Comparison of Eight Technologies to Determine Genotype at the UGT1A1 (TA)n Repeat Polymorphism: Potential Clinical Consequences of Genotyping Errors?

 , ,
, ,
Abstract
:1. Introduction
2. Results
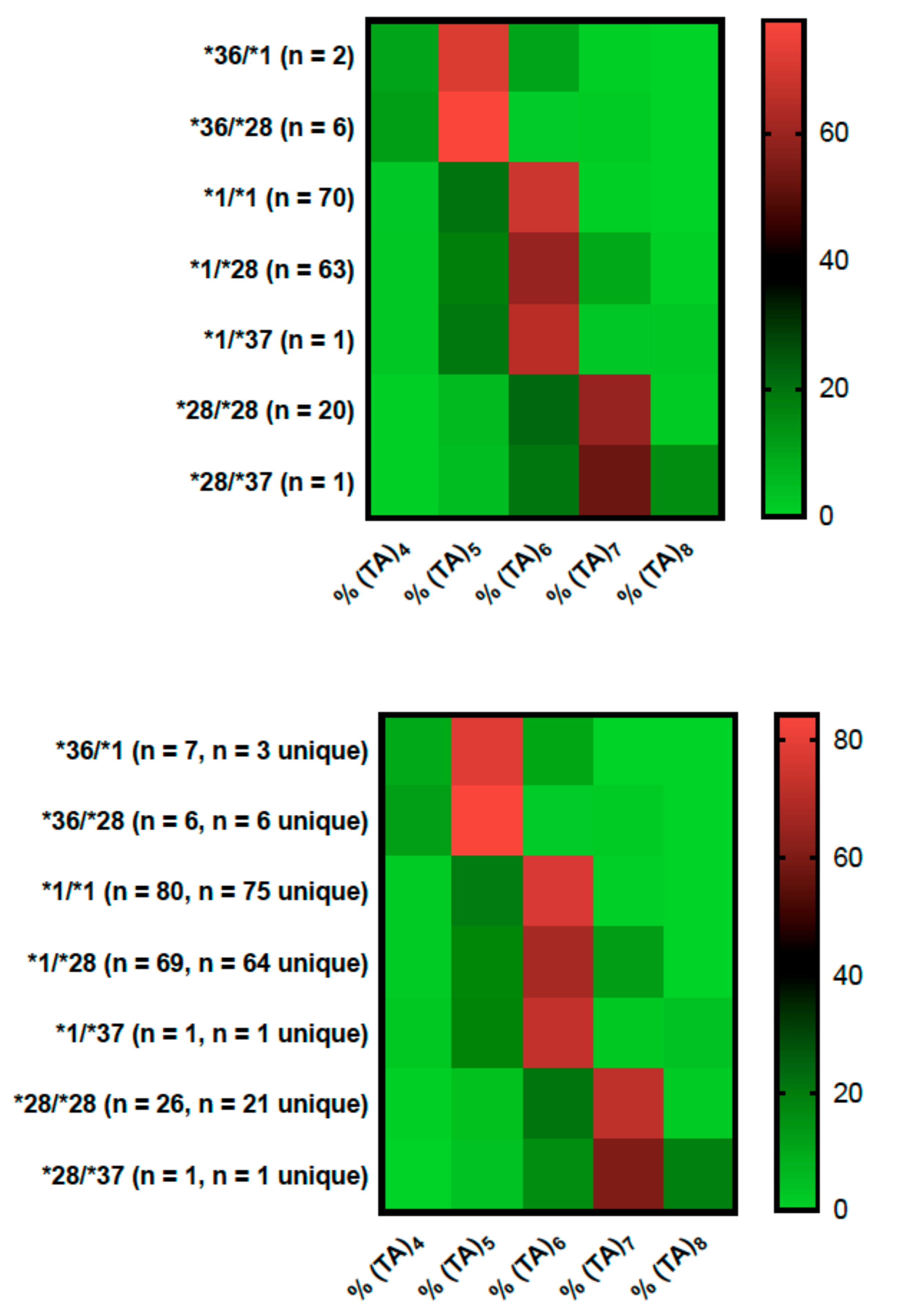
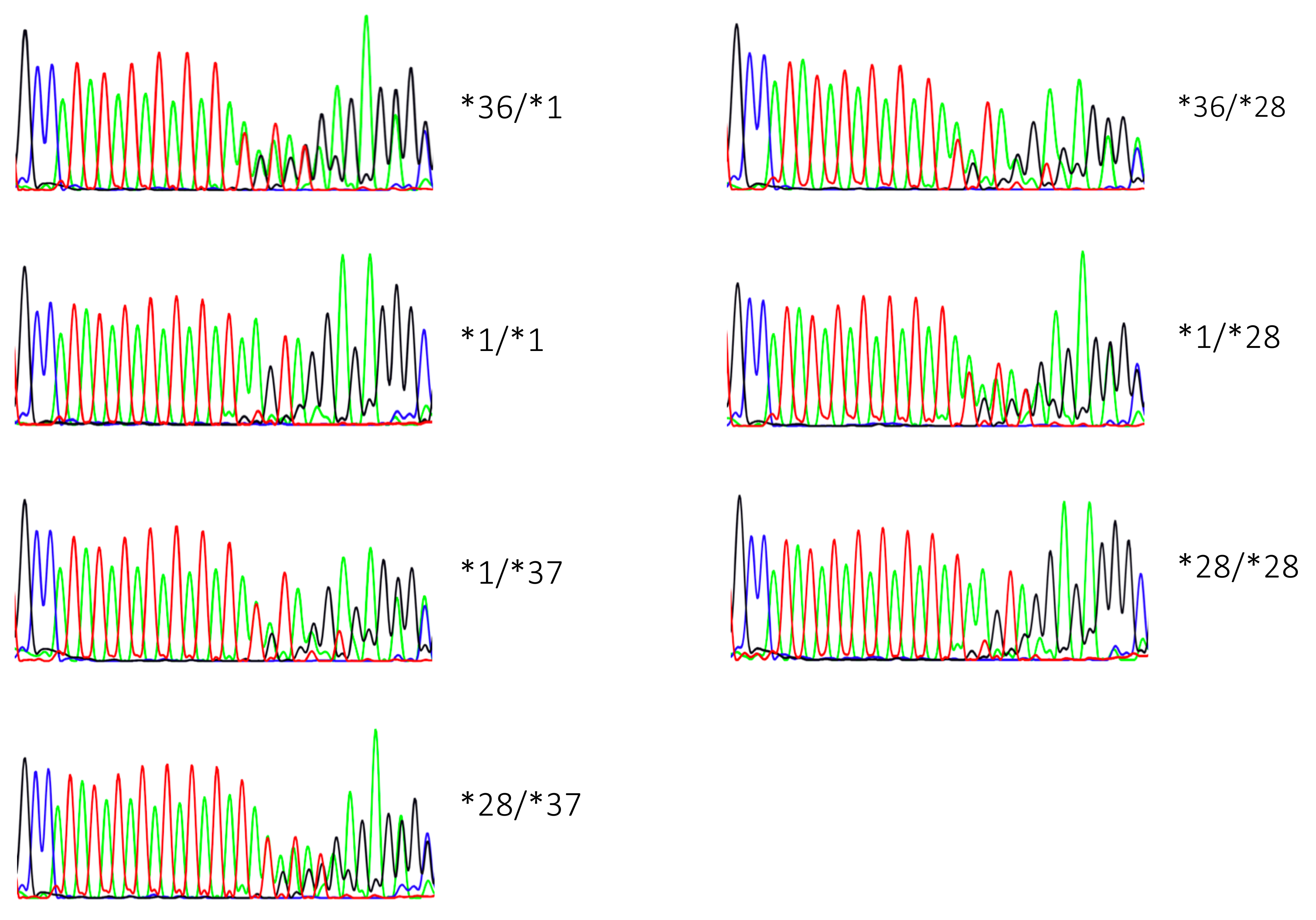
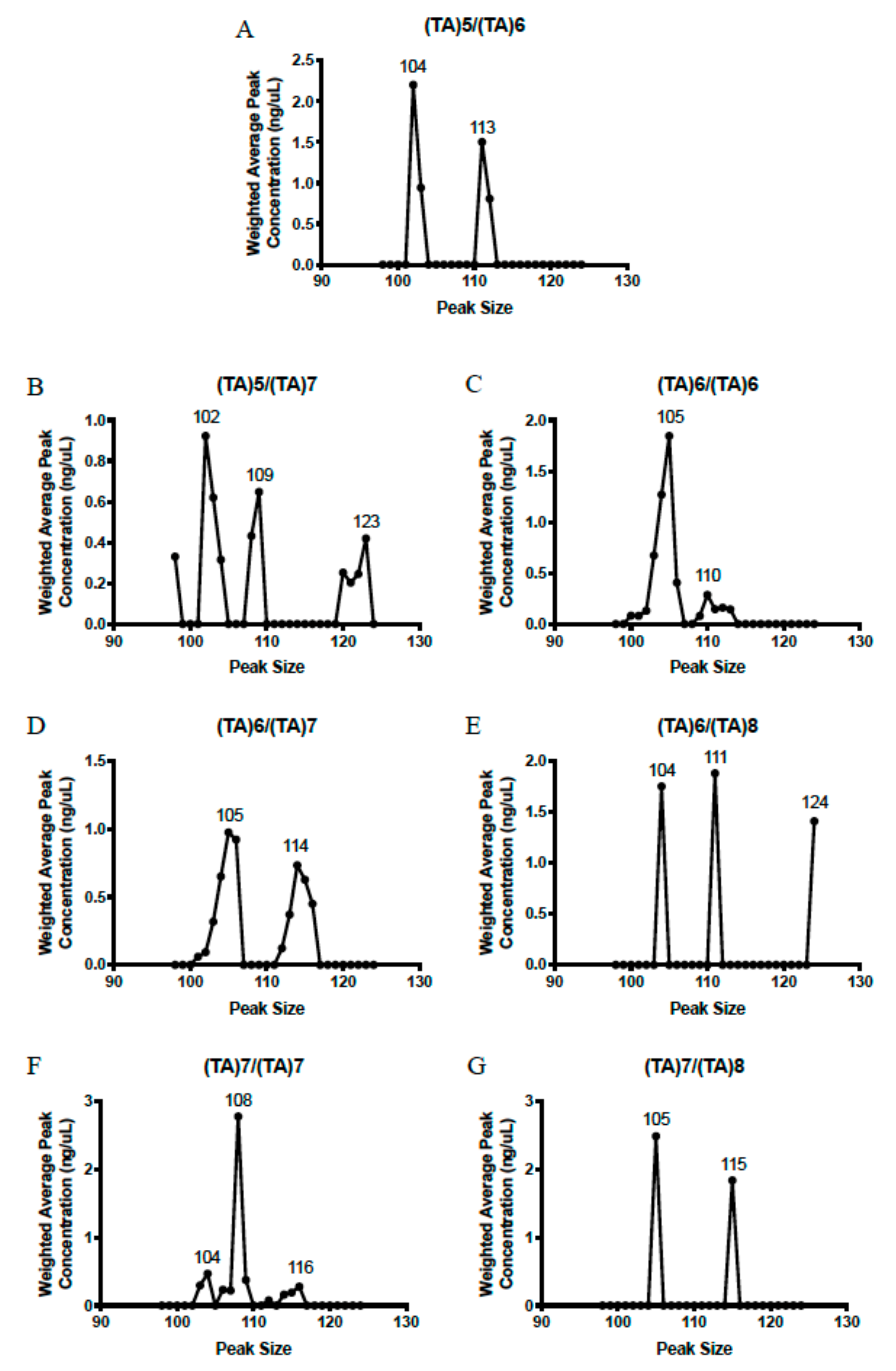
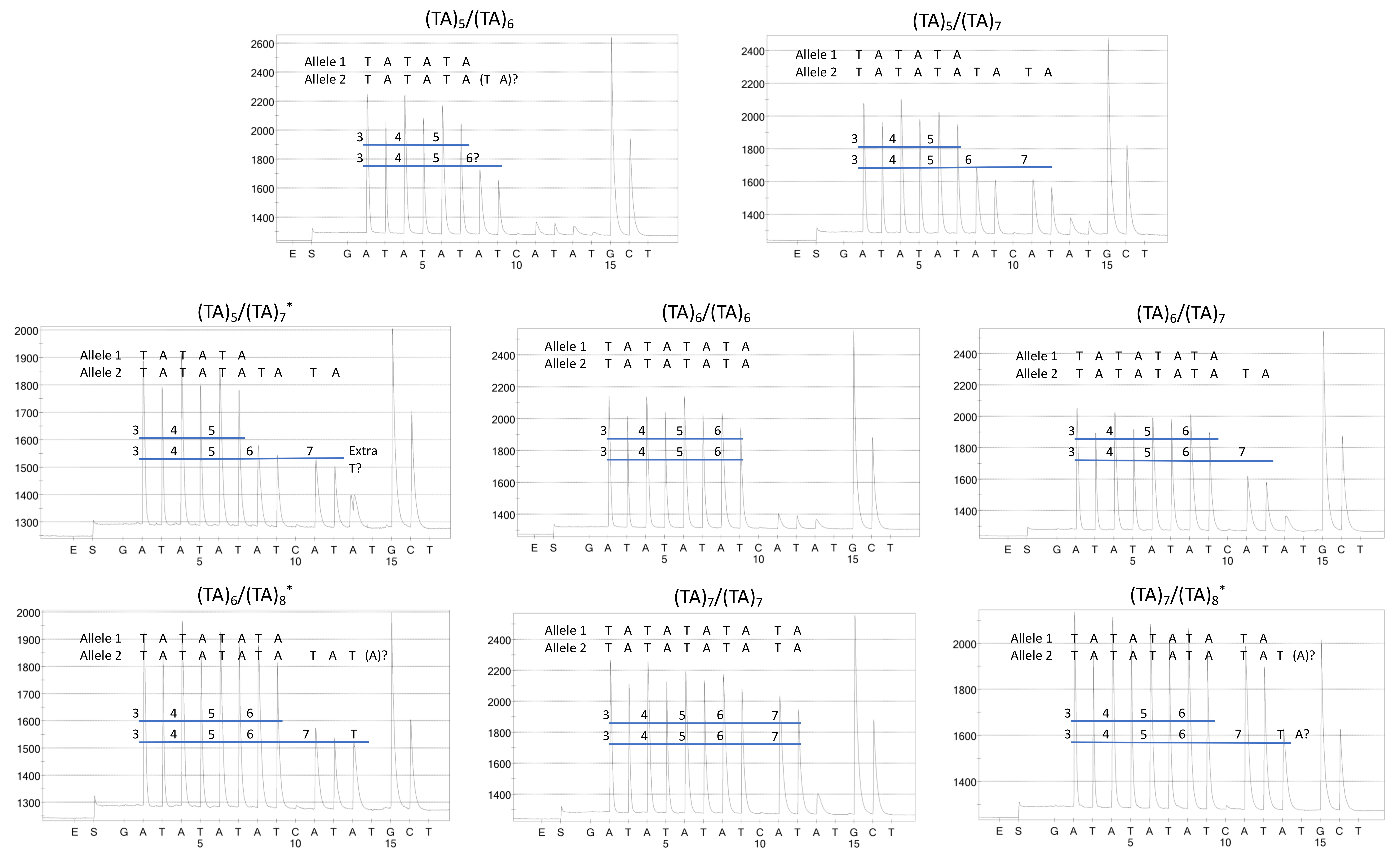
2.1. Genotyping Results
2.2. Genotype Concordance between Illumina Sequencing, Fragment Analysis, and Fluorescent PCR
