The Pentose Phosphate Pathway and Its Involvement in Cisplatin Resistance



Abstract
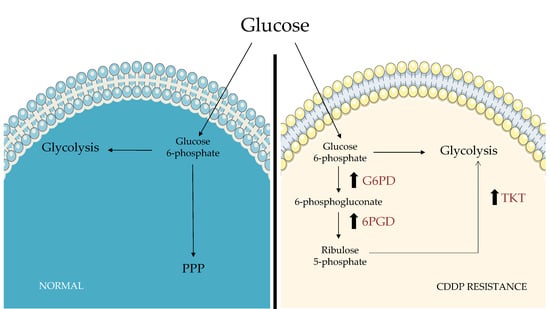
:
1. Introduction
2. Glucose Metabolism and the Pentose Phosphate Pathway in Tumor Cells
3. Targeting PPP Could Be a Strategy to Overcome Cisplatin Resistance
4. Drug Delivery Systems Targeting PPP to Overcome Cisplatin Resistance
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Dasari, S.; Bernard Tchounwou, P. Cisplatin in Cancer Therapy: Molecular Mechanisms of Action. Eur. J. Pharmacol. 2014, 740, 364–378. [Google Scholar] [CrossRef] [Green Version]
- Aldossary, S.A. Review on Pharmacology of Cisplatin: Clinical Use, Toxicity and Mechanism of Resistance of Cisplatin. Biomed. Pharmacol. J. 2019, 12, 9. [Google Scholar] [CrossRef]
- Eljack, N.D.; Ma, H.-Y.M.; Drucker, J.; Shen, C.; Hambley, T.W.; New, E.J.; Friedrich, T.; Clarke, R.J. Mechanisms of Cell Uptake and Toxicity of the Anticancer Drug Cisplatin. Metallomics 2014, 6, 2126–2133. [Google Scholar] [CrossRef] [Green Version]
- Gately, D.; Howell, S. Cellular Accumulation of the Anticancer Agent Cisplatin: A Review. Br. J. Cancer 1993, 67, 1171–1176. [Google Scholar] [CrossRef] [Green Version]
- Gómez-Ruiz, S.; Maksimović-Ivanić, D.; Mijatović, S.; Kaluđerović, G.N. On the Discovery, Biological Effects, and Use of Cisplatin and Metallocenes in Anticancer Chemotherapy. Bioinorg. Chem. Appl. 2012, 2012, 1–14. [Google Scholar] [CrossRef]
- Galluzzi, L.; Senovilla, L.; Vitale, I.; Michels, J.; Martins, I.; Kepp, O.; Castedo, M.; Kroemer, G. Molecular Mechanisms of Cisplatin Resistance. Oncogene 2012, 31, 1869–1883. [Google Scholar] [CrossRef] [Green Version]
- Miller, R.P.; Tadagavadi, R.K.; Ramesh, G.; Reeves, W.B. Mechanisms of Cisplatin Nephrotoxicity. Toxins 2010, 2, 2490–2518. [Google Scholar] [CrossRef] [Green Version]
- Kelland, L. The Resurgence of Platinum-Based Cancer Chemotherapy. Nat. Rev. Cancer 2007, 7, 573–584. [Google Scholar] [CrossRef]
- Siddik, Z.H. Cisplatin: Mode of Cytotoxic Action and Molecular Basis of Resistance. Oncogene 2003, 22, 7265–7279. [Google Scholar] [CrossRef] [Green Version]
- Damia, G.; Broggini, M. Platinum Resistance in Ovarian Cancer: Role of DNA Repair. Cancers 2019, 11, 119. [Google Scholar] [CrossRef] [Green Version]
- Galluzzi, L.; Vitale, I.; Michels, J.; Brenner, C.; Szabadkai, G.; Harel-Bellan, A.; Castedo, M.; Kroemer, G. Systems Biology of Cisplatin Resistance: Past, Present and Future. Cell Death Dis. 2014, 5, e1257. [Google Scholar] [CrossRef] [Green Version]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of Cancer Metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [Green Version]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Giannattasio, S.; Mirisola, M.G.; Mazzoni, C. Editorial: Cell Stress, Metabolic Reprogramming, and Cancer. Front. Oncol. 2018, 8, 236. [Google Scholar] [CrossRef]
- Butler, E.B.; Zhao, Y.; Munoz-Pinedo, C.; Lu, J.; Tan, M. Stalling the Engine of Resistance: Targeting Cancer Metabolism to Overcome Therapeutic Resistance. Cancer Res. 2013, 73, 2709–2717. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G. Targeting Cancer Metabolism: A Therapeutic Window Opens. Nat. Rev. Drug Discov. 2011, 10, 671–684. [Google Scholar] [CrossRef] [Green Version]
