Pisum sativum Defensin 1 Eradicates Mouse Metastatic Lung Nodules from B16F10 Melanoma Cells


 , , , and
, , , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
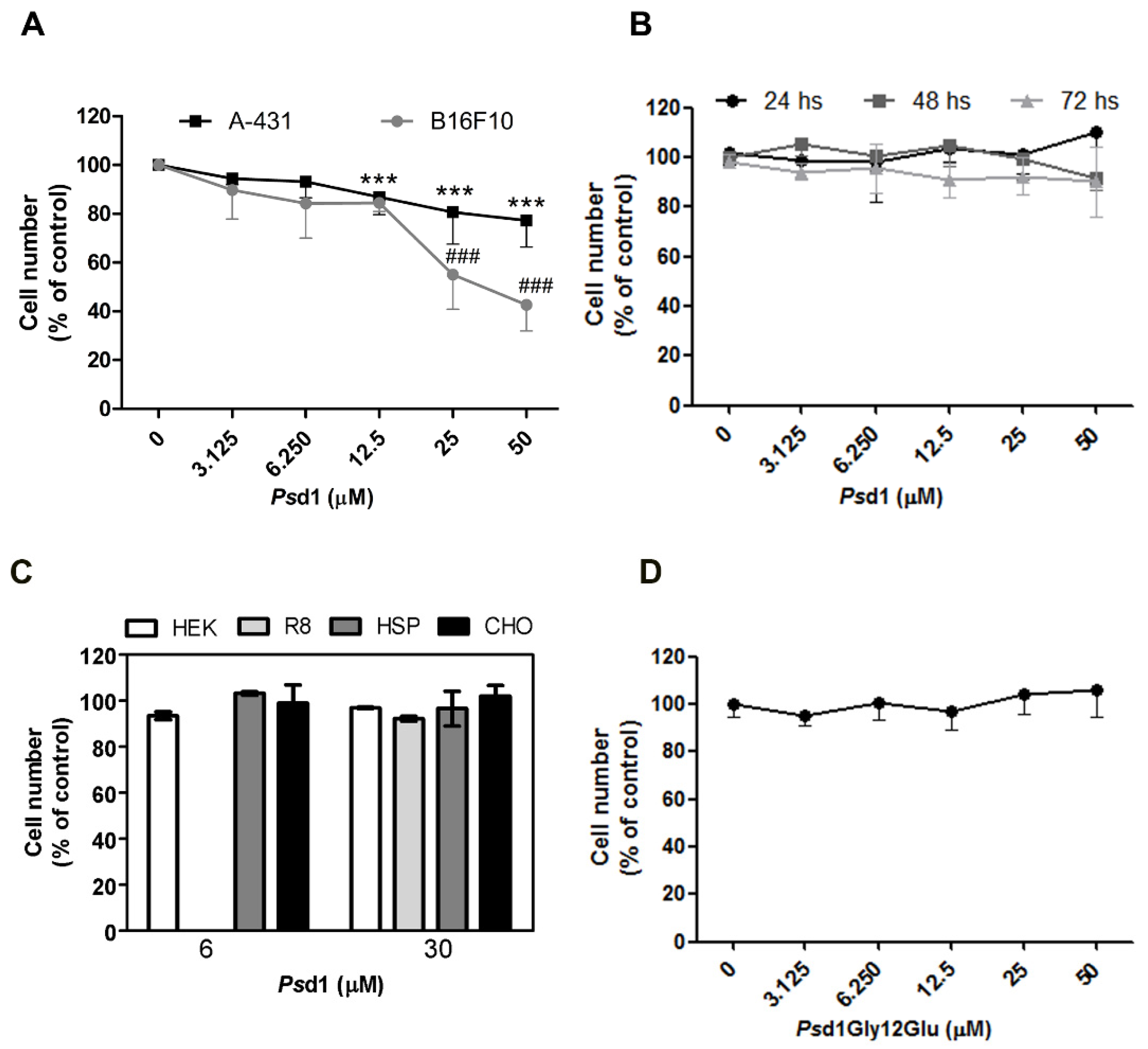
2.1. Psd1 Presents Selective Cytotoxic Effects against Tumor Cells In Vitro
2.2. Insights into the Action Mechanism of Psd1 Antitumor Activity
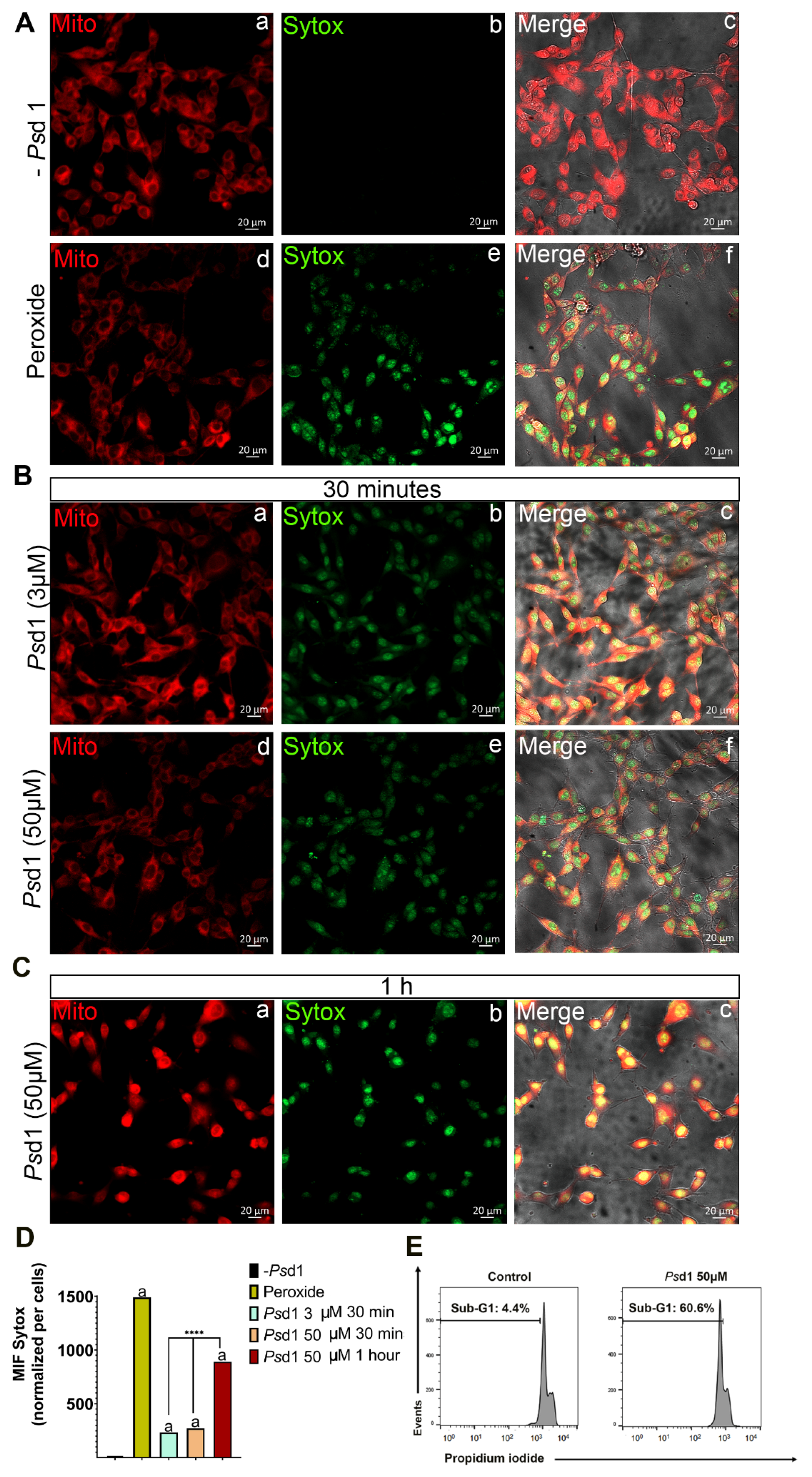
2.2.1. Psd1 Permeabilizes the Plasma Membrane and Induces Apoptosis in B16F10 Cells
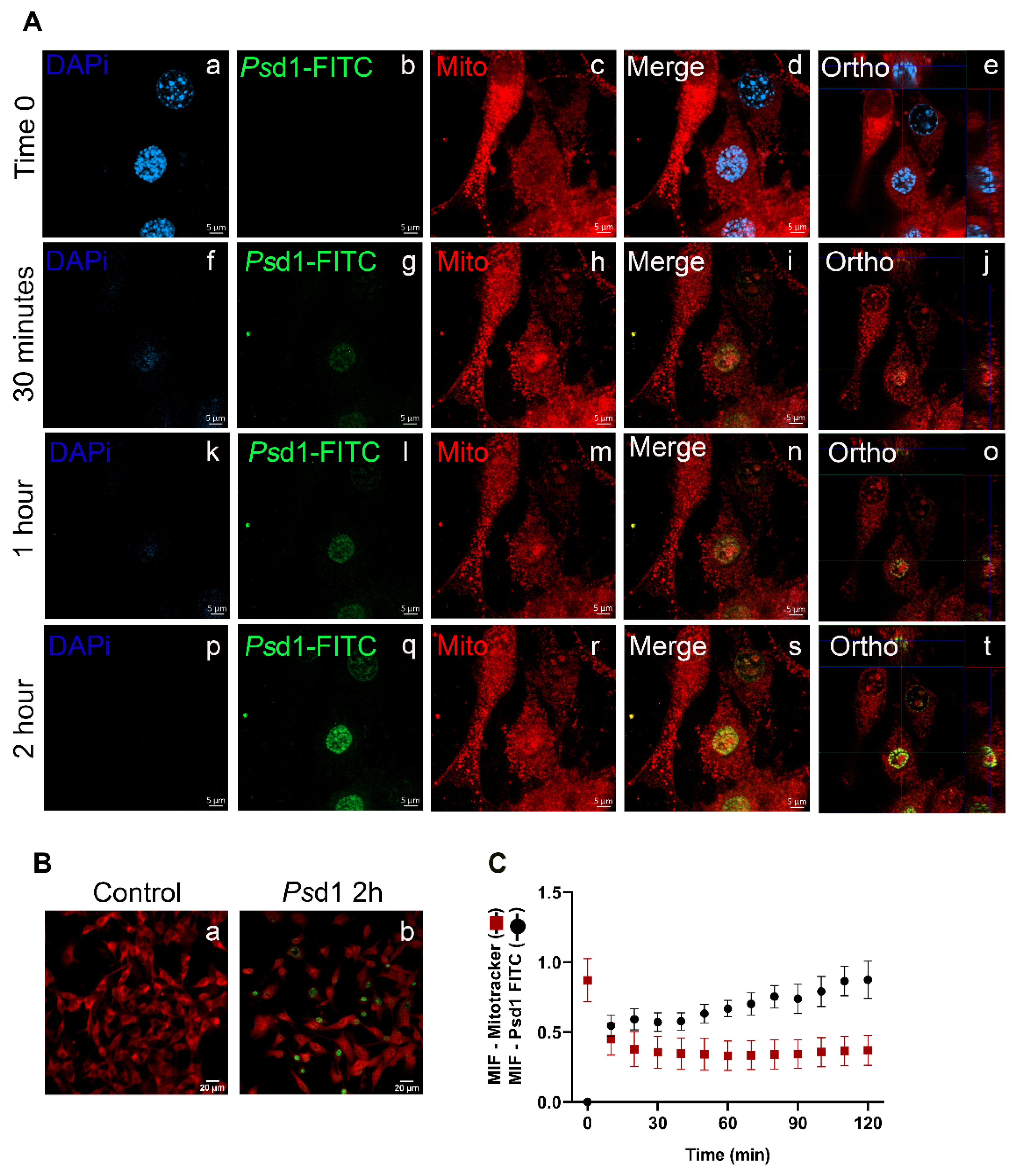
2.2.2. Psd1 Internalizes towards the Nucleus
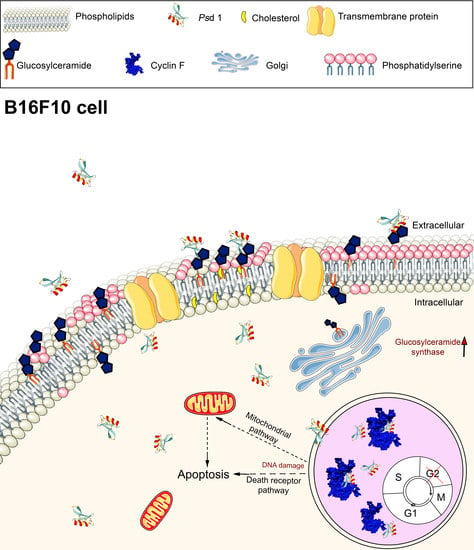
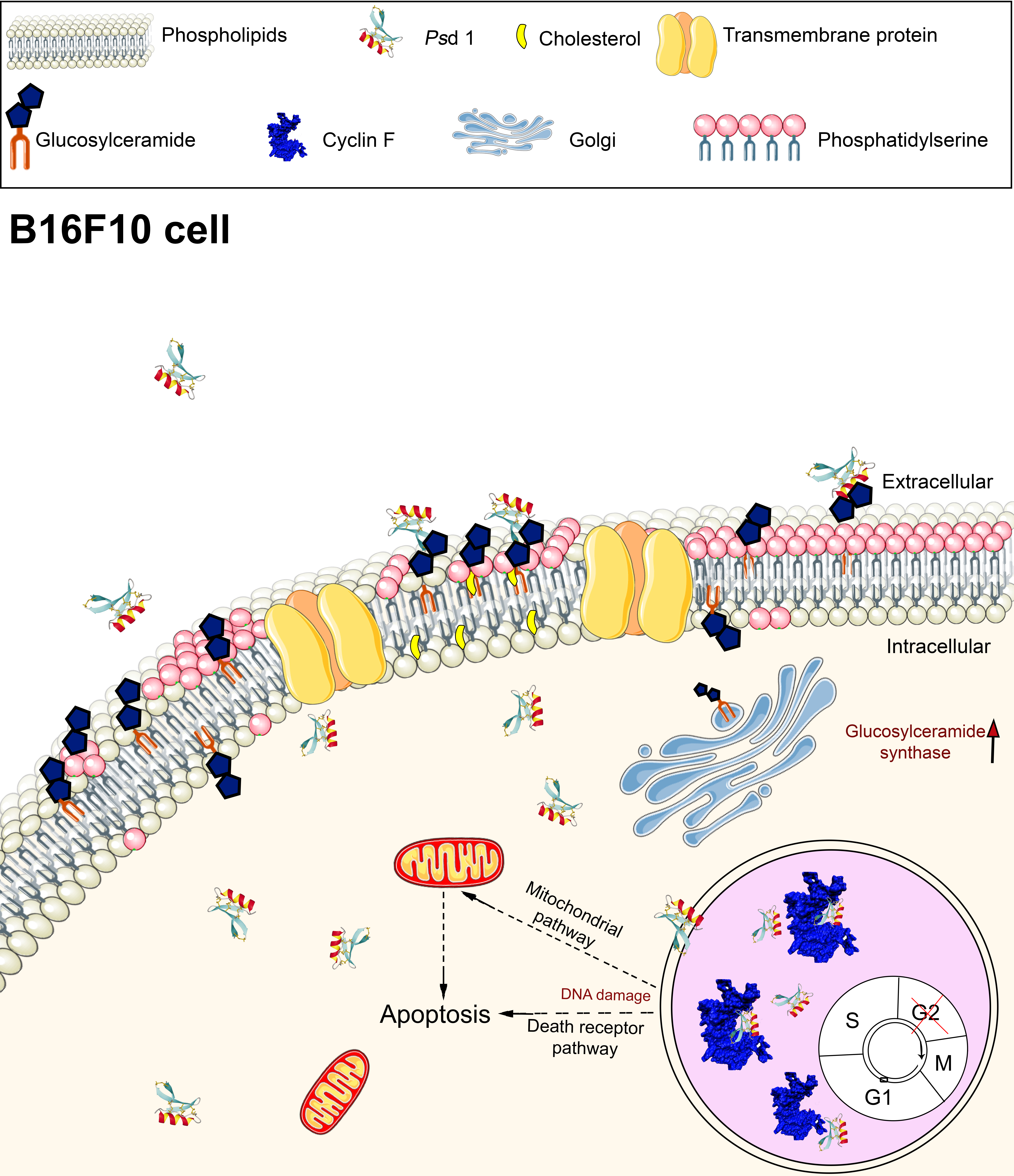
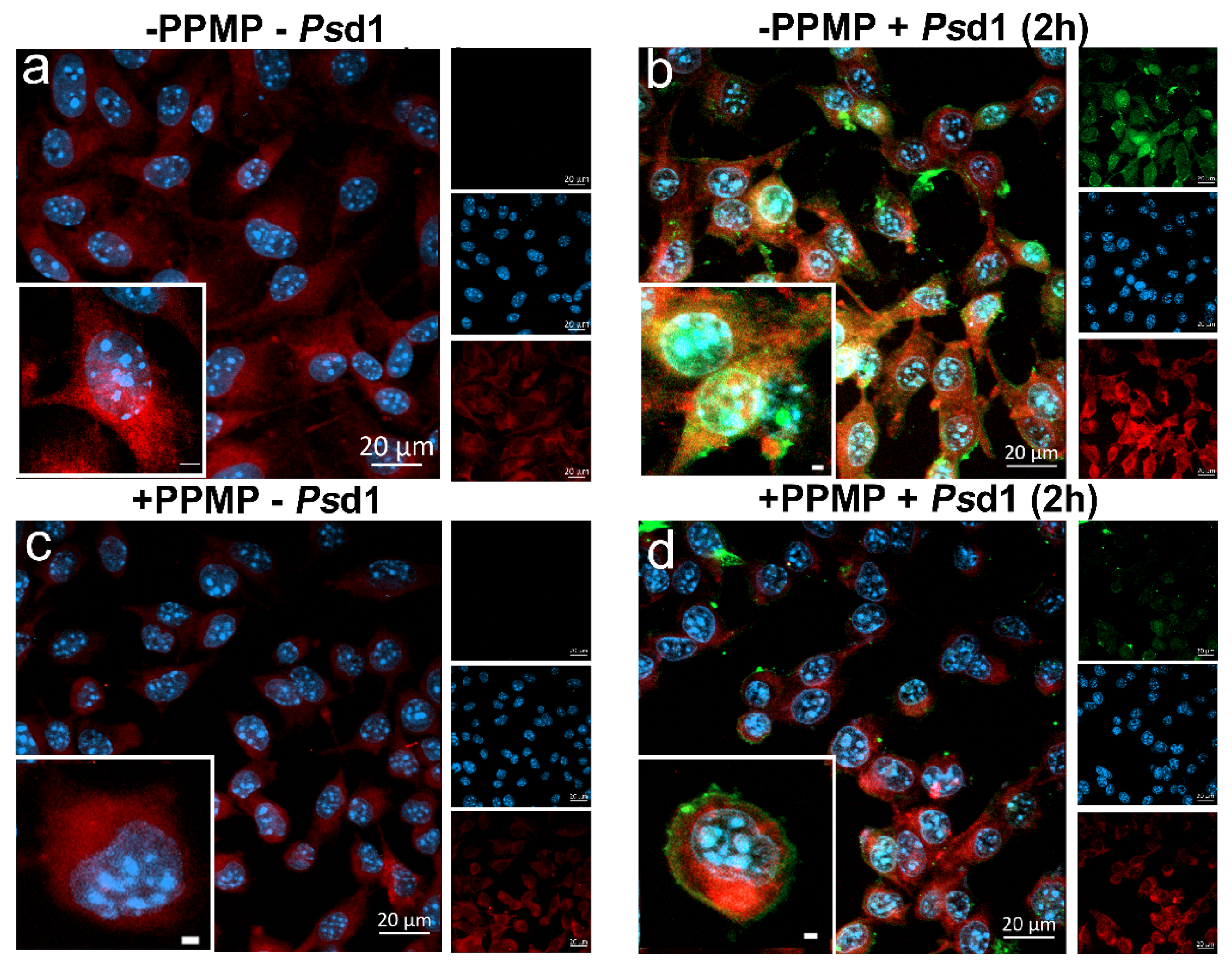
2.2.3. Psd1 Entrance in B16F10 Cells is Glucosylceramide-Dependent
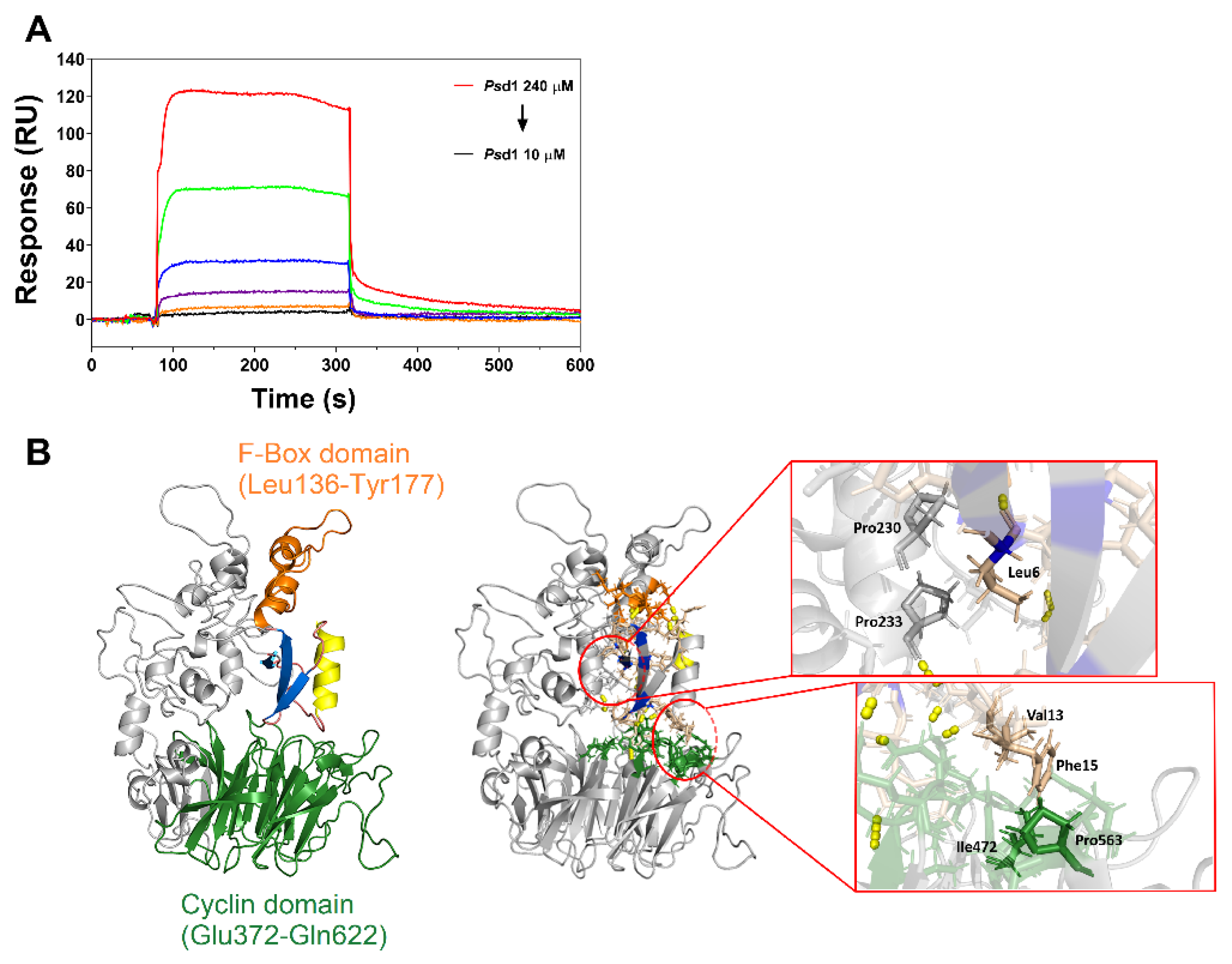
2.3. Psd1–Cyclin F Interaction by Surface Plasmon Resonance (SPR)
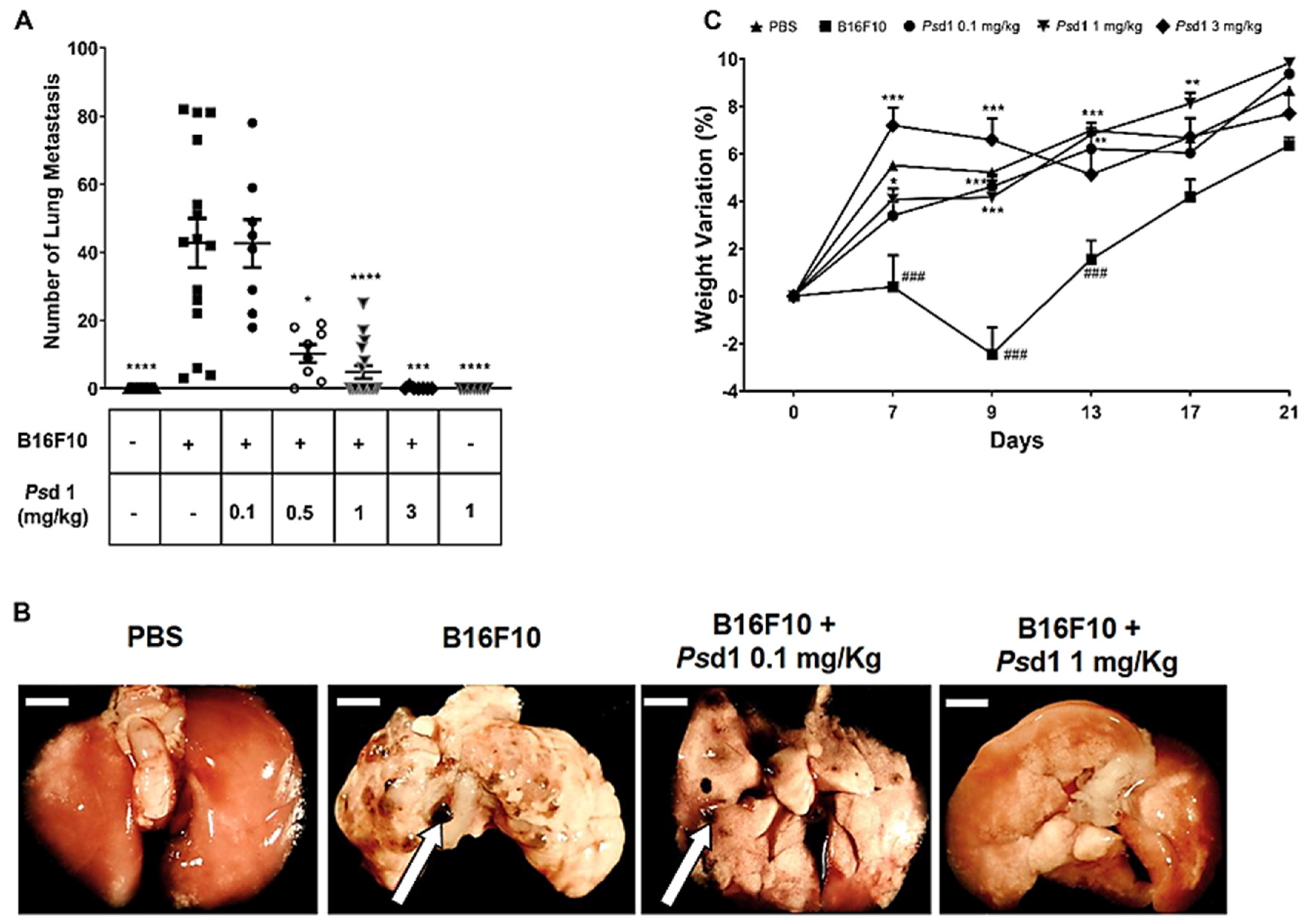
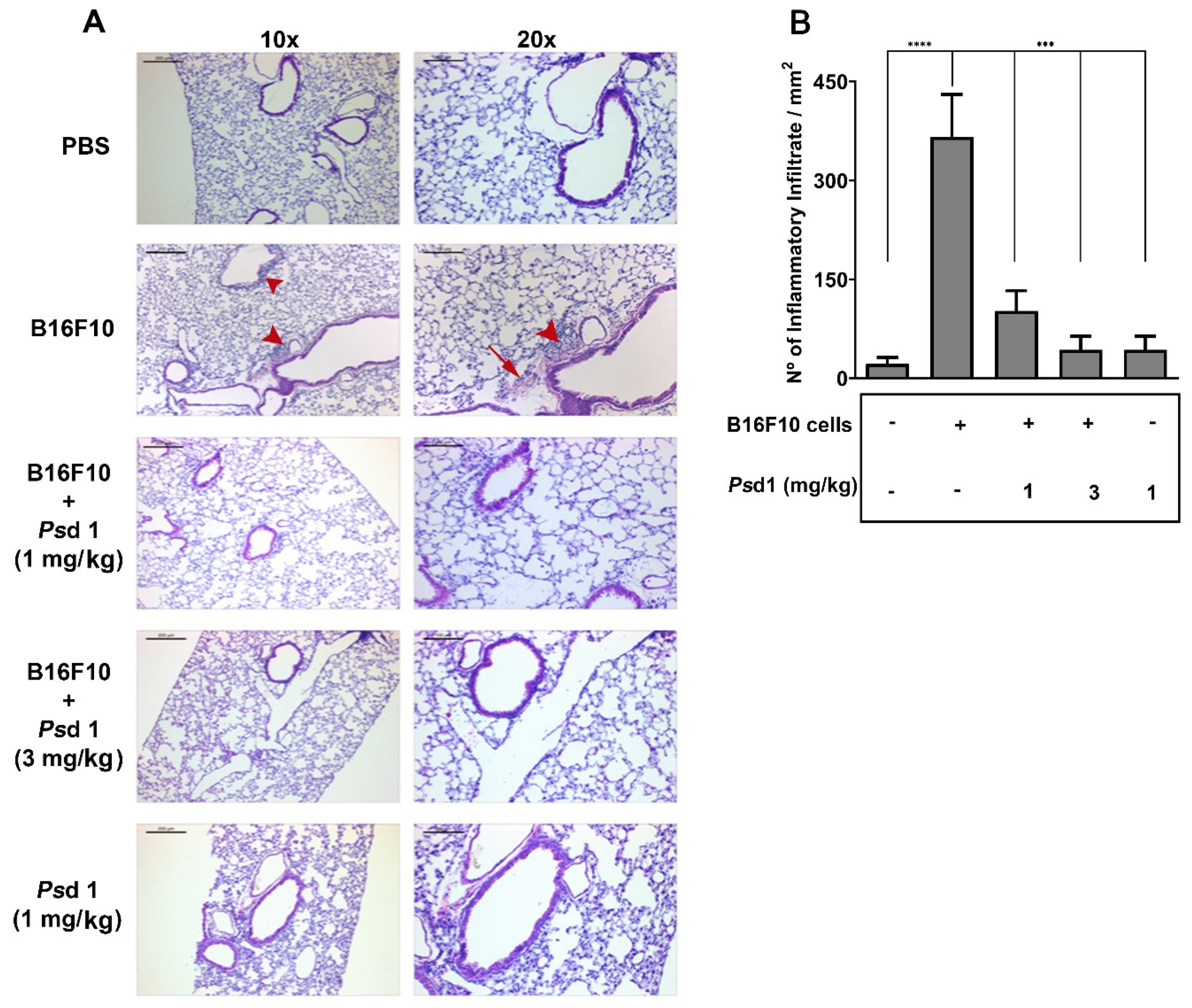
2.4. Psd1 Impaired the Establishment of B16F10 Tumor Metastasis In Vivo
3. Discussion
4. Materials and Methods
4.1. Expression and Purification of Psd1 and the Site-Directed Mutant Psd1 Gly12Glu
4.2. Cell Lines and Culture Conditions
4.3. Cell Viability Assays
4.4. Confocal Microscopy Scanning
4.5. SYTOXGreen (SG) Uptake Assay
4.6. Psd1 Localization Fluorescence Assays
4.7. Glucosylceramide Depletion Studies on Psd1 B16F10 Cell Entrance
4.8. Flow Cytometry-Based Apoptosis Detection
4.9. Surface Plasmon Resonance Studies
4.10. Psd1–Cyclin F Molecular Docking
4.11. Experimental Animals
4.12. Experimental B16F10 Melanoma Metastasis Assays
4.13. Histological Analysis
4.14. Data Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Cools, T.L.; Struyfs, C.; Cammue, B.P.; Thevissen, K. Antifungal plant defensins: Increased insight in their mode of action as a basis for their use to combat fungal infections. Future Microbiol. 2017, 12, 441–454. [Google Scholar] [CrossRef] [PubMed]
- Maroti, G.; Kereszt, A.; Kondorosi, E.; Mergaert, P. Natural roles of antimicrobial peptides in microbes, plants and animals. Res. Microbiol. 2011, 162, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Tam, J.P.; Wang, S.; Wong, K.H.; Tan, W.L. Antimicrobial Peptides from Plants. Pharmaceuticals 2015, 8, 711–757. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.S.; Cabral, K.M.; Zingali, R.B.; Kurtenbach, E. Characterization of two novel defense peptides from pea (Pisum sativum) seeds. Arch. Biochem. Biophys. 2000, 378, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Parisi, K.; Shafee, T.M.A.; Quimbar, P.; van der Weerden, N.L.; Bleackley, M.R.; Anderson, M.A. The evolution, function and mechanisms of action for plant defensins. Semin. Cell Dev. Biol. 2019, 88, 107–118. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro-Aguiar, R.; do Amaral, V.S.G.; Pereira, I.B.; Kurtenbach, E.; Almeida, F.C.L. Nuclear magnetic resonance solution structure of Pisum sativum defensin 2 provides evidence for the presence of hydrophobic surface-clusters. Proteins 2019, 88, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.S.; Cabral, K.M.; Kurtenbach, E.; Almeida, F.C.; Valente, A.P. Solution structure of Pisum sativum defensin 1 by high resolution NMR: Plant defensins, identical backbone with different mechanisms of action. J. Mol. Biol. 2002, 315, 749–757. [Google Scholar] [CrossRef]
