Ceramide Kinase Is Upregulated in Metastatic Breast Cancer Cells and Contributes to Migration and Invasion by Activation of PI 3-Kinase and Akt


Abstract
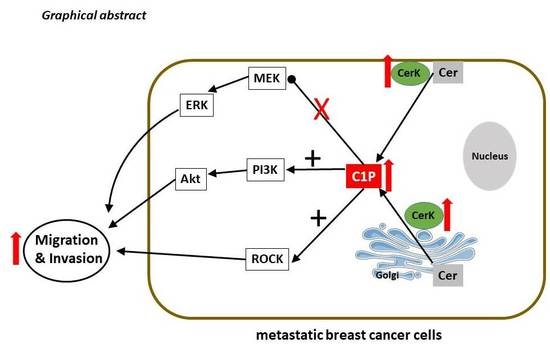
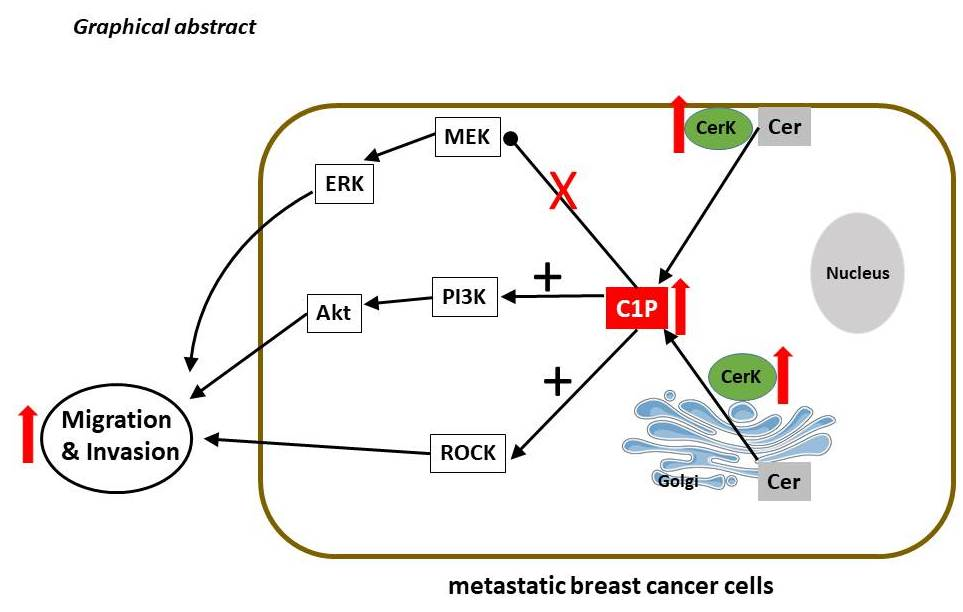
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
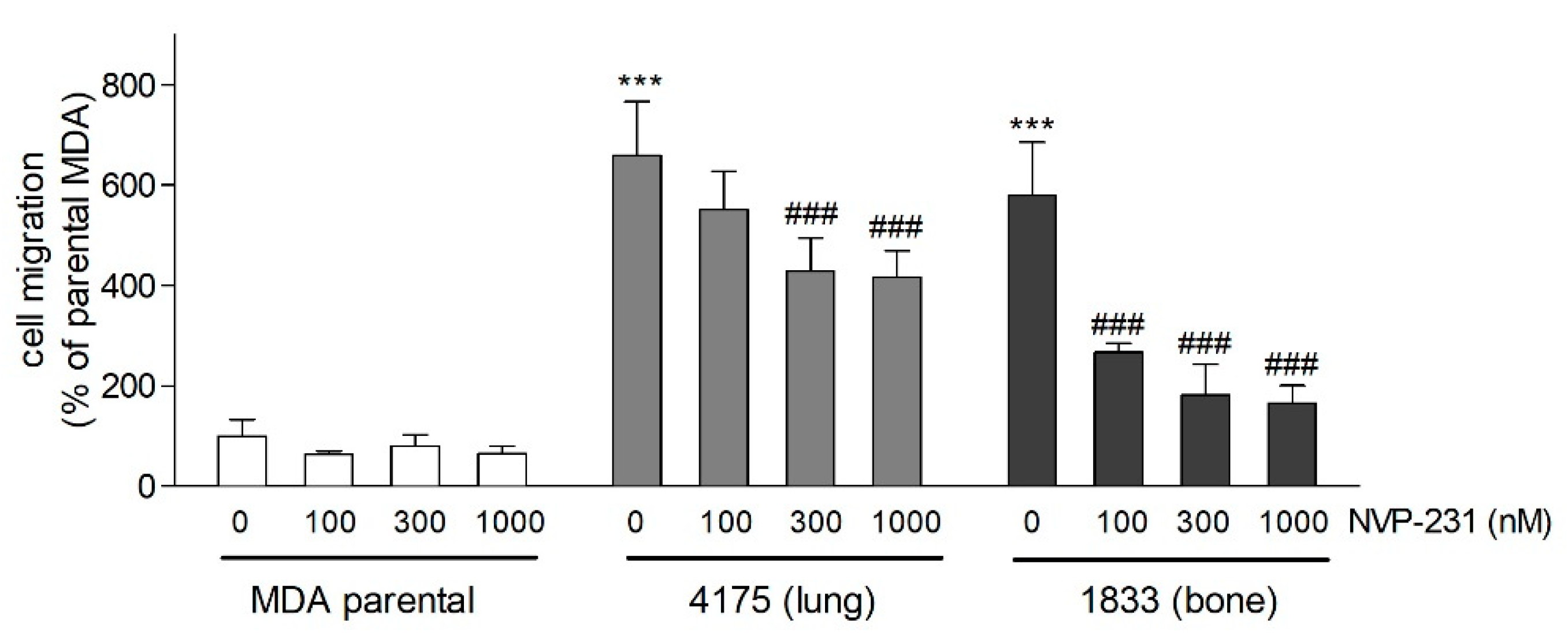
2. Results
3. Discussion
4. Materials and Methods
4.1. Chemicals
4.2. Cell Lines and Cell Culture Conditions
4.3. CerK Overexpression in Parental MDA-MB-231 and MCF-7 Cells
4.4. CerK Knockdown in the Metastatic Sublines 4175 and 1833
4.5. Cell Stimulation, Homogenization, and Western Blotting
4.6. Quantitative Real-Time PCR
4.7. CerK Activity Assay
4.8. Migration Assay
4.9. Invasion Assay
4.10. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| C1P | ceramide 1-phosphate |
| CerK | ceramide kinase |
| cPLA2 | cytosolic phospholipase A2 |
| DMEM | Dulbecco’s Modified Eagle Medium |
| ER | estrogen receptor |
| FBS | fetal bovine serum |
| kd | knockdown |
| PBS | phosphate-buffered saline |
| PGE2 | prostaglandin E2 |
| PH | pleckstrin homology domain |
| PI3K | phosphoinositide 3-kinase |
| PKC | protein kinase C |
| PPAR | peroxisome proliferator activated receptor |
| S1P | sphingosine 1-phosphate |
| SDS-PAGE | sodium dodecyl sulfate-polyacrylamide gel electrophoresis |
| shRNA | small hairpin RNA |
| TLC | thin layer chromatography |
References
- Bottos, A.; Hynes, N.E. Cancer staying together on the road to metastasis. Nature 2014, 514, 309–310. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.; Naishadham, D.; Jemal, A. Cancer statistics, 2013. CA-Cancer J. Clin. 2013, 63, 11–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newton, J.; Lima, S.; Maceyka, M.; Spiegel, S. Revisiting the sphingolipid rheostat: Evolving concepts in cancer therapy. Exp. Cell Res. 2015, 333, 195–200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huwiler, A.; Pfeilschifter, J. Altering the sphingosine-1-phosphate/ceramide balance: A promising approach for tumor therapy. Curr. Pharm. Des. 2006, 12, 4625–4635. [Google Scholar] [CrossRef] [PubMed]
- Huwiler, A.; Zangemeister-Wittke, U. Targeting the conversion of ceramide to sphingosine 1-phosphate as a novel strategy for cancer therapy. Crit. Rev. Oncol. Hematol. 2007, 63, 150–159. [Google Scholar] [CrossRef]
- Pyne, N.J.; Pyne, S. Sphingosine 1-phosphate and cancer. Nat. Rev. Cancer 2010, 10, 489–503. [Google Scholar] [CrossRef] [Green Version]