- Vander Heiden, M.G.; DeBerardinis, R.J. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017, 168, 657–669. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Y.; Butler, E.B.; Tan, M. Targeting Cellular Metabolism to Improve Cancer Therapeutics. Cell Death Dis. 2013, 4, e532. [Google Scholar] [CrossRef] [Green Version]
- Hay, N. Reprogramming Glucose Metabolism in Cancer: Can It Be Exploited for Cancer Therapy? Nat. Rev. Cancer 2016, 16, 635–649. [Google Scholar] [CrossRef] [Green Version]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does It Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Patra, K.C.; Hay, N. The Pentose Phosphate Pathway and Cancer. Trends Biochem. Sci. 2014, 39, 347–354. [Google Scholar] [CrossRef] [Green Version]
- Jin, L.; Zhou, Y. Crucial Role of the Pentose Phosphate Pathway in Malignant Tumors (Review). Oncol. Lett. 2019, 17, 4213–4221. [Google Scholar] [CrossRef] [Green Version]
- Tennant, D.A.; Durán, R.V.; Gottlieb, E. Targeting Metabolic Transformation for Cancer Therapy. Nat. Rev. Cancer 2010, 10, 267–277. [Google Scholar] [CrossRef]
- Jiang, P.; Du, W.; Wu, M. Regulation of the Pentose Phosphate Pathway in Cancer. Protein Cell 2014, 5, 592–602. [Google Scholar] [CrossRef] [Green Version]
- Catanzaro, D.; Gaude, E.; Orso, G.; Giordano, C.; Guzzo, G.; Rasola, A.; Ragazzi, E.; Caparrotta, L.; Frezza, C.; Montopoli, M. Inhibition of Glucose-6-Phosphate Dehydrogenase Sensitizes Cisplatin-Resistant Cells to Death. Oncotarget 2015, 6, 30102–30114. [Google Scholar] [CrossRef] [Green Version]
- Hong, W.; Cai, P.; Xu, C.; Cao, D.; Yu, W.; Zhao, Z.; Huang, M.; Jin, J. Inhibition of Glucose-6-Phosphate Dehydrogenase Reverses Cisplatin Resistance in Lung Cancer Cells via the Redox System. Front. Pharmacol. 2018, 9, 43. [Google Scholar] [CrossRef] [Green Version]
- Cho, E.S.; Cha, Y.H.; Kim, H.S.; Kim, N.H.; Yook, J.I. The Pentose Phosphate Pathway as a Potential Target for Cancer Therapy. Biomol. Ther. 2018, 26, 29–38. [Google Scholar] [CrossRef] [Green Version]
- Riganti, C.; Gazzano, E.; Polimeni, M.; Aldieri, E.; Ghigo, D. The Pentose Phosphate Pathway: An Antioxidant Defense and a Crossroad in Tumor Cell Fate. Free Radic. Biol. Med. 2012, 53, 421–436. [Google Scholar] [CrossRef]
- Lin, R.; Elf, S.; Shan, C.; Kang, H.-B.; Ji, Q.; Zhou, L.; Hitosugi, T.; Zhang, L.; Zhang, S.; Seo, J.H.; et al. 6-Phosphogluconate Dehydrogenase Links Oxidative PPP, Lipogenesis and Tumour Growth by Inhibiting LKB1–AMPK Signalling. Nat. Cell Biol. 2015, 17, 1484–1496. [Google Scholar] [CrossRef] [Green Version]
- Preuss, J.; Richardson, A.D.; Pinkerton, A.; Hedrick, M.; Sergienko, E.; Rahlfs, S.; Becker, K.; Bode, L. Identification and Characterization of Novel Human Glucose-6-Phosphate Dehydrogenase Inhibitors. J. Biomol. Screen. 2013, 18, 286–297. [Google Scholar] [CrossRef]
- Zhang, H.; Zhang, H.; Wang, S.; Ni, Z.; Wang, T. 1-Hydroxy-8-Methoxy-Anthraquinon Reverses Cisplatin Resistance by Inhibiting 6PGD in Cancer Cells. Open Life Sci. 2019, 14, 454–461. [Google Scholar] [CrossRef]
- Zheng, W.; Feng, Q.; Liu, J.; Guo, Y.; Gao, L.; Li, R.; Xu, M.; Yan, G.; Yin, Z.; Zhang, S.; et al. Inhibition of 6-Phosphogluconate Dehydrogenase Reverses Cisplatin Resistance in Ovarian and Lung Cancer. Front. Pharmacol. 2017, 8, 421. [Google Scholar] [CrossRef]
- Mullarky, E.; Cantley, L.C. Diverting Glycolysis to Combat Oxidative Stress. In Innovative Medicine; Nakao, K., Minato, N., Uemoto, S., Eds.; Springer: Tokyo, Japan, 2015; pp. 3–23. [Google Scholar]
