Myeloperoxidase and Septic Conditions Disrupt Sphingolipid Homeostasis in Murine Brain Capillaries In Vivo and Immortalized Human Brain Endothelial Cells In Vitro

 , , ,
, , ,  , ,
, ,
Abstract
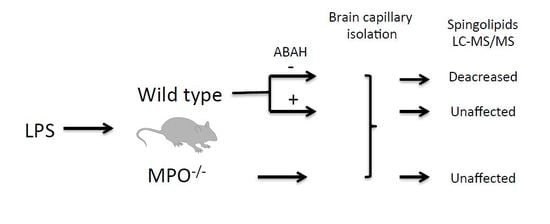
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Animals
4.3. Evans Blue (EB) Extravasation
4.4. Brain Capillary Isolation
4.5. Indirect Calorimetry (Energy Expenditure, Locomotor Activity)
4.6. Human Blood Samples
4.7. Cell Culture
4.8. Gas Chromatography (GC)
4.9. Liquid Chromatography-Electrospray Ionization-Mass Spectroscopy (LC-ESI-MS/MS)
4.10. ELISA
4.11. Electrical Cell-Substrate Impedance Sensing (ECIS)
4.12. Mitochondrial Respiration Measurement
4.13. qPCR Analysis
4.14. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| ABAH | 4-aminobenzoic acid hydrazide |
| ACER | Alkaline ceramidase |
| BBB | Blood–brain barrier |
| BMVEC | Brain microvascular endothelial cells |
| Cer | Ceramide |
| CerS | Ceramide synthase |
| CNS | Central nervous system |
| EAE | Experimental autoimmune encephalomyelitis |
| ECIS | Electric cell-substrate impedance sensing |
| FA | Fatty acid |
| FCCP | Carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone |
| HOCl | Hypochlorous acid |
| HPRT | Hypoxanthine-guanine phosphoribosyltransferase |
| LPP | Lipidphosphate phosphatase |
| LPS | Lipopolysaccharide |
| MPO | Myeloperoxidase |
| OCR | Oxygen consumption rate |
| RER | Respiratory exchange ratio |
| ROS | Reactive oxygen species |
| S1P | Sphingosine-1-phosphate |
| S1PR | Sphingosine-1-phosphate receptor |
| SGMS | Phosphatidylcholine:ceramide cholinephosphotransferase |
| SGPP | Sphingosine-1-phosphate phosphatase |
| SL | Sphingolipid |
| SM | Sphingomyelin |
| SMPD | Sphingomyelin phosphodiesterase |
| SOFA | Sepsis-associated organ failure |
| SPHK | Sphingosine kinase |
| SPT1-3 | Serine palmitoyltransferase 1-3 (gene name SPTLC1-3) |
| SPTSS | Serine palmitoyltransferase small subunit |
| Wt | Wild-type |
References
- Obermeier, B.; Daneman, R.; Ransohoff, R.M. Development, maintenance and disruption of the blood-brain barrier. Nat. Med. 2013, 19, 1584–1596. [Google Scholar] [CrossRef] [PubMed]
- Schenk, G.J.; de Vries, H.E. Altered blood-brain barrier transport in neuro-inflammatory disorders. Drug. Discov. Today Technol. 2016, 20, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Ben-Zvi, A.; Lacoste, B.; Kur, E.; Andreone, B.J.; Mayshar, Y.; Yan, H.; Gu, C. Mfsd2a is critical for the formation and function of the blood-brain barrier. Nature 2014, 509, 507–511. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreone, B.J.; Chow, B.W.; Tata, A.; Lacoste, B.; Ben-Zvi, A.; Bullock, K.; Deik, A.A.; Ginty, D.D.; Clish, C.B.; Gu, C. Blood-Brain Barrier Permeability Is Regulated by Lipid Transport-Dependent Suppression of Caveolae-Mediated Transcytosis. Neuron 2017, 94, 581–594.e5. [Google Scholar] [CrossRef] [Green Version]
- Kuperberg, S.J.; Wadgaonkar, R. Sepsis-Associated Encephalopathy: The Blood-Brain Barrier and the Sphingolipid Rheostat. Front. Immunol. 2017, 8, 597. [Google Scholar] [CrossRef] [Green Version]