- Medeiros, L.N.d.; Domitrovic, T.; Andrade, P.C.d.; Faria, J.; Barreto-Bergter, E.; Weissmüller, G.; Kurtenbach, E. Psd1 binding affinity toward fungal membrane components as assessed by SPR: The role of glucosylceramide in fungal recognition and entry. Pept. Sci. 2014, 102, 456–464. [Google Scholar] [CrossRef]
- Pushpanathan, M.; Gunasekaran, P.; Rajendhran, J. Antimicrobial peptides: Versatile biological properties. Int. J. Pept. 2013, 2013, 675391. [Google Scholar] [CrossRef] [Green Version]
- Amaral, V.S.G.; Fernandes, C.M.; Felício, M.R.; Valle, A.S.; Quintana, P.G.; Almeida, C.C.; Barreto-Bergter, E.; Gonçalves, S.; Santos, N.C.; Kurtenbach, E. Psd2 pea defensin shows a preference for mimetic membrane rafts enriched with glucosylceramide and ergosterol. Biochim. Et Biophys. Acta (Bba)-Biomembr. 2019, 1861, 713–728. [Google Scholar] [CrossRef]
- Fernandes, C.; de Castro, P.; Singh, A.; Fonseca, F.; Pereira, M.; Vila, T.; Atella, G.; Rozental, S.; Savoldi, M.; Del Poeta, M. Functional characterization of the Aspergillus nidulans glucosylceramide pathway reveals that LCB Δ8-desaturation and C9-methylation are relevant to filamentous growth, lipid raft localization and Psd1 defensin activity. Mol. Microbiol. 2016, 102, 488–505. [Google Scholar] [CrossRef] [Green Version]
- Ramamoorthy, V.; Cahoon, E.B.; Li, J.; Thokala, M.; Minto, R.E.; Shah, D.M. Glucosylceramide synthase is essential for alfalfa defensin-mediated growth inhibition but not for pathogenicity of Fusarium graminearum. Mol. Microbiol. 2007, 66, 771–786. [Google Scholar] [CrossRef] [PubMed]
- Thevissen, K.; Warnecke, D.C.; Francois, I.E.; Leipelt, M.; Heinz, E.; Ott, C.; Zähringer, U.; Thomma, B.P.; Ferket, K.K.; Cammue, B.P. Defensins from insects and plants interact with fungal glucosylceramides. J. Biol. Chem. 2004, 279, 3900–3905. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thevissen, K.; Cammue, B.P.; Lemaire, K.; Winderickx, J.; Dickson, R.C.; Lester, R.L.; Ferket, K.K.; Van Even, F.; Parret, A.H.; Broekaert, W.F. A gene encoding a sphingolipid biosynthesis enzyme determines the sensitivity of Saccharomyces cerevisiae to an antifungal plant defensin from dahlia (Dahlia merckii). Proc. Natl. Acad. Sci. USA 2000, 97, 9531–9536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medeiros, L.N.; Angeli, R.; Sarzedas, C.G.; Barreto-Bergter, E.; Valente, A.P.; Kurtenbach, E.; Almeida, F.C. Backbone dynamics of the antifungal Psd1 pea defensin and its correlation with membrane interaction by NMR spectroscopy. Biochim. Biophys. Acta (BBA)-Biomembr. 2010, 1798, 105–113. [Google Scholar] [CrossRef] [Green Version]