- Boros, L.G.; Lee, P.W.N.; Brandes, J.L.; Cascante, M.; Muscarella, P.; Schirmer, W.J.; Melvin, W.S.; Ellison, E.C. Nonoxidative Pentose Phosphate Pathways and Their Direct Role in Ribose Synthesis in Tumors: Is Cancer a Disease of Cellular Glucose Metabolism? Med. Hypotheses 1998, 50, 55–59. [Google Scholar] [CrossRef]
- Xu, Y.; Gao, W.; Zhang, Y.; Wu, S.; Liu, Y.; Deng, X.; Xie, L.; Yang, J.; Yu, H.; Su, J.; et al. ABT737 Reverses Cisplatin Resistance by Targeting Glucose Metabolism of Human Ovarian Cancer Cells. Int. J. Oncol. 2018, 53, 1055–1068. [Google Scholar] [CrossRef] [Green Version]
- Lucarelli, G.; Galleggiante, V.; Rutigliano, M.; Sanguedolce, F.; Cagiano, S.; Bufo, P.; Lastilla, G.; Maiorano, E.; Ribatti, D.; Giglio, A.; et al. Metabolomic Profile of Glycolysis and the Pentose Phosphate Pathway Identifies the Central Role of Glucose-6-Phosphate Dehydrogenase in Clear Cell-Renal Cell Carcinoma. Oncotarget 2015, 6, 13371–13386. [Google Scholar] [CrossRef] [Green Version]
- Zhang, R.; Tao, F.; Ruan, S.; Hu, M.; Hu, Y.; Fang, Z.; Mei, L.; Gong, C. The TGFβ1-FOXM1-HMGA1-TGFβ1 Positive Feedback Loop Increases the Cisplatin Resistance of Non-Small Cell Lung Cancer by Inducing G6PD Expression. Am. J. Transl. Res. 2019, 11, 6860–6876. [Google Scholar]
- Mele, L.; Paino, F.; Papaccio, F.; Regad, T.; Boocock, D.; Stiuso, P.; Lombardi, A.; Liccardo, D.; Aquino, G.; Barbieri, A.; et al. A New Inhibitor of Glucose-6-Phosphate Dehydrogenase Blocks Pentose Phosphate Pathway and Suppresses Malignant Proliferation and Metastasis in Vivo. Cell Death Dis. 2018, 9, 572. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Wu, X.-L.; Wu, K.-H.; Zhang, R.; Ju, L.-L.; Ji, Y.; Zhang, Y.-W.; Xue, S.-L.; Yang, Y.-F.; Yu, M.-M. MicroRNA-497 Regulates Cisplatin Chemosensitivity of Cervical Cancer by Targeting Transketolase. Am. J. Cancer Res. 2016, 6, 2690–2699. [Google Scholar]
- Dong, Y.; Wang, M. Knockdown of TKTL1 Additively Complements Cisplatin-Induced Cytotoxicity in Nasopharyngeal Carcinoma Cells by Regulating the Levels of NADPH and Ribose-5-Phosphate. Biomed. Pharmacother. 2017, 85, 672–678. [Google Scholar] [CrossRef]
- Zhang, D.; Zhou, C.; Jiang, X.; Chen, J.; Shi, B. Increased Expression of MiR-222 Is Associated with Poor Prognosis in Bladder Cancer. World J. Surg. Oncol. 2014, 12, 241. [Google Scholar] [CrossRef] [Green Version]
- Garofalo, M.; Quintavalle, C.; Romano, G.; Croce, C.M.; Condorelli, G. MiR221/222 in Cancer: Their Role in Tumor Progression and Response to Therapy. Curr. Mol. Med. 2012, 12, 27–33. [Google Scholar] [CrossRef]
- Zeng, L.-P.; Hu, Z.-M.; Li, K.; Xia, K. MiR-222 Attenuates Cisplatin-Induced Cell Death by Targeting the PPP2R2A/Akt/MTOR Axis in Bladder Cancer Cells. J. Cell. Mol. Med. 2016, 20, 559–567. [Google Scholar] [CrossRef]
- Bansal, A.; Simon, M.C. Glutathione Metabolism in Cancer Progression and Treatment Resistance. J. Cell Biol. 2018, 217, 2291–2298. [Google Scholar] [CrossRef] [Green Version]
- Simons, A.L.; Ahmad, I.M.; Mattson, D.M.; Dornfeld, K.J.; Spitz, D.R. 2-Deoxy-d-Glucose Combined with Cisplatin Enhances Cytotoxicity via Metabolic Oxidative Stress in Human Head and Neck Cancer Cells. Cancer Res. 2007, 67, 3364–3370. [Google Scholar] [CrossRef] [Green Version]
- Maeda, H.; Wu, J.; Sawa, T.; Matsumura, Y.; Hori, K. Tumor Vascular Permeability and the EPR Effect in Macromolecular Therapeutics: A Review. J. Controlled Release 2000, 65, 271–284. [Google Scholar] [CrossRef]
