.; Glassman, A.R.; Miller, K.M.; et al. Randomized trial evaluating ranibizumab plus prompt or deferred laser or triamcinolone plus prompt laser for diabetic macular edema. Ophthalmology 2010, 117, 1064–1077. [Google Scholar] [CrossRef] [Green Version]
- Klein, R.; Klein, B.E.; Moss, S.E.; Cruickshanks, K.J. The Wisconsin Epidemiologic Study of Diabetic Retinopathy. XV. The long-term incidence of macular edema. Ophthalmology 1995, 102, 7–16. [Google Scholar] [CrossRef]
- Kuiper, E.J.; Van Nieuwenhoven, F.A.; de Smet, M.D.; van Meurs, J.C.; Tanck, M.W.; Oliver, N.; Klaassen, I.; Van Noorden, C.J.; Goldschmeding, R.; Schlingemann, R.O. The angio-fibrotic switch of VEGF and CTGF in proliferative diabetic retinopathy. PLoS ONE 2008, 3, e2675. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, S.; Mehta, A.; Patel, A.; Das, A. Proliferative Diabetic Retinopathy and Diabetic Macular Edema: Are these two different disease processes? A Retrospective Cross-sectional Study. Investig. Ophthalmol. Vis. Sci. 2016, 57, 1592. [Google Scholar]
- Gao, X.; Gauderman, W.J.; Marjoram, P.; Torres, M.; Chen, Y.D.I.; Taylor, K.D.; Rotter, J.I.; Varma, R. Native American ancestry is associated with severe diabetic retinopathy in Latinos. Investig. Ophthalmol. Vis. Sci. 2014, 55, 6041–6045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, D.M.; Nguyen, Q.D.; Marcus, D.M.; Boyer, D.S.; Patel, S.; Feiner, L.; Schlottmann, P.G.; Rundle, A.C.; Zhang, J.; Rubio, R.G.; et al. Long-term outcomes of ranibizumab therapy for diabetic macular edema: The 36-month results from two phase III trials: RISE and RIDE. Ophthalmology 2013, 120, 2013–2022. [Google Scholar] [CrossRef] [Green Version]
- Elman, M.J.; Qin, H.; Aiello, L.P.; Beck, R.W.; Bressler, N.M.; Ferris, F.L., III; Glassman, A.R.; Maturi, R.K.; Melia, M. Intravitreal ranibizumab for diabetic macular edema with prompt versus deferred laser treatment: Three-year randomized trial results. Ophthalmology 2012, 119, 2312–2318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Korobelnik, J.F.; Do, D.V.; Schmidt-Erfurth, U.; Boyer, D.S.; Holz, F.G.; Heier, J.S.; Midena, E.; Kaiser, P.K.; Terasaki, H.; Marcus, D.M.; et al. Intravitreal aflibercept for diabetic macular edema. Ophthalmology 2014, 121, 2247–2254. [Google Scholar] [CrossRef]
- Aiello, L.P.; Avery, R.L.; Arrigg, P.G.; Keyt, B.A.; Jampel, H.D.; Shah, S.T.; Pasquale, L.R.; Thieme, H.; Iwamoto, M.A.; Park, J.E.; et al. Vascular endothelial growth factor in ocular fluid of patients with diabetic retinopathy and other retinal disorders. N. Engl. J. Med. 1994, 331, 1480–1487. [Google Scholar] [CrossRef]
- Funatsu, H.; Yamashita, H.; Ikeda, T.; Mimura, T.; Eguchi, S.; Hori, S. Vitreous levels of interleukin-6 and vascular endothelial growth factor are related to diabetic macular edema. Ophthalmology 2003, 110, 1690–1696. [Google Scholar] [CrossRef]
- DCCT/EDIC Research Group. Intensive diabetes therapy and ocular surgery in type 1 diabetes. N. Engl. J. Med. 2015, 372, 1722–1733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avery, R.L.; Pearlman, J.; Pieramici, D.J.; Rabena, M.D.; Castellarin, A.A.; Nasir, M.A.; Giust, M.J.; Wendel, R.; Patel, A. Intravitreal bevacizumab (Avastin) in the treatment of proliferative diabetic retinopathy. Ophthalmology 2006, 113, 1695–1705. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.A.; Glassman, A.R.; Ayala, A.R.; Jampol, L.M.; Aiello, L.P.; Antoszyk, A.N.; Arnold-Bush, B.; Baker, C.W.; Bressler, N.M.; Browning, D.J.; et al. Aflibercept, bevacizumab, or ranibizumab for diabetic macular edema. N. Engl. J. Med. 2015, 372, 1193–1203. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klein, R.; Klein, B.E.; Moss, S.E.; Cruickshanks, K.J. The Wisconsin Epidemiologic Study of Diabetic Retinopathy: XVII. The 14-year incidence and progression of diabetic retinopathy and associated risk factors in type 1 diabetes. Ophthalmology 1998, 105, 1801–1815. [Google Scholar] [CrossRef]
- El-Shazly, S.F.; El-Bradey, M.H.; Tameesh, M.K. Vascular endothelial growth factor gene polymorphism prevalence in patients with diabetic macular oedema and its correlation with anti-vascular endothelial growth factor treatment outcomes. Clin. Exp. Ophthalmol. 2014, 42, 369–378. [Google Scholar] [CrossRef]
- Hamman, R.F.; Mayer, E.J.; Moo-Young, G.A.; Hildebrandt, W.; Marshall, J.A.; Baxter, J. Prevalence and risk factors of diabetic retinopathy in non-Hispanic whites and Hispanics with NIDDM. San Luis Valley Diabetes Study. Diabetes 1989, 38, 1231–1237. [Google Scholar] [CrossRef]
- Harris, M.I.; Klein, R.; Cowie, C.C.; Rowland, M.; Byrd-Holt, D.D. Is the risk of diabetic retinopathy greater in non-Hispanic blacks and Mexican Americans than in non-Hispanic whites with type 2 diabetes? A U.S. population study. Diabetes Care 1998, 21, 1230–1235. [Google Scholar] [CrossRef]
- Tudor, S.M.; Hamman, R.F.; Baron, A.; Johnson, D.W.; Shetterly, S.M. Incidence and progression of diabetic retinopathy in Hispanics and non-Hispanic whites with type 2 diabetes. San Luis Valley Diabetes Study, Colorado. Diabetes Care 1998, 21, 53–61. [Google Scholar] [CrossRef]
- Imperatore, G.; Hanson, R.L.; Pettitt, D.J.; Kobes, S.; Bennett, P.H.; Knowler, W.C. Sib-pair linkage analysis for susceptibility genes for microvascular complications among Pima Indians with type 2 diabetes. Pima Diabetes Genes Group. Diabetes 1998, 47, 821–830. [Google Scholar] [CrossRef]
- Nagi, D.K.; Pettitt, D.J.; Bennett, P.H.; Klein, R.; Knowler, W.C. Diabetic retinopathy assessed by fundus photography in Pima Indians with impaired glucose tolerance and NIDDM. Diabet Med. 1997, 14, 449–456. [Google Scholar] [CrossRef]
- Summary Health Statistics for US Adults: National Health Interview Survey 2012; Center for Disease Control and Prevention: Atlanta, GA, USA, 2012.
- Chew, E.Y.; Klein, M.L.; Ferris, F.L., III; Remaley, N.A.; Murphy, R.P.; Chantry, K.; Hoogwerf, B.J.; Miller, D. Association of elevated serum lipid levels with retinal hard exudate in diabetic retinopathy. Early Treatment Diabetic Retinopathy Study (ETDRS) Report 22. Arch. Ophthalmol. 1996, 114, 1079–1084. [Google Scholar] [CrossRef] [PubMed]
- K Prospective Diabetes Study Group. Tight blood pressure control and risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 38. BMJ 1998, 317, 703–713. [CrossRef] [Green Version]
- Lachin, J.M.; Genuth, S.; Nathan, D.M.; Zinman, B.; Rutledge, B.N. Effect of glycemic exposure on the risk of microvascular complications in the diabetes control and complications trial—Revisited. Diabetes 2008, 57, 995–1001. [Google Scholar] [CrossRef] [Green Version]
- Yokomizo, H.; Maeda, Y.; Park, K.; Clermont, A.C.; Hernandez, S.L.; Fickweiler, W.; Li, Q.; Wang, C.H.; Paniagua, S.M.; Simao, F.; et al. Retinol binding protein 3 is increased in the retina of patients with diabetes resistant to diabetic retinopathy. Sci. Transl. Med. 2019, 11, eaau6627. [Google Scholar] [CrossRef]