- Hannun, Y.A.; Obeid, L.M. Sphingolipids and their metabolism in physiology and disease. Nat. Rev. Mol. Cell Biol. 2018, 19, 175–191. [Google Scholar] [CrossRef]
- Prager, B.; Spampinato, S.F.; Ransohoff, R.M. Sphingosine 1-phosphate signaling at the blood-brain barrier. Trends Mol. Med. 2015, 21, 354–363. [Google Scholar] [CrossRef]
- Castillo, S.S.; Levy, M.; Thaikoottathil, J.V.; Goldkorn, T. Reactive nitrogen and oxygen species activate different sphingomyelinases to induce apoptosis in airway epithelial cells. Exp. Cell Res. 2007, 313, 2680–2686. [Google Scholar] [CrossRef]
- Rapizzi, E.; Taddei, M.L.; Fiaschi, T.; Donati, C.; Bruni, P.; Chiarugi, P. Sphingosine 1-phosphate increases glucose uptake through trans-activation of insulin receptor. Cell Mol. Life Sci. 2009, 66, 3207–3218. [Google Scholar] [CrossRef]
- Jernigan, P.L.; Makley, A.T.; Hoehn, R.S.; Edwards, M.J.; Pritts, T.A. The role of sphingolipids in endothelial barrier function. Biol. Chem. 2015, 396, 681–691. [Google Scholar] [CrossRef]
- Vutukuri, R.; Brunkhorst, R.; Kestner, R.I.; Hansen, L.; Ferreiros Bouzas, N.; Pfeilschifter, J.; Devraj, K.; Pfeilschifter, W. Alteration of sphingolipid metabolism as a putative mechanism underlying LPS-induced BBB disruption. J. Neurochem 2017, 144, 172–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghezzi, P.; Jaquet, V.; Marcucci, F.; Schmidt, H. The oxidative stress theory of disease: Levels of evidence and epistemological aspects. Br. J. Pharmacol 2017, 174, 1784–1796. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M.; Schmeisser, S. Extending life span by increasing oxidative stress. Free Radic Biol. Med. 2011, 51, 327–336. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Patel, M. Targeting Oxidative Stress in Central Nervous System Disorders. Trends Pharmacol. Sci. 2016, 37, 768–778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, H.H.; Stocker, R.; Vollbracht, C.; Paulsen, G.; Riley, D.; Daiber, A.; Cuadrado, A. Antioxidants in Translational Medicine. Antioxid. Redox Signal. 2015, 23, 1130–1143. [Google Scholar] [CrossRef] [Green Version]
- Casas, A.I.; Dao, V.T.; Daiber, A.; Maghzal, G.J.; Di Lisa, F.; Kaludercic, N.; Leach, S.; Cuadrado, A.; Jaquet, V.; Seredenina, T.; et al. Reactive Oxygen-Related Diseases: Therapeutic Targets and Emerging Clinical Indications. Antioxid. Redox Signal. 2015, 23, 1171–1185. [Google Scholar] [CrossRef] [Green Version]
- Klebanoff, S.J. Myeloperoxidase: Friend and foe. J. Leukoc. Biol. 2005, 77, 598–625. [Google Scholar] [CrossRef]
- Palladino, E.N.D.; Hartman, C.L.; Albert, C.J.; Ford, D.A. The chlorinated lipidome originating from myeloperoxidase-derived HOCl targeting plasmalogens: Metabolism, clearance, and biological properties. Arch. Biochem. Biophys. 2018, 641, 31–38. [Google Scholar] [CrossRef]
- Brahmbhatt, V.V.; Hsu, F.F.; Kao, J.L.; Frank, E.C.; Ford, D.A. Novel carbonyl and nitrile products from reactive chlorinating species attack of lysosphingolipid. Chem. Phys. Lipids 2007, 145, 72–84. [Google Scholar] [CrossRef]