2.3. Genotype Concordance in Direct Sequencing, Gel Sizing, Pyrosequencing, DMET Plus, and Pharmacoscan
2.4. Phenotype Concordance
2.5. Comparisons with Previously Published Peer-Reviewed Data
3. Discussion
4. Materials and Methods
4.1. Patients and Samples
4.2. Illumina Sequencing (MiSeq)
4.3. Fragment Analysis
4.4. Direct Sequencing (in-House)
4.5. Pyrosequencing
4.6. DMET Plus
4.7. Statistical Considerations
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Sissung, T.M.; Cordes, L.M.; Figg, W.D. UGT1A1 Polymorphisms and Mutations Affect Anticancer Drug Therapy. In Handbook of Therapeutiv Biomarkers in Cancer; Yang, S.X., Dancey, J.E., Eds.; CRC Press: Boca Raton, FL, USA, 2018; in press. [Google Scholar]
- Liu, X.H.; Lu, J.; Duan, W.; Dai, Z.M.; Wang, M.; Lin, S.; Yang, P.T.; Tian, T.; Liu, K.; Zhu, Y.Y.; et al. Predictive Value of UGT1A1*28 Polymorphism In Irinotecan-based Chemotherapy. J. Cancer 2017, 8, 691–703. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Memon, N.; Weinberger, B.I.; Hegyi, T.; Aleksunes, L.M. Inherited disorders of bilirubin clearance. Pediatr. Res. 2016, 79, 378–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- FDA. Indacaterol Package Insert. 2012. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/022383s002lbl.pdf (accessed on 29 January 2020).