- ACCORD Study Group and ACCORD Eye Study Group. Effects of medical therapies on retinopathy progression in type 2 diabetes. N. Engl. J. Med. 2010, 363, 233–244. [Google Scholar] [CrossRef] [Green Version]
- Penman, A.; Hancock, H.; Papavasileiou, E.; James, M.; Idowu, O.; Riche, D.M.; Fernandez, M.; Brauner, S.; Smith, S.O.; Hoadley, S.; et al. Risk Factors for Proliferative Diabetic Retinopathy in African Americans with Type 2 Diabetes. Ophthalmic. Epidemiol. 2016, 23, 88–93. [Google Scholar] [CrossRef] [Green Version]
- Acan, D.; Calan, M.; Er, D.; Arkan, T.; Kocak, N.; Bayraktar, F.; Kaynak, S. The prevalence and systemic risk factors of diabetic macular edema: A cross-sectional study from Turkey. BMC Ophthalmol. 2018, 18, 91. [Google Scholar] [CrossRef] [Green Version]
- Bansal, A.S.; Khurana, R.N.; Wieland, M.R.; Wang, P.W.; Van Everen, S.A.; Tuomi, L. Influence of Glycosylated Hemoglobin on the Efficacy of Ranibizumab for Diabetic Macular Edema: A Post Hoc Analysis of the RIDE/RISE Trials. Ophthalmology 2015, 122, 1573–1579. [Google Scholar] [CrossRef] [Green Version]
- Peng, Y.J.; Tsai, M.J. Impact of metabolic control on macular thickness in diabetic macular oedema. Diab. Vasc. Dis. Res. 2018, 15, 165–168. [Google Scholar] [CrossRef] [Green Version]
- Hammes, H.P.; Welp, R.; Kempe, H.P.; Wagner, C.; Siegel, E.; Holl, R.W. DPV Initiative—German BMBF Competence Network. Risk Factors for Retinopathy and DME in Type 2 Diabetes-Results from the German/Austrian DPV Database. PLoS ONE 2015, 10, e0132492. [Google Scholar] [CrossRef] [PubMed]
- Singh, R.P.; Habbu, K.; Ehlers, J.P.; Lansang, M.C.; Hill, L.; Stoilov, I. The Impact of Systemic Factors on Clinical Response to Ranibizumab for Diabetic Macular Edema. Ophthalmology 2016, 123, 1581–1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- ADVANCE Collaborative Group. Intensive blood glucose control and vascular outcomes in patients with type 2 diabetes. N. Engl. J. Med. 2008, 358, 2560–2572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benarous, R.; Sasongko, M.B.; Qureshi, S.; Fenwick, E.; Dirani, M.; Wong, T.Y.; Lamoureux, E.L. Differential association of serum lipids with diabetic retinopathy and diabetic macular edema. Investig. Ophthalmol. Vis. Sci. 2011, 52, 7464–7469. [Google Scholar] [CrossRef]
- Das, R.; Kerr, R.; Chakravarthy, U.; Hogg, R.E. Dyslipidemia and Diabetic Macular Edema: A Systematic Review and Meta-Analysis. Ophthalmology 2015, 122, 1820–1827. [Google Scholar] [CrossRef]
- Pyke, D.A.; Tattersall, R.B. Diabetic retinopathy in identical twins. Diabetes 1973, 22, 613–618. [Google Scholar] [CrossRef]
- Haffner, S.M.; Fong, D.; Stern, M.P.; Pugh, J.A.; Hazuda, H.P.; Patterson, J.K.; van Heuven, W.A.; Klein, R. Diabetic retinopathy in Mexican Americans and non-Hispanic whites. Diabetes 1988, 37, 878–884. [Google Scholar] [CrossRef]
- Levy, S.E.; Myers, R.M. Advancements in Next-Generation Sequencing. Annu. Rev. Genom. Hum. Genet. 2016, 17, 95–115. [Google Scholar] [CrossRef] [Green Version]
- Abhary, S.; Burdon, K.P.; Gupta, A.; Lake, S.; Selva, D.; Petrovsky, N.; Craig, J.E. Common sequence variation in the VEGFA gene predicts risk of diabetic retinopathy. Investig. Ophthalmol. Vis. Sci. 2009, 50, 5552–5558. [Google Scholar] [CrossRef] [Green Version]