- Haass, N.K.; Nassif, N.; McGowan, E.M. Switching the sphingolipid rheostat in the treatment of diabetes and cancer comorbidity from a problem to an advantage. Biomed. Res. Int. 2015, 2015, 165105. [Google Scholar] [CrossRef] [Green Version]
- Presa, N.; Gomez-Larrauri, A.; Rivera, I.G.; Ordonez, M.; Trueba, M.; Gomez-Munoz, A. Regulation of cell migration and inflammation by ceramide 1-phosphate. Biochim. Biophys. Acta 2016, 1861, 402–409. [Google Scholar] [CrossRef]
- Bornancin, F. Ceramide kinase: The first decade. Cell Signal. 2011, 23, 999–1008. [Google Scholar] [CrossRef]
- Bajjalieh, S.M.; Martin, T.F.; Floor, E. Synaptic vesicle ceramide kinase. A calcium-stimulated lipid kinase that co-purifies with brain synaptic vesicles. J. Biol. Chem. 1989, 264, 14354–14360. [Google Scholar]
- Hinkovska-Galcheva, V.T.; Boxer, L.A.; Mansfield, P.J.; Harsh, D.; Blackwood, A.; Shayman, J.A. The formation of ceramide-1-phosphate during neutrophil phagocytosis and its role in liposome fusion. J. Biol. Chem. 1998, 273, 33203–33209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsutake, S.; Kim, T.J.; Inagaki, Y.; Kato, M.; Yamashita, T.; Igarashi, Y. Ceramide kinase is a mediator of calcium-dependent degranulation in mast cells. J. Biol. Chem. 2004, 279, 17570–17577. [Google Scholar] [CrossRef] [Green Version]
- Rivera, I.G.; Ordonez, M.; Presa, N.; Gangoiti, P.; Gomez-Larrauri, A.; Trueba, M.; Fox, T.; Kester, M.; Gomez-Munoz, A. Ceramide 1-phosphate regulates cell migration and invasion of human pancreatic cancer cells. Biochem. Pharmacol. 2016, 102, 107–119. [Google Scholar] [CrossRef] [PubMed]
- Pastukhov, O.; Schwalm, S.; Zangemeister-Wittke, U.; Fabbro, D.; Bornancin, F.; Japtok, L.; Kleuser, B.; Pfeilschifter, J.; Huwiler, A. The ceramide kinase inhibitor NVP-231 inhibits breast and lung cancer cell proliferation by inducing M phase arrest and subsequent cell death. Br. J. Pharmacol. 2014, 171, 5829–5844. [Google Scholar] [CrossRef] [Green Version]
- Pettus, B.J.; Bielawska, A.; Spiegel, S.; Roddy, P.; Hannun, Y.A.; Chalfant, C.E. Ceramide kinase mediates cytokine- and calcium ionophore-induced arachidonic acid release. J. Biol. Chem. 2003, 278, 38206–38213. [Google Scholar] [CrossRef] [Green Version]
