Impaired Innate Immunity Mechanisms in the Brain of Alzheimer’s Disease

 , and
, and
Abstract
:1. Introduction
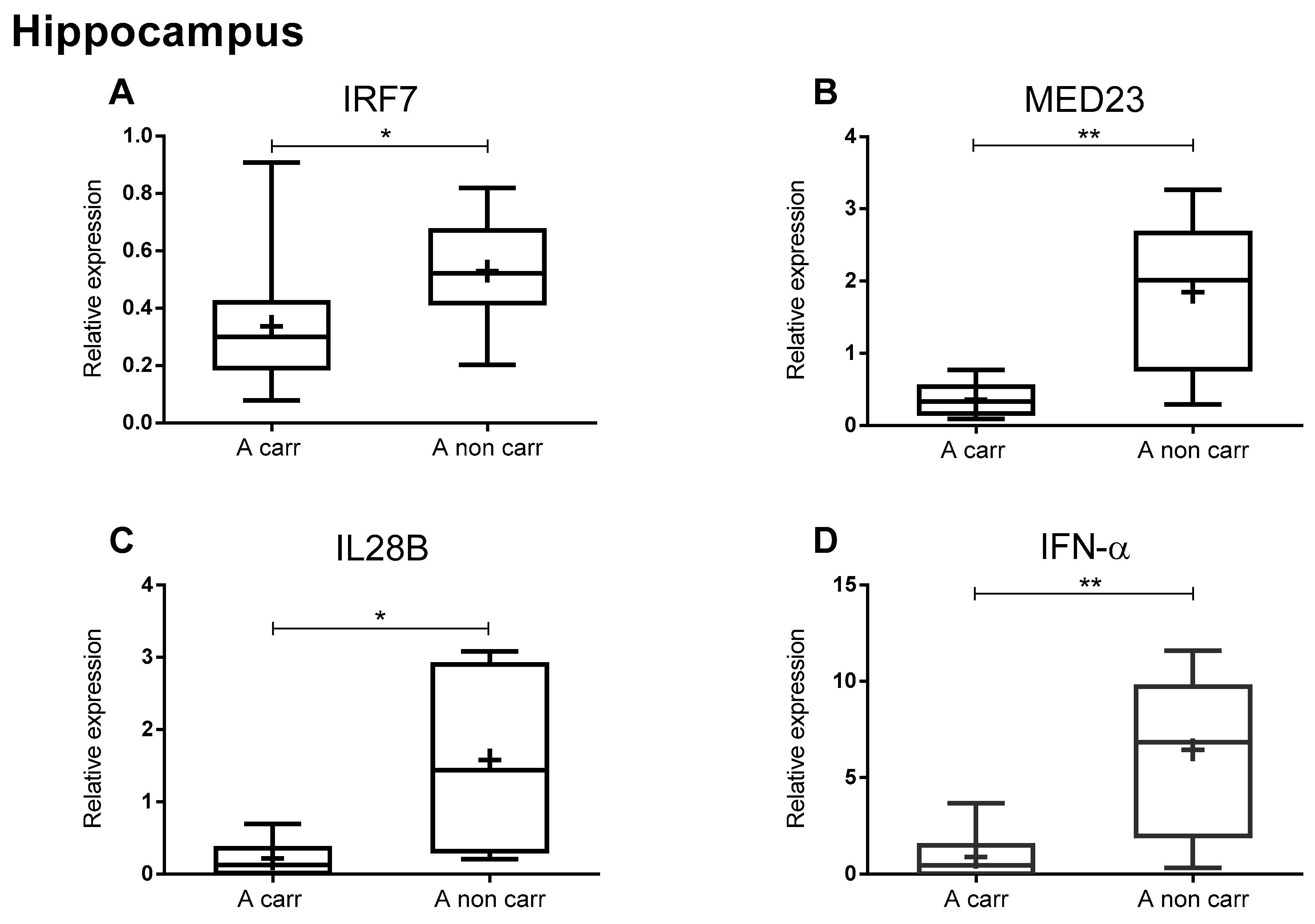
2. Results
3. Discussion
4. Materials and Methods
4.1. Post-Mortem Human Brain Tissue
4.2. Genomic DNA Isolation
4.3. RNA Isolation
4.4. Quantitative Reverse-Transcription Polymerase Chain Reaction
4.5. SNPs Detection
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AD | Alzheimer’s Disease |
| IRF | Interferon Regulatory Factor |
| MED23 | Mediator Complex 23 |
| IL | Interleukin |
| IFN | Interferon |
| APOE | Apolipoprotein |
| Aβ | Amyloid-β |
| AMPs | Antimicrobial Peptides |
| HSV-1 | Herpes Simplex Virus 1 |
| HHV | Human Herpes Virus |
| SNP | Single Nucleotide Polymorphism |
| EBV | Epstein-Barr Virus |
| DD | Disease Duration |
| PMI | Post-Mortem Interval |
| CXCL13 | Chemokine (C-X-C motif) Ligand 13 |
| NBB | Netherlands Brain Bank |
| MTA | Material Transfer Agreement |
| NTFs | NeuroFibrillary Tangles |
| CYC1 | Cytochrome C1 |
| EIF4A2 | Eukaryotic Initiation Factor 4A2 |
| q-PCR | Quantitative PCR assay |
References
- Katzman, R. Editorial: The prevalence and malignancy of Alzheimer disease. A major killer. Arch. Neurol. 1976, 33, 217–218. [Google Scholar] [CrossRef] [PubMed]
- Pawelec, G.; Derhovanessian, E.; Larbi, A.; Strindhall, J.; Wikby, A. Cytomegalovirus and human immunosenescence. Rev. Med. Virol. 2009, 19, 47–56. [Google Scholar] [CrossRef] [PubMed]
- Heppner, F.L.; Ransohoff, R.M.; Becher, B. Immune attack: The role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 2015, 16, 358–372. [Google Scholar] [CrossRef]
- Carbone, I.; Lazzarotto, T.; Ianni, M.; Porcellini, E.; Forti, P.; Masliah, E.; Gabrielli, L.; Licastro, F. Herpes virus in Alzheimer’s disease: Relation to progression of the disease. Neurobiol. Aging 2014, 35, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Licastro, F.; Carbone, I.; Ianni, M.; Porcellini, E. Gene signature in Alzheimer’s disease and environmental factors: The virus chronicle. J. Alzheimers Dis. 2011, 27, 809–817. [Google Scholar] [CrossRef] [PubMed]
- Soscia, S.J.; Kirby, J.E.; Washicosky, K.J.; Tucker, S.M.; Ingelsson, M.; Hyman, B.; Burton, M.A.; Goldstein, L.E.; Duong, S.; Tanzi, R.E.; et al. The Alzheimer’s disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS ONE 2010, 5, e9505. [Google Scholar] [CrossRef]
- Bourgade, K.; Garneau, H.; Giroux, G.; Le Page, A.Y.; Bocti, C.; Dupuis, G.; Frost, E.H.; Fulop, T.J. beta-Amyloid peptides display protective activity against the human Alzheimer’s disease-associated herpes simplex virus-1. Biogerontology 2015, 16, 85–98. [Google Scholar] [CrossRef]