- Nusshold, C.; Kollroser, M.; Kofeler, H.; Rechberger, G.; Reicher, H.; Ullen, A.; Bernhart, E.; Waltl, S.; Kratzer, I.; Hermetter, A.; et al. Hypochlorite modification of sphingomyelin generates chlorinated lipid species that induce apoptosis and proteome alterations in dopaminergic PC12 neurons in vitro. Free Radic Biol. Med. 2010, 48, 1588–1600. [Google Scholar] [CrossRef] [Green Version]
- Lewis, A.J.; Seymour, C.W.; Rosengart, M.R. Current Murine Models of Sepsis. Surg. Infect. (Larchmt) 2016, 17, 385–393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhat, O.M.; Yuan, X.; Li, G.; Lee, R.; Li, P.L. Sphingolipids and Redox Signaling in Renal Regulation and Chronic Kidney Diseases. Antioxid. Redox Signal. 2018. [Google Scholar] [CrossRef] [PubMed]
- Dantzer, R.; O’Connor, J.C.; Freund, G.G.; Johnson, R.W.; Kelley, K.W. From inflammation to sickness and depression: When the immune system subjugates the brain. Nat. Rev. Neurosci 2008, 9, 46–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maki, R.A.; Holzer, M.; Motamedchaboki, K.; Malle, E.; Masliah, E.; Marsche, G.; Reynolds, W.F. Human myeloperoxidase (hMPO) is expressed in neurons in the substantia nigra in Parkinson’s disease and in the hMPO-alpha-synuclein-A53T mouse model, correlating with increased nitration and aggregation of alpha-synuclein and exacerbation of motor impairment. Free Radic Biol. Med. 2019, 141, 115–140. [Google Scholar]
- Kalogiannis, M.; Delikatny, E.J.; Jeitner, T.M. Serotonin as a putative scavenger of hypohalous acid in the brain. BBA 2016, 1862, 651–661. [Google Scholar] [CrossRef]
- Becher, B.; Spath, S.; Goverman, J. Cytokine networks in neuroinflammation. Nat. Rev. Immunol. 2017, 17, 49–59. [Google Scholar] [CrossRef]
- Boveri, M.; Kinsner, A.; Berezowski, V.; Lenfant, A.M.; Draing, C.; Cecchelli, R.; Dehouck, M.P.; Hartung, T.; Prieto, P.; Bal-Price, A. Highly purified lipoteichoic acid from gram-positive bacteria induces in vitro blood-brain barrier disruption through glia activation: Role of pro-inflammatory cytokines and nitric oxide. Neuroscience 2006, 137, 1193–1209. [Google Scholar] [CrossRef]
- Doll, D.N.; Hu, H.; Sun, J.; Lewis, S.E.; Simpkins, J.W.; Ren, X. Mitochondrial crisis in cerebrovascular endothelial cells opens the blood-brain barrier. Stroke 2015, 46, 1681–1689. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Liang, Y.; Huang, Z.; Jones, D.W.; Pritchard, K.A., Jr.; Zhang, H. Inhibition of myeloperoxidase oxidant production by N-acetyl lysyltyrosylcysteine amide reduces brain damage in a murine model of stroke. J. Neuroinflamm. 2016, 13, 119. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Zheng, S.; Liang, Y.; Zhang, H. Inhibition of myeloperoxidase by N-acetyl lysyltyrosylcysteine amide reduces oxidative stress-mediated inflammation, neuronal damage, and neural stem cell injury in a murine model of stroke. J. Pharmacol. Exp. Ther. 2017, 364, 311–322. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Ray, A.; Miller, N.M.; Hartwig, D.; Pritchard, K.A., Jr.; Dittel, B.N. Inhibition of Myeloperoxidase at the Peak of Experimental Autoimmune Encephalomyelitis Restores Blood-Brain-Barrier Integrity and Ameliorates Disease Severity. J. Neurochem. 