- Sugiura, M.; Kono, K.; Liu, H.; Shimizugawa, T.; Minekura, H.; Spiegel, S.; Kohama, T. Ceramide kinase, a novel lipid kinase. Molecular cloning and functional characterization. J. Biol. Chem. 2002, 277, 23294–23300. [Google Scholar] [CrossRef] [Green Version]
- Rovina, P.; Schanzer, A.; Graf, C.; Mechtcheriakova, D.; Jaritz, M.; Bornancin, F. Subcellular localization of ceramide kinase and ceramide kinase-like protein requires interplay of their pleckstrin homology domain-containing N-terminal regions together with C-terminal domains. Biochim. Biophys. Acta 2009, 1791, 1023–1030. [Google Scholar] [CrossRef]
- Kim, T.J.; Mitsutake, S.; Igarashi, Y. The interaction between the pleckstrin homology domain of ceramide kinase and phosphatidylinositol 4,5-bisphosphate regulates the plasma membrane targeting and ceramide 1-phosphate levels. Biochem. Biophys. Res. Commun. 2006, 342, 611–617. [Google Scholar] [CrossRef]
- Lamour, N.F.; Stahelin, R.V.; Wijesinghe, D.S.; Maceyka, M.; Wang, E.; Allegood, J.C.; Merrill, A.H., Jr.; Cho, W.; Chalfant, C.E. Ceramide kinase uses ceramide provided by ceramide transport protein: Localization to organelles of eicosanoid synthesis. J. Lipid Res. 2007, 48, 1293–1304. [Google Scholar] [CrossRef] [Green Version]
- Mishra, S.K.; Gao, Y.G.; Deng, Y.; Chalfant, C.E.; Hinchcliffe, E.H.; Brown, R.E. Cptp: A sphingolipid transfer protein that regulates autophagy and inflammasome activation. Autophagy 2018, 14, 862–879. [Google Scholar] [CrossRef] [Green Version]
- Ruckhaberle, E.; Karn, T.; Rody, A.; Hanker, L.; Gatje, R.; Metzler, D.; Holtrich, U.; Kaufmann, M. Gene expression of ceramide kinase, galactosyl ceramide synthase and ganglioside GD3 synthase is associated with prognosis in breast cancer. J. Cancer Res. Clin. Oncol. 2009, 135, 1005–1013. [Google Scholar] [CrossRef] [PubMed]
- Payne, A.W.; Pant, D.K.; Pan, T.C.; Chodosh, L.A. Ceramide kinase promotes tumor cell survival and mammary tumor recurrence. Cancer Res. 2014, 74, 6352–6363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Minn, A.J.; Gupta, G.P.; Siegel, P.M.; Bos, P.D.; Shu, W.P.; Giri, D.D.; Viale, A.; Olshen, A.B.; Gerald, W.L.; Massague, J. Genes that mediate breast cancer metastasis to lung. Nature 2005, 436, 518–524. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.B.; Siegel, P.M.; Shu, W.P.; Drobnjak, M.; Kakonen, S.M.; Cordon-Cardo, C.; Guise, T.A.; Massague, J. A multigenic program mediating breast cancer metastasis to bone. Cancer Cell 2003, 3, 537–549. [Google Scholar] [CrossRef] [Green Version]