- Readhead, B.; Haure-Mirande, J.V.; Funk, C.C.; Richards, M.A.; Shannon, P.; Haroutunian, V.; Sano, M.; Liang, W.S.; Beckmann, N.D.; Price, N.D.; et al. Multiscale Analysis of Independent Alzheimer’s Cohorts Finds Disruption of Molecular, Genetic, and Clinical Networks by Human Herpesvirus. Neuron 2018. [Google Scholar] [CrossRef] [Green Version]
- Pestka, S. The interferons: 50 years after their discovery, there is much more to learn. J. Biol. Chem. 2007, 282, 20047–20051. [Google Scholar] [CrossRef] [Green Version]
- Egli, A.; Santer, D.M.; O’Shea, D.; Tyrrell, D.L.; Houghton, M. The impact of the interferon-lambda family on the innate and adaptive immune response to viral infections. Emerg. Microbes Infect. 2014, 3, e51. [Google Scholar] [CrossRef]
- Lazear, H.M.; Nice, T.J.; Diamond, M.S. Interferon-lambda: Immune Functions at Barrier Surfaces and Beyond. Immunity 2015, 43, 15–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hermant, P.; Michiels, T. Interferon-lambda in the context of viral infections: Production, response and therapeutic implications. J. Innate Immun. 2014, 6, 563–574. [Google Scholar] [CrossRef]
- Kotenko, S.V. IFN-lambdas. Curr. Opin. Immunol. 2011, 23, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Sheppard, P.; Kindsvogel, W.; Xu, W.; Henderson, K.; Schlutsmeyer, S.; Whitmore, T.E.; Kuestner, R.; Garrigues, U.; Birks, C.; Roraback, J.; et al. IL-28, IL-29 and their class II cytokine receptor IL-28R. Nat. Immunol. 2003, 4, 63–68. [Google Scholar] [CrossRef] [PubMed]
- Bibert, S.; Roger, T.; Calandra, T.; Bochud, M.; Cerny, A.; Semmo, N.; Duong, F.H.; Gerlach, T.; Malinverni, R.; Moradpour, D.; et al. IL28B expression depends on a novel TT/-G polymorphism which improves HCV clearance prediction. J. Exp. Med. 2013, 210, 1109–1116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prokunina-Olsson, L.; Muchmore, B.; Tang, W.; Pfeiffer, R.M.; Park, H.; Dickensheets, H.; Hergott, D.; Porter-Gill, P.; Mumy, A.; Kohaar, I.; et al. A variant upstream of IFNL3 (IL28B) creating a new interferon gene IFNL4 is associated with impaired clearance of hepatitis C virus. Nat. Genet. 2013, 45, 164–171. [Google Scholar] [CrossRef] [PubMed]
- Sommereyns, C.; Paul, S.; Staeheli, P.; Michiels, T. IFN-lambda (IFN-lambda) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS Pathog. 2008, 4, e1000017. [Google Scholar] [CrossRef]
- Ousman, S.S.; Wang, J.; Campbell, I.L. Differential regulation of interferon regulatory factor (IRF)-7 and IRF-9 gene expression in the central nervous system during viral infection. J. Virol. 2005, 79, 7514–7527. [Google Scholar] [CrossRef] [Green Version]