- Gonçalves, S.; Teixeira, A.; Abade, J.; de Medeiros, L.N.; Kurtenbach, E.; Santos, N.C. Evaluation of the membrane lipid selectivity of the pea defensin Psd1. Biochim. Biophys. Acta (BBA)-Biomembr. 2012, 1818, 1420–1426. [Google Scholar] [CrossRef] [Green Version]
- Goncalves, S.; Silva, P.M.; Felicio, M.R.; de Medeiros, L.N.; Kurtenbach, E.; Santos, N.C. Psd1 Effects on Candida albicans Planktonic Cells and Biofilms. Front. Cell. Infect. Microbiol. 2017, 7, 249. [Google Scholar] [CrossRef]
- Lobo, D.S.; Pereira, I.B.; Fragel-Madeira, L.; Medeiros, L.N.; Cabral, L.M.; Faria, J.; Bellio, M.; Campos, R.C.; Linden, R.; Kurtenbach, E. Antifungal Pisum sativum defensin 1 interacts with Neurospora crassa cyclin F related to the cell cycle. Biochemistry 2007, 46, 987–996. [Google Scholar] [CrossRef]
- D’Angiolella, V.; Donato, V.; Forrester, F.M.; Jeong, Y.-T.; Pellacani, C.; Kudo, Y.; Saraf, A.; Florens, L.; Washburn, M.P.; Pagano, M. Cyclin F-mediated degradation of ribonucleotide reductase M2 controls genome integrity and DNA repair. Cell 2012, 149, 1023–1034. [Google Scholar] [CrossRef] [Green Version]
- D’Angiolella, V.; Esencay, M.; Pagano, M. A cyclin without cyclin-dependent kinases: Cyclin F controls genome stability through ubiquitin-mediated proteolysis. Trends Cell Biol. 2013, 23, 135–140. [Google Scholar] [CrossRef] [Green Version]
- Cardozo, T.; Pagano, M. The SCF ubiquitin ligase: Insights into a molecular machine. Nat. Rev. Mol. Cell Biol. 2004, 5, 739–751. [Google Scholar] [CrossRef]
- Clijsters, L.; Hoencamp, C.; Calis, J.J.; Marzio, A.; Handgraaf, S.M.; Cuitino, M.C.; Rosenberg, B.R.; Leone, G.; Pagano, M. Cyclin F controls cell-cycle transcriptional outputs by directing the degradation of the three activator E2Fs. Mol. Cell 2019, 74, 1264–1277.e7. [Google Scholar] [CrossRef]
- Guaní-Guerra, E.; Santos-Mendoza, T.; Lugo-Reyes, S.O.; Terán, L.M. Antimicrobial peptides: General overview and clinical implications in human health and disease. Clin. Immunol. 2010, 135, 1–11. [Google Scholar] [CrossRef]
- Kruse, T.; Kristensen, H.H. Using antimicrobial host defense peptides as anti-infective and immunomodulatory agents. Expert Rev. Anti Infect. Ther. 2008, 6, 887–895. [Google Scholar] [CrossRef]
- Riedl, S.; Zweytick, D.; Lohner, K. Membrane-active host defense peptides-challenges and perspectives for the development of novel anticancer drugs. Chem. Phys. Lipids 2011, 164, 766–781. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gaspar, D.; Veiga, A.S.; Castanho, M.A. From antimicrobial to anticancer peptides. A review. Front. Microbiol. 2013, 4, 294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beloribi-Djefaflia, S.; Vasseur, S.; Guillaumond, F. Lipid metabolic reprogramming in cancer cells. Oncogenesis 2016, 5, e189. [Google Scholar] [CrossRef] [PubMed]
- Utsugi, T.; Schroit, A.J.; Connor, J.; Bucana, C.D.; Fidler, I.J. Elevated expression of phosphatidylserine in the outer membrane leaflet of human tumor cells and recognition by activated human blood monocytes. Cancer Res. 1991, 51, 3062–3066. [Google Scholar]