- Bozzuto, G.; Condello, M.; Molinari, A. Migratory behaviour of tumour cells: A scanning electron microscopy study. Annali dell’Istituto Superiore di Sanita 2015, 51, 139–147. [Google Scholar] [PubMed]
- Filipenko, I.; Schwalm, S.; Reali, L.; Pfeilschifter, J.; Fabbro, D.; Huwiler, A.; Zangemeister-Wittke, U. Upregulation of the S1P3 receptor in metastatic breast cancer cells increases migration and invasion by induction of PGE2 and EP2/EP4 activation. Biochim. Biophys. Acta 2016, 1861, 1840–1851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Graf, C.; Klumpp, M.; Habig, M.; Rovina, P.; Billich, A.; Baumruker, T.; Oberhauser, B.; Bornancin, F. Targeting ceramide metabolism with a potent and specific ceramide kinase inhibitor. Mol. Pharmacol. 2008, 74, 925–932. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Zheng, H.M.; Zhan, Y.T.; Fan, S.Q. An emerging tumor invasion mechanism about the collective cell migration. Am. J. Transl. Res. 2019, 11, 5301–5312. [Google Scholar]
- Yilmaz, M.; Christofori, G. Mechanisms of motility in metastasizing cells. Mol. Cancer Res. 2010, 8, 629–642. [Google Scholar] [CrossRef] [Green Version]
- Hait, N.C.; Maiti, A. The role of sphingosine-1-phosphate and ceramide-1-phosphate in inflammation and cancer. Mediators Inflamm. 2017, 2017, 4806541. [Google Scholar] [CrossRef] [PubMed]
- Granado, M.H.; Gangoiti, P.; Ouro, A.; Arana, L.; Gonzalez, M.; Trueba, M.; Gomez-Munoz, A. Ceramide 1-phosphate (C1P) promotes cell migration involvement of a specific C1P receptor. Cell Signal. 2009, 21, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Stahelin, R.V.; Subramanian, P.; Vora, M.; Cho, W.; Chalfant, C.E. Ceramide-1-phosphate binds group iva cytosolic phospholipase A2 via a novel site in the C2 domain. J. Biol. Chem. 2007, 282, 20467–20474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lien, E.C.; Dibble, C.C.; Toker, A. PI3K signaling in cancer: Beyond Akt. Curr. Opin. Cell Biol. 2017, 45, 62–71. [Google Scholar] [CrossRef]
- Brown, J.S.; Banerji, U. Maximising the potential of Akt inhibitors as anti-cancer treatments. Pharmacol. Ther. 2017, 172, 101–115. [Google Scholar] [CrossRef]
- Tian, G.; Wang, X.; Zhang, F.; Geng, H.; Hou, W.; Chen, L.; Guo, H.; Zhang, N. Downregulation of cPLA2gamma expression inhibits EGF-induced chemotaxis of human breast cancer cells through Akt pathway. Biochem. Biophys. Res. Commun. 2011, 409, 506–512. [Google Scholar] [CrossRef]