- Khorooshi, R.; Owens, T. Injury-induced type I IFN signaling regulates inflammatory responses in the central nervous system. J. Immunol. 2010, 185, 1258–1264. [Google Scholar] [CrossRef] [Green Version]
- Griffiths, S.J.; Koegl, M.; Boutell, C.; Zenner, H.L.; Crump, C.M.; Pica, F.; Gonzalez, O.; Friedel, C.C.; Barry, G.; Martin, K.; et al. A systematic analysis of host factors reveals a Med23-interferon-lambda regulatory axis against herpes simplex virus type 1 replication. PLoS Pathog. 2013, 9, e1003514. [Google Scholar] [CrossRef] [Green Version]
- Ning, S.; Pagano, J.S.; Barber, G.N. IRF7: Activation, regulation, modification and function. Genes Immun. 2011, 12, 399–414. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osterlund, P.I.; Pietila, T.E.; Veckman, V.; Kotenko, S.V.; Julkunen, I. IFN regulatory factor family members differentially regulate the expression of type III IFN (IFN-lambda) genes. J. Immunol. 2007, 179, 3434–3442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Licastro, F.; Raschi, E.; Carbone, I.; Porcellini, E. Variants in Antiviral Genes are Risk Factors for Cognitive Decline and Dementia. J. Alzheimers Dis. 2015, 46, 655–663. [Google Scholar] [CrossRef] [PubMed]
- Ransohoff, R.M. How neuroinflammation contributes to neurodegeneration. Science 2016, 353, 777–783. [Google Scholar] [CrossRef]
- Fulop, T.; Larbi, A.; Pawelec, G. Human T cell aging and the impact of persistent viral infections. Front. Immunol. 2013, 4. [Google Scholar] [CrossRef] [Green Version]
- Hollingworth, P.; Harold, D.; Sims, R.; Gerrish, A.; Lambert, J.C.; Carrasquillo, M.M.; Abraham, R.; Hamshere, M.L.; Pahwa, J.S.; Moskvina, V.; et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer’s disease. Nat. Genet. 2011, 43, 429–435. [Google Scholar] [CrossRef] [Green Version]
- Lambert, J.C.; Heath, S.; Even, G.; Campion, D.; Sleegers, K.; Hiltunen, M.; Combarros, O.; Zelenika, D.; Bullido, M.J.; Tavernier, B.; et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer’s disease. Nat. Genet. 2009, 41, 1094–1099. [Google Scholar] [CrossRef]
- Licastro, F.; Porcellini, E. Persistent infections, immune-senescence and Alzheimer’s disease. Oncoscience 2016, 3, 135–142. [Google Scholar] [CrossRef] [Green Version]
- Costa Sa, A.C.; Madsen, H.; Brown, J.R. Shared Molecular Signatures Across Neurodegenerative Diseases and Herpes Virus Infections Highlights Potential Mechanisms for Maladaptive Innate Immune Responses. Sci. Rep. 2019, 9, 8795. [Google Scholar] [CrossRef]