- Balasubbu, S.; Sundaresan, P.; Rajendran, A.; Ramasamy, K.; Govindarajan, G.; Perumalsamy, N.; Hejtmancik, J.F. Association analysis of nine candidate gene polymorphisms in Indian patients with type 2 diabetic retinopathy. BMC Med. Genet. 2010, 11, 158. [Google Scholar] [CrossRef] [Green Version]
- Hallman, D.M.; Boerwinkle, E.; Gonzalez, V.H.; Klein, B.E.; Klein, R.; Hanis, C.L. A genome-wide linkage scan for diabetic retinopathy susceptibility genes in Mexican Americans with type 2 diabetes from Starr County, Texas. Diabetes 2007, 56, 1167–1173. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Louey, J.W.; Choy, K.W.; Liu, D.T.; Chan, W.M.; Chan, Y.M.; Fung, N.S.; Fan, B.J.; Baum, L.; Chan, J.C.; et al. EDN1 Lys198Asn is associated with diabetic retinopathy in type 2 diabetes. Mol. Vis. 2008, 14, 1698–1704. [Google Scholar] [PubMed]
- Looker, H.C.; Nelson, R.G.; Chew, E.; Klein, R.; Klein, B.E.; Knowler, W.C.; Hanson, R.L. Genome-wide linkage analyses to identify Loci for diabetic retinopathy. Diabetes 2007, 56, 1160–1166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gresham, D.; Ruderfer, D.M.; Pratt, S.C.; Schacherer, J.; Dunham, M.J.; Botstein, D.; Kruglyak, L. Genome-wide detection of polymorphisms at nucleotide resolution with a single DNA microarray. Science 2006, 311, 1932–1936. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Homer, N.; Szelinger, S.; Redman, M.; Duggan, D.; Tembe, W.; Muehling, J.; Pearson, J.V.; Stephan, D.A.; Nelson, S.F.; Craig, D.W. Resolving individuals contributing trace amounts of DNA to highly complex mixtures using high-density SNP genotyping microarrays. PLoS Genet. 2008, 4. [Google Scholar] [CrossRef] [Green Version]
- Pollack, S.; Igo, R.P.; Jensen, R.A.; Christiansen, M.; Li, X.; Cheng, C.Y.; Ng, M.C.; Smith, A.V.; Rossin, E.J.; Segrè, A.V.; et al. Multiethnic genome-wide association study of diabetic retinopathy using liability threshold modeling of duration of diabetes and glycemic control. Diabetes 2019, 68, 441–456. [Google Scholar] [CrossRef] [Green Version]
- Cho, H.; Sobrin, L. Genetics of diabetic retinopathy. Curr. Diab. Rep. 2014, 14, 515. [Google Scholar] [CrossRef]
- Kuo, J.Z.; Wong, T.Y.; Rotter, J.I. Challenges in elucidating the genetics of diabetic retinopathy. JAMA Ophthalmol. 2014, 132, 96–107. [Google Scholar] [CrossRef] [Green Version]
- Mishra, B.; Swaroop, A.; Kandpal, R.P. Genetic components in diabetic retinopathy. Indian J. Ophthalmol. 2016, 64, 55–61. [Google Scholar] [CrossRef]
- Fong, D.S.; Aiello, L.; Gardner, T.W.; King, G.L.; Blankenship, G.; Cavallerano, J.D.; Ferris, F.L., III; Klein, R. Diabetic retinopathy. Diabetes Care 2003, 26 (Suppl. 1), S99–S102. [Google Scholar] [CrossRef] [Green Version]
- Krolewski, A.S.; Skupien, J.; Rossing, P.; Warram, J.H. Fast renal decline to end-stage renal disease: An unrecognized feature of nephropathy in diabetes. Kidney Int. 2017, 91, 1300–1311. [Google Scholar] [CrossRef] [PubMed]
- Mogensen, C.E.; Christensen, C.K.; Vittinghus, E. The stages in diabetic renal disease. With emphasis on the stage of incipient diabetic nephropathy. Diabetes 1983, 32 (Suppl. 2), 64–78. [Google Scholar] [CrossRef]