2015, 136, 826–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haddadi, R.; Mohajjel Nayebi, A.; Brooshghalan, S.E. Pre-treatment with silymarin reduces brain myeloperoxidase activity and inflammatory cytokines in 6-OHDA hemi-parkinsonian rats. Neurosci. Lett. 2013, 555, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Wei, Y.; Lee, J.Y.; Wu, Y.; Zheng, Y.; Moskowitz, M.A.; Chen, J.W. Myeloperoxidase Inhibition Increases Neurogenesis after Ischemic Stroke. J. Pharmacol. Exp. Ther. 2016, 359, 262–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forghani, R.; Kim, H.J.; Wojtkiewicz, G.R.; Bure, L.; Wu, Y.; Hayase, M.; Wei, Y.; Zheng, Y.; Moskowitz, M.A.; Chen, J.W. Myeloperoxidase propagates damage and is a potential therapeutic target for subacute stroke. J. Cereb. Blood Flow Metab. 2015, 35, 485–493. [Google Scholar] [CrossRef]
- Kubota, K.; Saiwai, H.; Kumamaru, H.; Maeda, T.; Ohkawa, Y.; Aratani, Y.; Nagano, T.; Iwamoto, Y.; Okada, S. Myeloperoxidase exacerbates secondary injury by generating highly reactive oxygen species and mediating neutrophil recruitment in experimental spinal cord injury. Spine (Phila Pa 1976) 2012, 37, 1363–1369. [Google Scholar] [CrossRef]
- Ullen, A.; Singewald, E.; Konya, V.; Fauler, G.; Reicher, H.; Nusshold, C.; Hammer, A.; Kratky, D.; Heinemann, A.; Holzer, P.; et al. Myeloperoxidase-derived oxidants induce blood-brain barrier dysfunction in vitro and in vivo. PLoS ONE 2013, 8, e64034. [Google Scholar] [CrossRef]
- Phuah, C.L.; Dave, T.; Malik, R.; Raffeld, M.R.; Ayres, A.M.; Goldstein, J.N.; Viswanathan, A.; Greenberg, S.M.; Jagiella, J.M.; Hansen, B.M.; et al. Genetic variants influencing elevated myeloperoxidase levels increase risk of stroke. Brain 2017, 140, 2663–2672. [Google Scholar] [CrossRef] [Green Version]
- Meyer, N.J.; Reilly, J.P.; Feng, R.; Christie, J.D.; Hazen, S.L.; Albert, C.J.; Franke, J.D.; Hartman, C.L.; McHowat, J.; Ford, D.A. Myeloperoxidase-derived 2-chlorofatty acids contribute to human sepsis mortality via acute respiratory distress syndrome. JCI Insight 2017, 2, e96432. [Google Scholar] [CrossRef] [Green Version]
- Brown, K.A.; Brain, S.D.; Pearson, J.D.; Edgeworth, J.D.; Lewis, S.M.; Treacher, D.F. Neutrophils in development of multiple organ failure in sepsis. Lancet 2006, 368, 157–169. [Google Scholar] [CrossRef]
- Carr, A.C.; Spencer, E.; Hoskin, T.S.; Rosengrave, P.; Kettle, A.J.; Shaw, G. Circulating myeloperoxidase is elevated in septic shock and is associated with systemic organ failure and mortality in critically ill patients. Free Radic. Biol. Med. 2019, S0891-5849(19)31559-X. [Google Scholar] [CrossRef]
- Demaret, J.; Venet, F.; Friggeri, A.; Cazalis, M.A.; Plassais, J.; Jallades, L.; Malcus, C.; Poitevin-Later, F.; Textoris, J.; Lepape, A.; et al. Marked alterations of neutrophil functions during sepsis-induced immunosuppression. J. Leukoc. Biol. 2015, 98, 1081–1090. [Google Scholar] [CrossRef] [PubMed]