- FDA. Arformoterol Package Insert. 2014. Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&varApplNo=021912 (accessed on 29 January 2020).
- FDA. Irinotecan Package Insert. 2014. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/020571s048lbl.pdf (accessed on 29 January 2020).
- FDA. Belinostat Package Insert. 2017. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/206256s002lbl.pdf (accessed on 29 January 2020).
- FDA. Dolutegravir Package Insert. 2017. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/204790s014lbl.pdf (accessed on 29 January 2020).
- FDA. Paxopanib Package Insert. 2017. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/022465s024s025lbl.pdf (accessed on 29 January 2020).
- FDA. Nilotinib Package Insert. 2018. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/022068s027lbl.pdf (accessed on 29 January 2020).
- FDA. Atazanavir Package Insert. 2018. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/021567s042,206352s007lbl.pdf (accessed on 29 January 2020).
- Sissung, T.M.; McKeeby, J.W.; Patel, J.; Lertora, J.J.; Kumar, P.; Flegel, W.A.; Adams, S.D.; Eckes, E.J.; Mickey, F.; Plona, T.M.; et al. Pharmacogenomics Implementation at the National Institutes of Health Clinical Center. J. Clin. Pharmacol. 2017, 57 Suppl 10, S67–S77. [Google Scholar] [CrossRef]
- Marques, S.C.; Ikediobi, O.N. The clinical application of UGT1A1 pharmacogenetic testing: gene-environment interactions. Hum. Genomics 2010, 4, 238–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perera, M.A.; Innocenti, F.; Ratain, M.J. Pharmacogenetic testing for uridine diphosphate glucuronosyltransferase 1A1 polymorphisms: are we there yet? Pharmacotherapy 2008, 28, 755–768. [Google Scholar] [CrossRef] [PubMed]
- Gammal, R.S.; Court, M.H.; Haidar, C.E.; Iwuchukwu, O.F.; Gaur, A.H.; Alvarellos, M.; Guillemette, C.; Lennox, J.L.; Whirl-Carrillo, M.; Brummel, S.S.; et al. Clinical Pharmacogenetics Implementation, C. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for UGT1A1 and Atazanavir Prescribing. Clin. Pharmacol. Ther. 2016, 99, 363–369. [Google Scholar] [CrossRef] [PubMed]
- Lampe, J.W.; Bigler, J.; Horner, N.K.; Potter, J.D. UDP-glucuronosyltransferase (UGT1A1*28 and UGT1A6*2) polymorphisms in Caucasians and Asians: relationships to serum bilirubin concentrations. Pharmacogenetics 1999, 9, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Innocenti, F.; Grimsley, C.; Das, S.; Ramirez, J.; Cheng, C.; Kuttab-Boulos, H.; Ratain, M.J.; Di Rienzo, A. Haplotype structure of the UDP-glucuronosyltransferase 1A1 promoter in different ethnic groups. Pharmacogenetics 2002, 12, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Marsh, S.; King, C.R.; Van Booven, D.J.; Revollo, J.Y.; Gilman, R.H.; McLeod, H.L. Pharmacogenomic assessment of Mexican and Peruvian populations. Pharmacogenomics 2015, 16, 441–448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baudhuin, L.M.; Highsmith, W.E.; Skierka, J.; Holtegaard, L.; Moore, B.E.; O’Kane, D.J. Comparison of three methods for genotyping the UGT1A1 (TA)n repeat polymorphism. Clin. Biochem. 2007, 40, 710–717. [Google Scholar] [CrossRef] [PubMed]