- Eimer, W.A.; Vijaya Kumar, D.K.; Navalpur Shanmugam, N.K.; Rodriguez, A.S.; Mitchell, T.; Washicosky, K.J.; Gyorgy, B.; Breakefield, X.O.; Tanzi, R.E.; Moir, R.D. Alzheimer’s Disease-Associated beta-Amyloid Is Rapidly Seeded by Herpesviridae to Protect against Brain Infection. Neuron 2018, 100, 1527–1532. [Google Scholar] [CrossRef] [Green Version]
- Harris, S.A.; Harris, E.A. Herpes Simplex Virus Type 1 and Other Pathogens are Key Causative Factors in Sporadic Alzheimer’s Disease. J. Alzheimers Dis. 2015, 48, 319–353. [Google Scholar] [CrossRef] [Green Version]
- Itzhaki, R.F.; Lathe, R.; Balin, B.J.; Ball, M.J.; Bearer, E.L.; Braak, H.; Bullido, M.J.; Carter, C.; Clerici, M.; Cosby, S.L.; et al. Microbes and Alzheimer’s Disease. J. Alzheimers Dis. 2016, 51, 979–984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, D.K.; Choi, S.H.; Washicosky, K.J.; Eimer, W.A.; Tucker, S.; Ghofrani, J.; Lefkowitz, A.; McColl, G.; Goldstein, L.E.; Tanzi, R.E.; et al. Amyloid-beta peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci. Transl. Med. 2016, 8, 340ra372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moir, R.D.; Lathe, R.; Tanzi, R.E. The antimicrobial protection hypothesis of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 1602–1614. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Geldmacher, D.S.; Herrup, K. DNA replication precedes neuronal cell death in Alzheimer’s disease. J. Neurosci. 2001, 21, 2661–2668. [Google Scholar] [CrossRef]
- Grainger, D.J.; Reckless, J.; McKilligin, E. Apolipoprotein E modulates clearance of apoptotic bodies in vitro and in vivo, resulting in a systemic proinflammatory state in apolipoprotein E-deficient mice. J. Immunol. 2004, 173, 6366–6375. [Google Scholar] [CrossRef] [Green Version]
- Laskowitz, D.T.; Lee, D.M.; Schmechel, D.; Staats, H.F. Altered immune responses in apolipoprotein E-deficient mice. J. Lipid Res. 2000, 41, 613–620. [Google Scholar]
- Akwa, Y.; Hassett, D.E.; Eloranta, M.L.; Sandberg, K.; Masliah, E.; Powell, H.; Whitton, J.L.; Bloom, F.E.; Campbell, I.L. Transgenic expression of IFN-alpha in the central nervous system of mice protects against lethal neurotropic viral infection but induces inflammation and neurodegeneration. J. Immunol. 1998, 161, 5016–5026. [Google Scholar]
- Esen, N.; Rainey-Barger, E.K.; Huber, A.K.; Blakely, P.K.; Irani, D.N. Type-I interferons suppress microglial production of the lymphoid chemokine, CXCL13. Glia 2014, 62, 1452–1462. [Google Scholar] [CrossRef] [Green Version]
- Kovarik, P.; Castiglia, V.; Ivin, M.; Ebner, F. Type I Interferons in Bacterial Infections: A Balancing Act. Front. Immunol. 2016, 7, 652. [Google Scholar] [CrossRef] [Green Version]