- Cabrera, A.P.; Monickaraj, F.; Rangasamy, S.; Hobbs, S.; McGuire, P.; Das, A. Do Genomic Factors Play a Role in Diabetic Retinopathy? J. Clin. Med. 2020, 9, 216. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, A.; Rangasamy, S.; Naymik, M.; Monickaraj, F.; Legendre, C.; Balak, C.; Duggan, D.; Schork, N.; McGuire, P. Novel genetic variants in extreme phenotypes of diabetic retinopathy: DRGen Study. Investig. Ophthalmol. Vis. Sci. 2018, 59, 1911. [Google Scholar]
- Akey, J.M.; Zhang, G.; Zhang, K.; Jin, L.; Shriver, M.D. Interrogating a high-density SNP map for signatures of natural selection. Genome Res. 2002, 12, 1805–1814. [Google Scholar] [CrossRef] [Green Version]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [Green Version]
- Horikawa, N.; Abiko, K.; Matsumura, N.; Hamanishi, J.; Baba, T.; Yamaguchi, K.; Yoshioka, Y.; Koshiyama, M.; Konishi, I. Expression of Vascular Endothelial Growth Factor in Ovarian Cancer Inhibits Tumor Immunity through the Accumulation of Myeloid-Derived Suppressor Cells. Clin. Cancer Res. 2017, 23, 587–599. [Google Scholar] [CrossRef] [Green Version]
- Iwanicki, M.P.; Brugge, J.S. Transcriptional regulation of metastatic [Id] entity by KLF17. Genome Biol. 2009, 10, 244. [Google Scholar] [CrossRef] [Green Version]
- Maystadt, I.; Rezsohazy, R.; Barkats, M.; Duque, S.; Vannuffel, P.; Remacle, S.; Lambert, B.; Najimi, M.; Sokal, E.; Munnich, A.; et al. The nuclear factor kappaB-activator gene PLEKHG5 is mutated in a form of autosomal recessive lower motor neuron disease with childhood onset. Am. J. Hum. Genet. 2007, 81, 67–76. [Google Scholar] [CrossRef] [Green Version]
- Murat, A.; Migliavacca, E.; Hussain, S.F.; Heimberger, A.B.; Desbaillets, I.; Hamou, M.F.; Ruegg, C.; Stupp, R.; Delorenzi, M.; Hegi, M.E. Modulation of angiogenic and inflammatory response in glioblastoma by hypoxia. PLoS ONE 2009, 4, e5947. [Google Scholar] [CrossRef]
- O’Reilly, M.S.; Boehm, T.; Shing, Y.; Fukai, N.; Vasios, G.; Lane, W.S.; Flynn, E.; Birkhead, J.R.; Olsen, B.R.; Folkman, J. Endostatin: An endogenous inhibitor of angiogenesis and tumor growth. Cell 1997, 88, 277–285. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; Hegarty, J.P.; Berg, A.; Chen, X.; West, G.; Kelly, A.A.; Wang, Y.; Poritz, L.S.; Koltun, W.A.; Lin, Z. NKX2-3 transcriptional regulation of endothelin-1 and VEGF signaling in human intestinal microvascular endothelial cells. PLoS ONE 2011, 6, e20454. [Google Scholar] [CrossRef] [PubMed]
- Lello, L.; Avery, S.G.; Tellier, L.; Vazquez, A.I.; de Los Campos, G.; Hsu, S.D.H. Accurate Genomic Prediction of Human Height. Genetics 2018, 210, 477–497. [Google Scholar] [CrossRef] [PubMed] [Green Version]










© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cabrera, A.P.; Mankad, R.N.; Marek, L.; Das, R.; Rangasamy, S.; Monickaraj, F.; Das, A. Genotypes and Phenotypes: A Search for Influential Genes in Diabetic Retinopathy. Int. J. Mol. Sci. 2020, 21, 2712. https://doi.org/10.3390/ijms21082712
Cabrera AP, Mankad RN, Marek L, Das R, Rangasamy S, Monickaraj F, Das A. Genotypes and Phenotypes: A Search for Influential Genes in Diabetic Retinopathy. International Journal of Molecular Sciences. 2020; 21(8):2712. https://doi.org/10.3390/ijms21082712
Chicago/Turabian StyleCabrera, Andrea P., Rushi N. Mankad, Lauren Marek, Ryan Das, Sampath Rangasamy, Finny Monickaraj, and Arup Das. 2020. "Genotypes and Phenotypes: A Search for Influential Genes in Diabetic Retinopathy" International Journal of Molecular Sciences 21, no. 8: 2712. https://doi.org/10.3390/ijms21082712