- Reber, L.L.; Gillis, C.M.; Starkl, P.; Jonsson, F.; Sibilano, R.; Marichal, T.; Gaudenzio, N.; Berard, M.; Rogalla, S.; Contag, C.H.; et al. Neutrophil myeloperoxidase diminishes the toxic effects and mortality induced by lipopolysaccharide. J. Exp. Med. 2017, 214, 1249–1258. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Liu, W.; Huang, Y.; Guo, J.; Zhao, R.; Yang, X. Lipopolysaccharide significantly influences the hepatic triglyceride metabolism in growing pigs. Lipids Health Dis. 2015, 14, 64. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feingold, K.R.; Staprans, I.; Memon, R.A.; Moser, A.H.; Shigenaga, J.K.; Doerrler, W.; Dinarello, C.A.; Grunfeld, C. Endotoxin rapidly induces changes in lipid metabolism that produce hypertriglyceridemia: Low doses stimulate hepatic triglyceride production while high doses inhibit clearance. J. Lipid Res. 1992, 33, 1765–1776. [Google Scholar] [PubMed]
- Khovidhunkit, W.; Kim, M.S.; Memon, R.A.; Shigenaga, J.K.; Moser, A.H.; Feingold, K.R.; Grunfeld, C. Effects of infection and inflammation on lipid and lipoprotein metabolism: Mechanisms and consequences to the host. J. Lipid Res. 2004, 45, 1169–1196. [Google Scholar] [CrossRef] [Green Version]
- Brown, J.A.; Codreanu, S.G.; Shi, M.; Sherrod, S.D.; Markov, D.A.; Neely, M.D.; Britt, C.M.; Hoilett, O.S.; Reiserer, R.S.; Samson, P.C.; et al. Metabolic consequences of inflammatory disruption of the blood-brain barrier in an organ-on-chip model of the human neurovascular unit. J. Neuroinflamm. 2016, 13, 306. [Google Scholar] [CrossRef] [Green Version]
- Clarke, C.J.; Truong, T.G.; Hannun, Y.A. Role for neutral sphingomyelinase-2 in tumor necrosis factor alpha-stimulated expression of vascular cell adhesion molecule-1 (VCAM) and intercellular adhesion molecule-1 (ICAM) in lung epithelial cells: p38 MAPK is an upstream regulator of nSMase2. J. Biol. Chem. 2007, 282, 1384–1396. [Google Scholar] [CrossRef] [Green Version]
- Thukkani, A.K.; McHowat, J.; Hsu, F.F.; Brennan, M.L.; Hazen, S.L.; Ford, D.A. Identification of alpha-chloro fatty aldehydes and unsaturated lysophosphatidylcholine molecular species in human atherosclerotic lesions. Circulation 2003, 108, 3128–3133. [Google Scholar] [CrossRef] [Green Version]
- Thukkani, A.K.; Martinson, B.D.; Albert, C.J.; Vogler, G.A.; Ford, D.A. Neutrophil-mediated accumulation of 2-ClHDA during myocardial infarction: 2-ClHDA-mediated myocardial injury. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H2955–H2964. [Google Scholar] [CrossRef]
- Ullen, A.; Fauler, G.; Kofeler, H.; Waltl, S.; Nusshold, C.; Bernhart, E.; Reicher, H.; Leis, H.J.; Wintersperger, A.; Malle, E.; et al. Mouse brain plasmalogens are targets for hypochlorous acid-mediated modification in vitro and in vivo. Free Radic Biol. Med. 2010, 49, 1655–1665. [Google Scholar] [CrossRef] [Green Version]
- Shadyro, O.; Lisovskaya, A.; Semenkova, G.; Edimecheva, I.; Amaegberi, N. Free-radical Destruction of Sphingolipids Resulting in 2-hexadecenal Formation. Lipid Insights 2015, 8, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haegens, A.; Heeringa, P.; van Suylen, R.J.; Steele, C.; Aratani, Y.; O’Donoghue, R.J.; Mutsaers, S.E.; Mossman, B.T.; Wouters, E.F.; Vernooy, J.H. Myeloperoxidase deficiency attenuates lipopolysaccharide-induced acute lung inflammation and subsequent cytokine and chemokine production. J. Immunol. 2009, 182, 7990–7996. [Google Scholar] [CrossRef] [PubMed]