- Deeken, J.F.; Cormier, T.; Price, D.K.; Sissung, T.M.; Steinberg, S.M.; Tran, K.; Liewehr, D.J.; Dahut, W.L.; Miao, X.; Figg, W.D. A pharmacogenetic study of docetaxel and thalidomide in patients with castration-resistant prostate cancer using the DMET genotyping platform. Pharmacogenomics J. 2010, 10, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Kaboli, P.J.; Glasgow, J.M.; Jaipaul, C.K.; Barry, W.A.; Strayer, J.R.; Mutnick, B.; Rosenthal, G.E. Identifying medication misadventures: poor agreement among medical record, physician, nurse, and patient reports. Pharmacotherapy 2010, 30, 529–538. [Google Scholar] [CrossRef] [PubMed]
- Ehmer, U.; Lankisch, T.O.; Erichsen, T.J.; Kalthoff, S.; Freiberg, N.; Wehmeier, M.; Manns, M.P.; Strassburg, C.P. Rapid allelic discrimination by TaqMan PCR for the detection of the Gilbert’s syndrome marker UGT1A1*28. J. Mol. Diagn 2008, 10, 549–552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillemette, C.; Millikan, R.C.; Newman, B.; Housman, D.E. Genetic polymorphisms in uridine diphospho-glucuronosyltransferase 1A1 and association with breast cancer among African Americans. Cancer Res. 2000, 60, 950–956. [Google Scholar] [PubMed]
- Bryc, K.; Durand, E.Y.; Macpherson, J.M.; Reich, D.; Mountain, J.L. The genetic ancestry of African Americans, Latinos, and European Americans across the United States. Am. J. Hum. Genet. 2015, 96, 37–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Royal Dutch Pharmacists Association (KNMP), D.P.W.G.D. Pharmacogenetic Guidelines: Irinotecan UGT1A1. Available online: http://kennisbank.knmp.nl (accessed on 29 January 2020).
- Quaranta, S.; Thomas, F. Pharmacogenetics of anti-cancer drugs: State of the art and implementation - recommendations of the French National Network of Pharmacogenetics. Therapie 2017, 72, 205–215. [Google Scholar] [CrossRef] [PubMed]
- Sissung, T.M.; Rajan, A.; Blumenthal, G.M.; Liewehr, D.J.; Steinberg, S.M.; Berman, A.; Giaccone, G.; Figg, W.D. Reproducibility of pharmacogenetics findings for paclitaxel in a heterogeneous population of patients with lung cancer. PLoS ONE 2018, in press. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sukasem, C.; Atasilp, C.; Chansriwong, P.; Chamnanphon, M.; Puangpetch, A.; Sirachainan, E. Development of Pyrosequencing Method for Detection of UGT1A1 Polymorphisms in Thai Colorectal Cancers. J. Clin. Lab. Anal. 2016, 30, 84–89. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Star Nomenclature | (TA)n | Expression/Function * |
|---|---|---|
| UGT1A1*36 | 5 | Highest |
| UGT1A1*1 | 6 | High |
| UGT1A1*28 | 7 | Low |
| UGT1A1*37 | 8 | Lowest |