- Lambert, J.C.; Grenier-Boley, B.; Chouraki, V.; Heath, S.; Zelenika, D.; Fievet, N.; Hannequin, D.; Pasquier, F.; Hanon, O.; Brice, A.; et al. Implication of the immune system in Alzheimer’s disease: Evidence from genome-wide pathway analysis. J. Alzheimers Dis. 2010, 20, 1107–1118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerriero, F.; Sgarlata, C.; Francis, M.; Maurizi, N.; Faragli, A.; Perna, S.; Rondanelli, M.; Rollone, M.; Ricevuti, G. Neuroinflammation, immune system and Alzheimer disease: Searching for the missing link. Aging Clin. Exp. Res. 2016. [Google Scholar] [CrossRef] [PubMed]
- Miklossy, J. Handbook of Infection and Alzheimer’s Disease. In Advances in Alzheimer’s Disease Ser 5; Fairfax: Amsterdam, The Netherlands, 2017. [Google Scholar]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef] [PubMed]
- Braak, H.; Braak, E. Staging of Alzheimer’s disease-related neurofibrillary changes. Neurobiol. Aging 1995, 16, 271–278. [Google Scholar] [CrossRef]
- Penna, I.; Vella, S.; Gigoni, A.; Russo, C.; Cancedda, R.; Pagano, A. Selection of candidate housekeeping genes for normalization in human postmortem brain samples. Int. J. Mol. Sci. 2011, 12, 5461–5470. [Google Scholar] [CrossRef] [Green Version]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| ID | Age (Years) | Gender | Clinical Diagnosis | DD | PMI (h) | BW (gr) | Cause of Death | Brain Area |
|---|---|---|---|---|---|---|---|---|
| 1 | 78 | F | MID | 7 | 4.35 | 1043 | Pneumonia, cachexia, decubitus | Hip; Temp |
| 2 | 78 | F | AD | 10 | 4.50 | 1105 | Gastrointestinal bleeding | Hip; Temp |
| 3 | 81 | M | AD | 5 | 4.50 | 1253 | Probable CVA and sepsis with unknown underlying disease | Hip |
| 4 | 95 | F | MID possibly mixed with AD | 7 | 3.40 | 1032 | Aspiration pneumonia | Hip |
| 5 | 86 | M | MID | 6 | 3.40 | 1206 | Metabolic disturbances, dehydration, metastasized prostate carcinoma | Hip; Temp |
| 6 | 84 | F | MID | na | 3.40 | 1109 | Dehydration, cachexia | Hip; Temp |
| 7 | 92 | F | VD | 3 | 3.40 | 1115 | Bleeding gastric ulcer | Hip |
| 8 | 73 | F | AD | 12 | 3.40 | 1082 | Sepsis, cachexia | Hip |
| 9 | 67 | F | presenile AD | 15 | 3.40 | 945 | Cachexia | Hip; Temp |
| 10 | 93 | M | AD | 1 | 3.40 | 1210 | na | Hip |
| 11 | 93 | M | MID | 9 | 3.40 | 1220 | Cardiac failure caused by pneumonia | Hip |
| 12 | 69 | M | na | na | 3.40 | 1241 | Dehydration, cachexia | Hip |
| 13 | 89 | F | AD | 11 | 3.40 | 1211 | na | Hip |
| 14 | 84 | F | AD | na | 3.40 | 1098 | Dehydration | Hip |
| 15 | 91 | F | AD | 5 | 3.40 | 1101 | Dehydration, cachexia, and probable CVA | Hip; Temp |
| 16 | 93 | F | AD | 4 | 2.30 | 1045 | Cachexia | Hip; Temp |