- Riedl, S.; Rinner, B.; Asslaber, M.; Schaider, H.; Walzer, S.; Novak, A.; Lohner, K.; Zweytick, D. In search of a novel target - phosphatidylserine exposed by non-apoptotic tumor cells and metastases of malignancies with poor treatment efficacy. Biochim. Biophys. Acta 2011, 1808, 2638–2645. [Google Scholar] [CrossRef] [Green Version]
- Lavie, Y.; Cao, H.; Bursten, S.L.; Giuliano, A.E.; Cabot, M.C. Accumulation of glucosylceramides in multidrug-resistant cancer cells. J. Biol. Chem. 1996, 271, 19530–19536. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.-Y.; Han, T.-Y.; Giuliano, A.E.; Cabot, M.C. Expression of glucosylceramide synthase, converting ceramide to glucosylceramide, confers adriamycin resistance in human breast cancer cells. J. Biol. Chem. 1999, 274, 1140–1146. [Google Scholar] [CrossRef] [Green Version]
- Morjani, H.; Aouali, N.; Belhoussine, R.; Veldman, R.J.; Levade, T.; Manfait, M. Elevation of glucosylceramide in multidrug-resistant cancer cells and accumulation in cytoplasmic droplets. Int. J. Cancer 2001, 94, 157–165. [Google Scholar] [CrossRef]
- Saito, K.; Takakuwa, N.; Ohnishi, M.; Oda, Y. Presence of glucosylceramide in yeast and its relation to alkali tolerance of yeast. Appl. Microbiol. Biotechnol. 2006, 71, 515–521. [Google Scholar] [CrossRef]
- Borradaile, N.M.; Han, X.; Harp, J.D.; Gale, S.E.; Ory, D.S.; Schaffer, J.E. Disruption of endoplasmic reticulum structure and integrity in lipotoxic cell death. J. Lipid Res. 2006, 47, 2726–2737. [Google Scholar] [CrossRef] [Green Version]
- Li, J.-F.; Zheng, S.-J.; Wang, L.-L.; Liu, S.; Ren, F.; Chen, Y.; Bai, L.; Liu, M.; Duan, Z.P. Glucosylceramide synthase regulates the proliferation and apoptosis of liver cells in vitro by Bcl-2/Bax pathway. Mol. Med. Rep. 2017, 16, 7355–7360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Oliveira, A.R.; da Silva, I.D.C.G.; Lo Turco, E.G.; Júnior, H.A.M.; Chauffaille, M.d.L.L.F. Initial analysis of lipid metabolomic profile reveals differential expression features in myeloid malignancies. J. Cancer Ther. 2015, 6, 1262–1272. [Google Scholar] [CrossRef] [Green Version]
- Jeon, J.H.; Kim, S.K.; Kim, H.J.; Chang, J.; Ahn, C.M.; Chang, Y.S. Lipid raft modulation inhibits NSCLC cell migration through delocalization of the focal adhesion complex. Lung Cancer 2010, 69, 165–171. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Bi, J.; Ampah, K.K.; Zhang, C.; Li, Z.; Jiao, Y.; Wang, X.; Ba, X.; Zeng, X. Lipid raft regulates the initial spreading of melanoma A375 cells by modulating β1 integrin clustering. Int. J. Biochem. Cell B 2013, 45, 1679–1689. [Google Scholar] [CrossRef] [PubMed]
- Al-Benna, S.; Shai, Y.; Jacobsen, F.; Steinstraesser, L. Oncolytic activities of host defense peptides. Int. J. Mol. Sci. 2011, 12, 8027–8051. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Sorvina, A.; Bader, C.A.; Darby, J.R.; Lock, M.C.; Soo, J.Y.; Johnson, I.R.; Caporale, C.; Voelcker, N.H.; Stagni, S.; Massi, M. Mitochondrial imaging in live or fixed tissues using a luminescent iridium complex. Sci. Rep. 2018, 8, 1–8. [Google Scholar] [CrossRef]