- Jeitner, T.M.; Kalogiannis, M.; Krasnikov, B.F.; Gomolin, I.; Peltier, M.R.; Moran, G.R. Linking Inflammation and Parkinson Disease: Hypochlorous Acid Generates Parkinsonian Poisons. Toxicol. Sci. 2016, 151, 388–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Samanta, S.; Govindaraju, T. Unambiguous Detection of Elevated Levels of Hypochlorous Acid in APP/PS1 Mouse Brain. ACS Chem. Neurosci. 2019, 10, 4847–4853. [Google Scholar] [CrossRef] [PubMed]
- Alexander, J.J.; Jacob, A.; Cunningham, P.; Hensley, L.; Quigg, R.J. TNF is a key mediator of septic encephalopathy acting through its receptor, TNF receptor-1. Neurochem. Int. 2008, 52, 447–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantle, J.L.; Lee, K.H. A differentiating neural stem cell-derived astrocytic population mitigates the inflammatory effects of TNF-alpha and IL-6 in an iPSC-based blood-brain barrier model. Neurobiol. Dis. 2018, 119, 113–120. [Google Scholar] [CrossRef]
- Cheng, Y.; Desse, S.; Martinez, A.; Worthen, R.J.; Jope, R.S.; Beurel, E. TNFalpha disrupts blood brain barrier integrity to maintain prolonged depressive-like behavior in mice. Brain Behav. Immun. 2018, 69, 556–567. [Google Scholar] [CrossRef]
- Wegner, M.S.; Schiffmann, S.; Parnham, M.J.; Geisslinger, G.; Grosch, S. The enigma of ceramide synthase regulation in mammalian cells. Prog. Lipid Res. 2016, 63, 93–119. [Google Scholar] [CrossRef]
- Chan, R.B.; Oliveira, T.G.; Cortes, E.P.; Honig, L.S.; Duff, K.E.; Small, S.A.; Wenk, M.R.; Shui, G.; Di Paolo, G. Comparative lipidomic analysis of mouse and human brain with Alzheimer disease. J. Biol. Chem. 2012, 287, 2678–2688. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, S.E.; Harikumar, K.B.; Hait, N.C.; Allegood, J.; Strub, G.M.; Kim, E.Y.; Maceyka, M.; Jiang, H.; Luo, C.; Kordula, T.; et al. Sphingosine-1-phosphate is a missing cofactor for the E3 ubiquitin ligase TRAF2. Nature 2010, 465, 1084–1088. [Google Scholar] [CrossRef] [Green Version]
- Danielski, L.G.; Giustina, A.D.; Badawy, M.; Barichello, T.; Quevedo, J.; Dal-Pizzol, F.; Petronilho, F. Brain Barrier Breakdown as a Cause and Consequence of Neuroinflammation in Sepsis. Mol. Neurobiol. 2018, 55, 1045–1053. [Google Scholar] [CrossRef] [PubMed]
- Hernandez-Corbacho, M.J.; Salama, M.F.; Canals, D.; Senkal, C.E.; Obeid, L.M. Sphingolipids in mitochondria. Biochim. Biophys. Acta Mol. Cell. Biol. Lipids 2017, 1862, 56–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, N.U.; Linzer, R.W.; Truman, J.P.; Gurevich, M.; Hannun, Y.A.; Senkal, C.E.; Obeid, L.M. Decreased ceramide underlies mitochondrial dysfunction in Charcot-Marie-Tooth 2F. FASEB J. 2018, 32, 1716–1728. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullen, A.; Fauler, G.; Bernhart, E.; Nusshold, C.; Reicher, H.; Leis, H.J.; Malle, E.; Sattler, W. Phloretin ameliorates 2-chlorohexadecanal-mediated brain microvascular endothelial cell dysfunction in vitro. Free Radic Biol. Med. 2012, 53, 1770–1781. [Google Scholar] [CrossRef] [Green Version]