- Pasut, G. Grand Challenges in Nano-Based Drug Delivery. Front. Med. Technol. 2019, 1, 1. [Google Scholar] [CrossRef] [Green Version]
- Catanzaro, D.; Nicolosi, S.; Cocetta, V.; Salvalaio, M.; Pagetta, A.; Ragazzi, E.; Montopoli, M.; Pasut, G. Cisplatin Liposome and 6-Amino Nicotinamide Combination to Overcome Drug Resistance in Ovarian Cancer Cells. Oncotarget 2018, 9, 16847–16860. [Google Scholar] [CrossRef] [Green Version]
- Ravera, M.; Gabano, E.; Zanellato, I.; Gallina, A.; Perin, E.; Arrais, A.; Cantamessa, S.; Osella, D. Cisplatin and valproate released from the bifunctional [Pt(IV)Cl2(NH3)2(valproato)2] antitumor prodrug or from liposome formulations: Who does what? Dalton Trans. 2017, 46, 1559–1566. [Google Scholar] [CrossRef]
- Santucci, L.; Mencarelli, A.; Renga, B.; Pasut, G.; Veronese, F.; Zacheo, A.; Germani, A.; Fiorucci, S. Nitric Oxide Modulates Proapoptotic and Antiapoptotic Properties of Chemotherapy Agents: The Case of NO-Pegylated Epirubicin. FASEB J. 2006, 20, 765–767. [Google Scholar] [CrossRef]
- Pasut, G.; Greco, F.; Mero, A.; Mendichi, R.; Fante, C.; Green, R.J.; Veronese, F.M. Polymer–Drug Conjugates for Combination Anticancer Therapy: Investigating the Mechanism of Action. J. Med. Chem. 2009, 52, 6499–6502. [Google Scholar] [CrossRef]
- Santucci, L.; Mencarelli, A.; Renga, B.; Ceccobelli, D.; Pasut, G.; Veronese, F.M.; Distrutti, E.; Fiorucci, F. Cardiac safety and antitumoral activity of a new nitric oxide derivative of pegylated epirubicin in mice. Anticancer Drugs. 2007, 18, 1081–1091. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Enzymes | Therapeutic/Molecular Strategies | Experimental Model | References |
|---|---|---|---|
| G6PD overexpression | 6-aminonicotinamide (6-AN) (competitive inhibitor) | Ovarian cancer cells: C13, IGROV Pt and SKOV3DDP | [25,35,48] |
| Renal cancer cells: ccRCC | [36] | ||
| Non-small-cell lung cancer: A459/DDP | [26] | ||
| Dehydroepiandrosterone (DHEA) (uncompetitive inhibitor) | Ovarian cancer cells: SKOV3DDP | [35] | |
| Polydatin (natural inhibitor) | Orthotopic xenografts model of oral cancer | [38] | |
| Genetic silencing | Non-small-cell lung cancer: A459/DDP | [26] | |
| 6PGD overexpression | 1-hydroxy-8-methoxy-anthraquinon (inhibitor of cancer cell proliferation and growth) | Non-small-cell lung cancer: A459/DDP Ovarian cancer cells: C13 | [31] [31] |
| Physcion (natural dihydroxyanthraquinone) | Non-small-cell lung cancer: A459/DDP Ovarian cancer cells: C13 | [32] [32] | |
| Transfection with miR-206 or miR-613 | Non-small-cell lung cancer: A459/DDP Ovarian cancer cells: C13 | [32] [32] | |
| TKT overexpression | Genetic silencing or miR-497 treatment | Cervical cancer cells: HeLa | [39] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giacomini, I.; Ragazzi, E.; Pasut, G.; Montopoli, M. The Pentose Phosphate Pathway and Its Involvement in Cisplatin Resistance. Int. J. Mol. Sci. 2020, 21, 937. https://doi.org/10.3390/ijms21030937
Giacomini I, Ragazzi E, Pasut G, Montopoli M. The Pentose Phosphate Pathway and Its Involvement in Cisplatin Resistance. International Journal of Molecular Sciences. 2020; 21(3):937. https://doi.org/10.3390/ijms21030937
Chicago/Turabian StyleGiacomini, Isabella, Eugenio Ragazzi, Gianfranco Pasut, and Monica Montopoli. 2020. "The Pentose Phosphate Pathway and Its Involvement in Cisplatin Resistance" International Journal of Molecular Sciences 21, no. 3: 937. https://doi.org/10.3390/ijms21030937