- Tunset, H.M.; Feuerherm, A.J.; Selvik, L.M.; Johansen, B.; Moestue, S.A. Cytosolic phospholipase A2 alpha regulates TLR signaling and migration in metastatic 4T1 cells. Int. J. Mol. Sci. 2019, 20, 4800. [Google Scholar] [CrossRef] [Green Version]
- Niwa, S.; Graf, C.; Bornancin, F. Ceramide kinase deficiency impairs microendothelial cell angiogenesis in vitro. Microvasc. Res. 2009, 77, 389–393. [Google Scholar] [CrossRef]
- Kim, C.; Schneider, G.; Abdel-Latif, A.; Mierzejewska, K.; Sunkara, M.; Borkowska, S.; Ratajczak, J.; Morris, A.J.; Kucia, M.; Ratajczak, M.Z. Ceramide-1-phosphate regulates migration of multipotent stromal cells and endothelial progenitor cells-implications for tissue regeneration. Stem Cells 2013, 31, 500–510. [Google Scholar] [CrossRef] [Green Version]
- Wijesinghe, D.S.; Brentnall, M.; Mietla, J.A.; Hoeferlin, L.A.; Diegelmann, R.F.; Boise, L.H.; Chalfant, C.E. Ceramide kinase is required for a normal eicosanoid response and the subsequent orderly migration of fibroblasts. J. Lipid Res. 2014, 55, 1298–1309. [Google Scholar] [CrossRef] [Green Version]
- Pastukhov, O.; Schwalm, S.; Romer, I.; Zangemeister-Wittke, U.; Pfeilschifter, J.; Huwiler, A. Ceramide kinase contributes to proliferation but not to prostaglandin E2 formation in renal mesangial cells and fibroblasts. Cell Physiol. Biochem. 2014, 34, 119–133. [Google Scholar] [CrossRef] [Green Version]
- Tsuji, K.; Mitsutake, S.; Yokose, U.; Sugiura, M.; Kohama, T.; Igarashi, Y. Role of ceramide kinase in peroxisome proliferator-activated receptor beta-induced cell survival of mouse keratinocytes. FEBS J. 2008, 275, 3815–3826. [Google Scholar] [CrossRef] [PubMed]
- Euskirchen, G.; Royce, T.E.; Bertone, P.; Martone, R.; Rinn, J.L.; Nelson, F.K.; Sayward, F.; Luscombe, N.M.; Miller, P.; Gerstein, M.; et al. CREB binds to multiple loci on human chromosome 22. Mol. Cell Biol. 2004, 24, 3804–3814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zygmunt, M.; Hahn, D.; Munstedt, K.; Bischof, P.; Lang, U. Invasion of cytotrophoblastic JEG-3 cells is stimulated by HCG in vitro. Placenta 1998, 19, 587–593. [Google Scholar] [CrossRef]