| *36/*36 | *36/*1 | *1/*1 | *36/*28 | *1/*28 | *1/*37 | *28/*28 | *28/*37 | Not Called | |
|---|---|---|---|---|---|---|---|---|---|
| Platform | n = (%) | n = (%) | n = (%) | n = (%) | n = (%) | n = (%) | n = (%) | n = (%) | n = (%) |
| (TA)n/(TA)n | 5/5 | 5/6 | 6/6 | 5/6 | 6/7 | 6/8 | 7/7 | 7/8 | |
| Metabolism Status | Normal | Intermediate | Poor | ||||||
| Illumina (n = 163) | |||||||||
| Female (n = 63) | 2 (3.2) | 24 (38.1) | 29 (46) | 1 (1.6) | 7 (11.1) | ||||
| Male (n = 100) | 46 (46) | 6 (6) | 34 (34) | 13 (13) | 1 (1) | ||||
| Caucasian (non-Hispanic) (n = 105) | 56 (49.1) | 48 (42.1) | 10 (8.8) | ||||||
| Black or African American (n = 32) | 1 (3.1) | 7 (21.9) | 6 (18.8) | 8 (25) | 1 (3.1) | 8 (25) | 1 (3.1) | ||
| Hispanic or Latino (n = 5) | 1 (20) | 2 (40) | 2 (40) | ||||||
| Asian or Pacific Islander (n = 9) | 5 (55.6) | 4 (44.4) | |||||||
| Other or Unknown (n = 3) | 1 (33.3) | 2 (66.7) | |||||||
| Fragment Analysis (n = 163) | |||||||||
| Female (n = 63) | 2 (3.2) | 24 (38.1) | 29 (46) | 1 (1.6) | 7 (11.1) | ||||
| Male (n = 100) | 46 (46) | 6 (6) | 34 (34) | 13 (13) | 1 (1) | ||||
| Caucasian (non-Hispanic) (n = 105) | 56 (49.1) | 48 (42.1) | 10 (8.8) | ||||||
| Black or African American (n = 32) | 1 (3.1) | 7 (21.9) | 6 (18.8) | 8 (25) | 1 (3.1) | 8 (25) | 1 (3.1) | ||
| Hispanic or Latino (n = 5) | 1 (20) | 2 (40) | 2 (40) | ||||||
| Asian or Pacific Islander (n = 9) | 5 (55.6) | 4 (44.4) | |||||||
| Other or Unknown (n = 3) | 1 (33.3) | 2 (66.7) | |||||||
| fPCR (n = 9) 1 | |||||||||
| Female (n = 6) | 2 (33.3) | 1 (16.7) | 1 (16.7) | 1 (16.7) | 1 (16.7) | ||||
| Male (n = 7) | 1 (14.3) | 2 (28.6) | 2 (28.6) | 1 (14.3) | 1 (14.3) | ||||
| Caucasian (non-Hispanic) (n = 6) | 2 (33.3) | 2 (33.3) | 2 (33.3) | ||||||
| Black or African American (n = 6) | 1 (16.7) | 2 (33.3) | 1 (16.7) | 1 (16.7) | 1 (16.7) | ||||
| Hispanic or Latino (n = 1) | 1 (100) | ||||||||
| Pyrosequencing (n = 162) | |||||||||
| Female (n = 63) | 2 (3.2) | 23 (36.5) | 29 (46) | 6 (9.5) | 3 (4.8) | ||||
| Male (n = 99) | 45 (45.5) | 5 (5.1) | 32 (32.3) | 13 (13.1) | 4 (4) | ||||
| Caucasian (non-Hispanic) (n = 113) | 54 (47.8) | 47 (41.6) | 9 (8) | 3 (2.7) | |||||
| Black or African American (n = 32) | 1 (3.1) | 7 (21.9) | 5 (15.6) | 7 (21.9) | 8 (25) | 4 (12.5) | |||
| Hispanic or Latino (n = 5) | 1 (20) | 2 (40) | 2 (40) | ||||||
| Asian or Pacific Islander (n = 9) | 5 (55.6) | 4 (44.4) | |||||||
| Other or Unknown (n = 3) | 1 (33.3) | 2 (66.7) | |||||||
| Pyromark Gel (n = 162) | |||||||||
| Female (n = 63) | 2 (3.2) | 24 (38.1) | 29 (46) | 7 (11.1) | 1 (1.6) | ||||
| Male (n = 99) | 46 (46.5) | 5 (5.1) | 33 (33.3) | 13 (13.1) | 2 (2) | ||||
| Caucasian (non-Hispanic) (n = 113) | 56 (49.6) | 47 (41.6) | 10 (8.8) | ||||||
| Black or African American (n = 32) | 1 (3.1) | 7 (21.9) | 5 (15.6) | 8 (25) | 8 (25) | 3 (9.4) | |||
| Hispanic or Latino (n = 5) | 1 (20) | 2 (40) | 2 (40) | ||||||