- Guimaraes, C.A.; Linden, R. Programmed cell deaths. Apoptosis and alternative deathstyles. Eur. J. Biochem. 2004, 271, 1638–1650. [Google Scholar] [CrossRef]
- Ablan, S.; Rawat, S.S.; Blumenthal, R.; Puri, A. Entry of influenza virus into a glycosphingolipid-deficient mouse skin fibroblast cell line. Arch. Virol. 2001, 146, 2227–2238. [Google Scholar] [CrossRef]
- Chan, S.Y.; Hilchie, A.L.; Brown, M.G.; Anderson, R.; Hoskin, D.W. Apoptosis induced by intracellular ceramide accumulation in MDA-MB-435 breast carcinoma cells is dependent on the generation of reactive oxygen species. Exp. Mol. Pathol. 2007, 82, 1–11. [Google Scholar] [CrossRef]
- Lee, H.S.; Park, C.B.; Kim, J.M.; Jang, S.A.; Park, I.Y.; Kim, M.S.; Cho, J.H.; Kim, S.C. Mechanism of anticancer activity of buforin IIb, a histone H2A-derived peptide. Cancer Lett. 2008, 271, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Inokuchi, J.; Usuki, S.; Jimbo, M. Stimulation of glycosphingolipid biosynthesis by L-threo-1-phenyl-2-decanoylamino-1-propanol and its homologs in B16 melanoma cells. J. Biochem. 1995, 117, 766–773. [Google Scholar] [CrossRef] [PubMed]
- Coelho, V.d.M.; Nguyen, D.; Giri, B.; Bunbury, A.; Schaffer, E.; Taub, D.D. Quantitative differences in lipid raft components between murine CD4+ and CD8+ T cells. BMC Immunol. 2004, 5, 2. [Google Scholar]
- Nichols, B.J.; Kenworthy, A.K.; Polishchuk, R.S.; Lodge, R.; Roberts, T.H.; Hirschberg, K.; Phair, R.D.; Lippincott-Schwartz, J. Rapid cycling of lipid raft markers between the cell surface and Golgi complex. J. Cell Biol. 2001, 153, 529–542. [Google Scholar] [CrossRef] [PubMed]
- Merritt, E.A.; Sarfaty, S.; Akker, F.V.D.; L’hoir, C.; Martial, J.A.; Hol, W.G. Crystal structure of cholera toxin B-pentamer bound to receptor GM1 pentasaccharide. Protein Sci. 1994, 3, 166–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calorini, L.; Fallani, A.; Tombaccini, D.; Mugnai, G.; Ruggieri, S. Lipid composition of cultured B16 melanoma cell variants with different lung-colonizing potential. Lipids 1987, 22, 651–656. [Google Scholar] [CrossRef]
- Diaz-Rohrer, B.B.; Levental, K.R.; Simons, K.; Levental, I. Membrane raft association is a determinant of plasma membrane localization. Proc. Natl. Acad. Sci. USA 2014, 111, 8500–8505. [Google Scholar] [CrossRef] [Green Version]
- Diaz-Rohrer, B.; Levental, K.R.; Levental, I. Rafting through traffic: Membrane domains in cellular logistics. Biochim. Biophys. Acta 2014, 1838, 3003–3013. [Google Scholar] [CrossRef] [Green Version]
- Bhatia, S.; Tykodi, S.S.; Thompson, J.A. Treatment of metastatic melanoma: An overview. Oncology 2009, 23, 488–496. [Google Scholar]
- Wong, J.H.; Ng, T.B. Sesquin, a potent defensin-like antimicrobial peptide from ground beans with inhibitory activities toward tumor cells and HIV-1 reverse transcriptase. Peptides 2005, 26, 1120–1126. [Google Scholar] [CrossRef]