- Chen, W.Q.; Graf, C.; Zimmel, D.; Rovina, P.; Krapfenbauer, K.; Jaritz, M.; Parker, P.J.; Lubec, G.; Bornancin, F. Ceramide kinase profiling by mass spectrometry reveals a conserved phosphorylation pattern downstream of the catalytic site. J. Proteome Res. 2010, 9, 420–429. [Google Scholar] [CrossRef]
- Takahashi, H.; Ashikawal, H.; Nakamura, H.; Murayama, T. Phosphorylation and inhibition of ceramide kinase by protein kinase C-beta: Their changes by serine residue mutations. Cell Signal. 2019, 54, 59–68. [Google Scholar] [CrossRef]
- Dowling, C.M.; Phelan, J.; Callender, J.A.; Cathcart, M.C.; Mehigan, B.; McCormick, P.; Dalton, T.; Coffey, J.C.; Newton, A.C.; O’Sullivan, J.; et al. Protein kinase C beta II suppresses colorectal cancer by regulating IGF-1 mediated cell survival. Oncotarget 2016, 7, 20919–20933. [Google Scholar] [CrossRef] [Green Version]
- Boath, A.; Graf, C.; Lidome, E.; Ullrich, T.; Nussbaumer, P.; Bornancin, F. Regulation and traffic of ceramide 1-phosphate produced by ceramide kinase - comparative analysis to glucosylceramide and sphingomyelin. J. Biol. Chem. 2008, 283, 8517–8526. [Google Scholar] [CrossRef] [Green Version]
- Carre, A.; Graf, C.; Stora, S.; Mechtcheriakova, D.; Csonga, R.; Urtz, N.; Billich, A.; Baumruker, T.; Bornancin, F. Ceramide kinase targeting and activity determined by its N-terminal pleckstrin homology domain. Biochem. Biophys. Res. Commun. 2004, 324, 1215–1219. [Google Scholar] [CrossRef]







© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schwalm, S.; Erhardt, M.; Römer, I.; Pfeilschifter, J.; Zangemeister-Wittke, U.; Huwiler, A. Ceramide Kinase Is Upregulated in Metastatic Breast Cancer Cells and Contributes to Migration and Invasion by Activation of PI 3-Kinase and Akt. Int. J. Mol. Sci. 2020, 21, 1396. https://doi.org/10.3390/ijms21041396
Schwalm S, Erhardt M, Römer I, Pfeilschifter J, Zangemeister-Wittke U, Huwiler A. Ceramide Kinase Is Upregulated in Metastatic Breast Cancer Cells and Contributes to Migration and Invasion by Activation of PI 3-Kinase and Akt. International Journal of Molecular Sciences. 2020; 21(4):1396. https://doi.org/10.3390/ijms21041396
Chicago/Turabian StyleSchwalm, Stephanie, Martin Erhardt, Isolde Römer, Josef Pfeilschifter, Uwe Zangemeister-Wittke, and Andrea Huwiler. 2020. "Ceramide Kinase Is Upregulated in Metastatic Breast Cancer Cells and Contributes to Migration and Invasion by Activation of PI 3-Kinase and Akt" International Journal of Molecular Sciences 21, no. 4: 1396. https://doi.org/10.3390/ijms21041396