- Singer, M.; Deutschman, C.S.; Seymour, C.W.; Shankar-Hari, M.; Annane, D.; Bauer, M.; Bellomo, R.; Bernard, G.R.; Chiche, J.D.; Coopersmith, C.M.; et al. The Third International Consensus Definitions for Sepsis and Septic Shock (Sepsis-3). JAMA 2016, 315, 801–810. [Google Scholar] [CrossRef]
- Nusshold, C.; Ullen, A.; Kogelnik, N.; Bernhart, E.; Reicher, H.; Plastira, I.; Glasnov, T.; Zangger, K.; Rechberger, G.; Kollroser, M.; et al. Assessment of electrophile damage in a human brain endothelial cell line utilizing a clickable alkyne analog of 2-chlorohexadecanal. Free Radic Biol. Med. 2016, 90, 59–74. [Google Scholar] [CrossRef] [Green Version]
- Sattler, W.; Puhl, H.; Hayn, M.; Kostner, G.M.; Esterbauer, H. Determination of fatty acids in the main lipoprotein classes by capillary gas chromatography: BF3/methanol transesterification of lyophilized samples instead of Folch extraction gives higher yields. Anal. Biochem. 1991, 198, 184–190. [Google Scholar] [CrossRef]
- Knittelfelder, O.L.; Weberhofer, B.P.; Eichmann, T.O.; Kohlwein, S.D.; Rechberger, G.N. A versatile ultra-high performance LC-MS method for lipid profiling. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2014, 951–952, 119–128. [Google Scholar] [CrossRef] [Green Version]
- Weksler, B.B.; Subileau, E.A.; Perriere, N.; Charneau, P.; Holloway, K.; Leveque, M.; Tricoire-Leignel, H.; Nicotra, A.; Bourdoulous, S.; Turowski, P.; et al. Blood-brain barrier-specific properties of a human adult brain endothelial cell line. FASEB J. 2005, 19, 1872–1874. [Google Scholar] [CrossRef]






© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Goeritzer, M.; Bernhart, E.; Plastira, I.; Reicher, H.; Leopold, C.; Eichmann, T.O.; Rechberger, G.; Madreiter-Sokolowski, C.T.; Prasch, J.; Eller, P.; et al. Myeloperoxidase and Septic Conditions Disrupt Sphingolipid Homeostasis in Murine Brain Capillaries In Vivo and Immortalized Human Brain Endothelial Cells In Vitro. Int. J. Mol. Sci. 2020, 21, 1143. https://doi.org/10.3390/ijms21031143
Goeritzer M, Bernhart E, Plastira I, Reicher H, Leopold C, Eichmann TO, Rechberger G, Madreiter-Sokolowski CT, Prasch J, Eller P, et al. Myeloperoxidase and Septic Conditions Disrupt Sphingolipid Homeostasis in Murine Brain Capillaries In Vivo and Immortalized Human Brain Endothelial Cells In Vitro. International Journal of Molecular Sciences. 2020; 21(3):1143. https://doi.org/10.3390/ijms21031143
Chicago/Turabian StyleGoeritzer, Madeleine, Eva Bernhart, Ioanna Plastira, Helga Reicher, Christina Leopold, Thomas O. Eichmann, Gerald Rechberger, Corina T. Madreiter-Sokolowski, Jürgen Prasch, Philipp Eller, and et al. 2020. "Myeloperoxidase and Septic Conditions Disrupt Sphingolipid Homeostasis in Murine Brain Capillaries In Vivo and Immortalized Human Brain Endothelial Cells In Vitro" International Journal of Molecular Sciences 21, no. 3: 1143. https://doi.org/10.3390/ijms21031143