- Wong, J.H.; Ng, T.B. Lunatusin, a trypsin-stable antimicrobial peptide from lima beans (Phaseolus lunatus L.). Peptides 2005, 26, 2086–2092. [Google Scholar] [CrossRef]
- Wong, J.H.; Ng, T. Limenin, a defensin-like peptide with multiple exploitable activities from shelf beans. J. Pept. Sci. 2006, 12, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.; Wong, J.H.; Ng, T.B. A defensin with highly potent antipathogenic activities from the seeds of purple pole bean. Biosci. Rep. 2009, 30, 101–109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ngai, P.H.; Ng, T.B. Phaseococcin, an antifungal protein with antiproliferative and anti-HIV-1 reverse transcriptase activities from small scarlet runner beans. Biochem. Cell Biol. 2005, 83, 212–220. [Google Scholar] [CrossRef] [PubMed]
- Ngai, P.H.; Ng, T. Coccinin, an antifungal peptide with antiproliferative and HIV-1 reverse transcriptase inhibitory activities from large scarlet runner beans. Peptides 2004, 25, 2063–2068. [Google Scholar] [CrossRef] [PubMed]
- Anaya-Lopez, J.L.; Lopez-Meza, J.E.; Baizabal-Aguirre, V.M.; Cano-Camacho, H.; Ochoa-Zarzosa, A. Fungicidal and cytotoxic activity of a Capsicum chinense defensin expressed by endothelial cells. Biotechnol. Lett. 2006, 28, 1101–1108. [Google Scholar] [CrossRef] [PubMed]
- Pendergrass, W.; Wolf, N.; Poot, M. Efficacy of MitoTracker Green™ and CMXrosamine to measure changes in mitochondrial membrane potentials in living cells and tissues. Cytom. A 2004, 61, 162–169. [Google Scholar] [CrossRef] [PubMed]
- Lemasters, J.J. Charpter 1—Molecular Mechanisms of Cell Death. In Molecular Pathology; Academic Press: Cambridge, MA, USA, 2018; pp. 1–24. [Google Scholar] [CrossRef]
- Liu, X.; Cheng, J.C.; Turner, L.S.; Elojeimy, S.; Beckham, T.H.; Bielawska, A.; Keane, T.E.; Hannun, Y.A.; Norris, J.S. Acid ceramidase upregulation in prostate cancer: Role in tumor development and implications for therapy. Expert Opin. Ther. Targets 2009, 13, 1449–1458. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Cui, X.; Zhang, N.; Li, M.; Bai, Y.; Han, X.; Shi, Y.; Liu, H. Comprehensive lipid profiling of plasma in patients with benign breast tumor and breast cancer reveals novel biomarkers. Anal. Bioanal. Chem. 2015, 407, 5065–5077. [Google Scholar] [CrossRef]
- Birge, R.; Boeltz, S.; Kumar, S.; Carlson, J.; Wanderley, J.; Calianese, D.; Barcinski, M.; Brekken, R.; Huang, X.; Hutchins, J. Phosphatidylserine is a global immunosuppressive signal in efferocytosis, infectious disease, and cancer. Cell Death Differ. 2016, 23, 962–978. [Google Scholar] [CrossRef] [Green Version]
- Li, A.; Yao, L.; Fang, Y.; Yang, K.; Jiang, W.; Huang, W.; Cai, Z. Specifically blocking the fatty acid synthesis to inhibit the malignant phenotype of bladder cancer. Int. J. Biol. Sci. 2019, 15, 1610–1617. [Google Scholar] [CrossRef] [Green Version]
- Menendez, J.A.; Lupu, R. Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 2007, 7, 763–777. [Google Scholar] [CrossRef] [PubMed]