| Asian or Pacific Islander (n = 9) | 5 (55.6) | 4 (44.4) | |||||||
| Other or Unknown (n = 3) | 1 (33.3) | 2 (66.7) | |||||||
| DMET (n = 168) 2 | |||||||||
| Female (n = 65) | 28 (43.1) | 30 (46.2) | 7 (10.8) | ||||||
| Male (n = 103) | 55 (53.4) | 34 (33) | 14 (13.6) | ||||||
| Caucasian (non-Hispanic) (n = 117) | 60 (51.3) | 47 (40.2) | 10 (8.5) | ||||||
| Black or African American (n = 32) | 13 (40.6) | 10 (31.3) | 9 (28.1) | ||||||
| Hispanic or Latino (n = 5) | 3 (60) | 2 (40) | |||||||
| Asian or Pacific Islander (n = 11) | 7 (63.6) | 4 (36.4) | |||||||
| Other or Unknown (n = 3) | 1 (33.3) | 2 (66.7) | |||||||
| Pharmacoscan (n = 21) 1,3 | |||||||||
| Female (n = 5) | 1 (20) | 2 (40) | 1 (20) | 1 (20) | |||||
| Male (n = 16) | 2 (12.5) | 5 (31.3) | 6 (37.5) | 3 (18.8) | |||||
| Caucasian (non-Hispanic) (n = 11) | 3 (27.3) | 5 (45.5) | 3 (27.3) | ||||||
| Black or African American (n = 9) | 1 (11.1) | 5 (55.6) | 2 (22.2) | 1 (11.1) | |||||
| Hispanic or Latino (n = 1) | 1 (100) | ||||||||
| Illumina Fraction (%) | Fragment Analysis Fraction (%) | DMET Plus Fraction (%) | Pyrosequencing Fraction (%) | |
|---|---|---|---|---|
| Miscalls only | ||||
| Illumina (n = 163) | x | 163/163 (100) | 154/163 (94.5) | 155/155 (100) |
| Fragment Analysis (n = 163) | x | 154/163 (94.5) | 155/155 (100) | |
| DMET Plus (n = 168) | x | 146/155 (94.2) | ||
| Pyrosequencing (n = 155) * | x | |||
| Miscalls and ambiguous calls | ||||
| Illumina (n = 163) | x | 163/163 (100) | 159/163 (97.5) | 155/162 (95.7) |
| Fragment Analysis (n = 163) | x | 159/163 (97.5) | 155/162 (95.7) | |
| DMET Plus (n = 168) | x | 146/162 (90.1) | ||
| Pyrosequencing (n = 162) * | x |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sissung, T.M.; Barbier, R.H.; Price, D.K.; Plona, T.M.; Pike, K.M.; Mellott, S.D.; Baugher, R.N.; Whiteley, G.R.; Soppet, D.R.; Venzon, D.; et al. Comparison of Eight Technologies to Determine Genotype at the UGT1A1 (TA)n Repeat Polymorphism: Potential Clinical Consequences of Genotyping Errors? Int. J. Mol. Sci. 2020, 21, 896. https://doi.org/10.3390/ijms21030896
Sissung TM, Barbier RH, Price DK, Plona TM, Pike KM, Mellott SD, Baugher RN, Whiteley GR, Soppet DR, Venzon D, et al. Comparison of Eight Technologies to Determine Genotype at the UGT1A1 (TA)n Repeat Polymorphism: Potential Clinical Consequences of Genotyping Errors? International Journal of Molecular Sciences. 2020; 21(3):896. https://doi.org/10.3390/ijms21030896
Chicago/Turabian StyleSissung, Tristan M., Roberto H. Barbier, Douglas K. Price, Teri M. Plona, Kristen M. Pike, Stephanie D. Mellott, Ryan N. Baugher, Gordon R. Whiteley, Daniel R. Soppet, David Venzon, and et al. 2020. "Comparison of Eight Technologies to Determine Genotype at the UGT1A1 (TA)n Repeat Polymorphism: Potential Clinical Consequences of Genotyping Errors?" International Journal of Molecular Sciences 21, no. 3: 896. https://doi.org/10.3390/ijms21030896