- Qi, H.; Priyadarsini, S.; Nicholas, S.E.; Sarker-Nag, A.; Allegood, J.; Chalfant, C.E.; Mandal, N.A.; Karamichos, D. Analysis of sphingolipids in human corneal fibroblasts from normal and keratoconus patients. J. Lipid Res. 2017, 58, 636–648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Meer, G.; Voelker, D.R.; Feigenson, G.W. Membrane lipids: Where they are and how they behave. Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [Google Scholar] [CrossRef] [PubMed]
- Van Meer, G.; de Kroon, A.I. Lipid map of the mammalian cell. J. Cell Sci. 2011, 124, 5–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryland, L.K.; Fox, T.E.; Liu, X.; Loughran, T.P.; Kester, M. Dysregulation of sphingolipid metabolism in cancer. Cancer Biol. Ther. 2011, 11, 138–149. [Google Scholar] [CrossRef] [PubMed]
- Ternes, P.; Wobbe, T.; Schwarz, M.; Albrecht, S.; Feussner, K.; Riezman, I.; Cregg, J.M.; Heinz, E.; Riezman, H.; Feussner, I.; et al. Two pathways of sphingolipid biosynthesis are separated in the yeast Pichia pastoris. J. Biol. Chem. 2011, 286, 11401–11414. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Tong, T.-T.; Yau, L.-F.; Chen, C.-Y.; Mi, J.-N.; Wang, J.-R.; Jiang, Z.-H. LC-MS based sphingolipidomic study on A549 human lung adenocarcinoma cell line and its taxol-resistant strain. BMC Cancer 2018, 18, 799. [Google Scholar] [CrossRef]
- Machado, L.; De Paula, V.S.; Pustovalova, Y.; Bezsonova, I.; Valente, A.P.; Korzhnev, D.M.; Almeida, F.C.L. Conformational dynamics of a cysteine-stabilized plant defensin reveals an evolutionary mechanism to expose hydrophobic residues. Biochemistry 2018, 57, 5797–5806. [Google Scholar] [CrossRef]
- Choudhury, R.; Bonacci, T.; Wang, X.; Truong, A.; Arceci, A.; Zhang, Y.; Mills, C.A.; Kernan, J.L.; Liu, P.; Emanuele, M.J. The E3 ubiquitin ligase SCF (cyclin F) transmits AKT signaling to the cell-cycle machinery. Cell Rep. 2017, 20, 3212–3222. [Google Scholar] [CrossRef] [Green Version]
- Bai, C.; Richman, R.; Elledge, S.J. Human cyclin F. EMBO J. 1994, 13, 6087–6098. [Google Scholar] [CrossRef]
- Zheng, N.; Zhou, Q.; Wang, Z.; Wei, W. Recent advances in SCF ubiquitin ligase complex: Clinical implications. BBA-Rev. Cancer 2016, 1866, 12–22. [Google Scholar] [CrossRef] [Green Version]
- D’Angiolella, V.; Donato, V.; Vijayakumar, S.; Saraf, A.; Florens, L.; Washburn, M.P.; Dynlacht, B.; Pagano, M. SCF Cyclin F controls centrosome homeostasis and mitotic fidelity through CP110 degradation. Nature 2010, 466, 138–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emanuele, M.J.; Elia, A.E.; Xu, Q.; Thoma, C.R.; Izhar, L.; Leng, Y.; Guo, A.; Chen, Y.-N.; Rush, J.; Hsu, P.W.-C. Global identification of modular cullin-RING ligase substrates. Cell 2011, 147, 459–474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, D.; Hoffmann, S.; Komseli, E.-S.; Rappsilber, J.; Gorgoulis, V.; Sorensen, C.S. SCF Cyclin F-dependent degradation of CDC6 suppresses DNA re-replication. Nat. Commun. 2016, 7, 10530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elia, A.E.; Boardman, A.P.; Wang, D.C.; Huttlin, E.L.; Everley, R.A.; Dephoure, N.; Zhou, C.; Koren, I.; Gygi, S.P.; Elledge, S.J. Quantitative proteomic atlas of ubiquitination and acetylation in the DNA damage response. Mol. Cell 2015, 59, 867–881. [Google Scholar] [CrossRef] [Green Version]
- Dankert, J.F.; Rona, G.; Clijsters, L.; Geter, P.; Skaar, J.R.; Bermudez-Hernandez, K.; Sassani, E.; Fenyö, D.; Ueberheide, B.; Schneider, R. Cyclin F-mediated degradation of SLBP limits H2A. X accumulation and apoptosis upon genotoxic stress in G2. Mol. Cell 2016, 64, 507–519. [Google Scholar] [CrossRef] [Green Version]
- Jones, K.B.; Su, L.; Jin, H.; Lenz, C.; Randall, R.L.; Underhill, T.M.; Nielsen, T.O.; Sharma, S.; Capecchi, M.R. SS18-SSX2 and the mitochondrial apoptosis pathway in mouse and human synovial sarcomas. Oncogene 2013, 32, 2365–2375.e5. [Google Scholar] [CrossRef] [Green Version]
- Macheret, M.; Halazonetis, T.D. Intragenic origins due to short G1 phases underlie oncogene-induced DNA replication stress. Nature 2018, 555, 112. [Google Scholar] [CrossRef]
- Tchirkov, A.; Khalil, T.; Chautard, E.; Mokhtari, K.; Veronese, L.; Irthum, B.; Vago, P.; Kémény, J.; Verrelle, P. Interleukin-6 gene amplification and shortened survival in glioblastoma patients. Br. J. Cancer 2007, 96, 474–476. [Google Scholar] [CrossRef] [Green Version]
- Roudi, R.; Syn, N.L.; Roudbary, M. Antimicrobial peptides as biologic and immunotherapeutic agents against cancer: A comprehensive overview. Front. Immunol. 2017, 8, 1320. [Google Scholar] [CrossRef] [Green Version]
- Lowry, O.H.; Rosebrough, N.J.; Farr, A.L.; Randall, R.J. Protein measurement with the Folin phenol reagent. J. Biol. Chem. 1951, 193, 265–275. [Google Scholar]
- Kozakov, D.; Hall, D.R.; Xia, B.; Porter, K.A.; Padhorny, D.; Yueh, C.; Beglov, D.; Vajda, S. The ClusPro web server for protein–protein docking. Nat. Protoc. 2017, 12, 255–278. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Amaral, V.S.G.d.; Santos, S.A.C.S.; de Andrade, P.C.; Nowatzki, J.; Júnior, N.S.; de Medeiros, L.N.; Gitirana, L.B.; Pascutti, P.G.; Almeida, V.H.; Monteiro, R.Q.; et al. Pisum sativum Defensin 1 Eradicates Mouse Metastatic Lung Nodules from B16F10 Melanoma Cells. Int. J. Mol. Sci. 2020, 21, 2662. https://doi.org/10.3390/ijms21082662
Amaral VSGd, Santos SACS, de Andrade PC, Nowatzki J, Júnior NS, de Medeiros LN, Gitirana LB, Pascutti PG, Almeida VH, Monteiro RQ, et al. Pisum sativum Defensin 1 Eradicates Mouse Metastatic Lung Nodules from B16F10 Melanoma Cells. International Journal of Molecular Sciences. 2020; 21(8):2662. https://doi.org/10.3390/ijms21082662
Chicago/Turabian StyleAmaral, Virginia Sara Grancieri do, Stephanie Alexia Cristina Silva Santos, Paula Cavalcante de Andrade, Jenifer Nowatzki, Nilton Silva Júnior, Luciano Neves de Medeiros, Lycia Brito Gitirana, Pedro Geraldo Pascutti, Vitor H. Almeida, Robson Q. Monteiro, and et al. 2020. "Pisum sativum Defensin 1 Eradicates Mouse Metastatic Lung Nodules from B16F10 Melanoma Cells" International Journal of Molecular Sciences 21, no. 8: 2662. https://doi.org/10.3390/ijms21082662