| 17 | 86 | F | AD | 10 | 5.05 | 998 | Uncontrolled anti-coagulation in combination with general deterioration after hip prosthesis | Hip |
| 18 | 94 | F | VD after CVA | 6 | 5.05 | 1170 | Cachexia and dehydration, decubitus | Hip |
| 19 | 75 | F | AD | 9 | 6.00 | 1129 | Dehydration | Hip |
| 20 | 89 | F | VD | 19 | 4.30 | 1185 | Pneumonia | Hip; Temp |
| 21 | 74 | M | AD | 2 | 5.35 | 1380 | Anemia, hepatic dysfunction, extensive lymphadenopathy by M. Kimura | Hip |
| 22 | 75 | F | AD | 6 | 15.0 | 1230 | Cardiac arrest | Hip |
| 23 | 94 | F | AD | 6 | 8.05 | 1053 | Cachexia | Hip |
| 24 | 90 | F | AD | 6 | 5.40 | 1100 | CVA, advanced AD | Hip |
| 25 | 88 | F | na | 1 | 6.45 | 1170 | Natural death | Hip |
| 26 | 66 | F | AD | 6 | 6.30 | 1190 | Acute heart failure by advanced dementia syndrome | Hip |
| 27 | 82 | F | VD | 6 | 5.55 | 1225 | Cardiac arrest with dehydration after MI | Hip |
| 28 | 99 | F | AD | 5 | 3.30 | 1150 | Airway infection | Hip |
| 29 | 60 | M | AD possibly of Lewy Body type | 3 | 6.15 | 1241 | Cachexia and dehydration by dementia syndrome | Hip; Temp |
| 30 | 81 | F | AD | 2 | 6.10 | 1295 | Cachexia and dehydration by dementia syndrome | Temp |
| 31 | 57 | M | presenile AD | 6 | 3.50 | 1055 | Aspiration pneumonia | Temp |
| 32 | 69 | M | VD, CI | 3 | 7.10 | 1173 | Aspiration pneumonia | Temp |
| 33 | 86 | M | AD | 6 | 6.15 | 1331 | Possible cardiac arrest after gastroenteritis by AD | Temp |
| 34 | 92 | F | AD | 5 | 3.25 | 1105 | Probable CVA | Temp |
| 35 | 88 | F | AD | 12 | 12.15 | 935 | Cachexia and decubitus | Temp |
| 36 | 85 | F | MID | 5 | 4.45 | 1310 | Shock due to acute abdomen | Temp |
| 37 | 94 | F | na | na | 5.00 | 1410 | Double side pneumonia | Temp |
| 38 | 80 | M | na | na | 4.00 | 1328 | Unknown | Temp |
| 39 | 85 | M | AD possibly in combination with MID | 5 | 4.25 | 1458 | Sudden death (suspected heart-failure) | Temp |
| ID | Pathological Diagnosis | Grade (Braak, NTF) | Grade (Thal, Aβ) |
|---|---|---|---|
| 1 | AD | 5 | C |
| 2 | AD | 5 | C |
| 3 | AD | 4 | C |
| 4 | AD; meningioma | 4 | C |
| 5 | AD | 3 | A/B |
| 6 | AD | 5 | C |
| 7 | AD; infarction | 4 | C |
| 8 | AD | 5 | C |
| 9 | AD | 6 | C |
| 10 | AD | 5 | C |
| 11 | AD; caa; arg | 4 | C |
| 12 | AD | 6 | C |
| 13 | AD | 5 | C |
| 14 | AD; infarction | 5 | C |
| 15 | AD | 4 | C |
| 16 | AD | 4 | C |
| 17 | AD | 4 | C |
| 18 | AD; infarction | 4 | C |
| 19 | AD; caa | 5 | C |
| 20 | AD | 6 | C |
| 21 | AD | 6 | C |
| 22 | AD | 5 | C |
| 23 | AD | 4 | C |
| 24 | AD; infarction | 6 | C |
| 25 | AD | 5 | C |
| 26 | AD | 5 | C |
| 27 | AD | 4 | C |
| 28 | AD | 4 | C |
| 29 | AD | 6 | C |
| 30 | AD | 5 | C |
| 31 | AD | 6 | C |
| 32 | AD; infarction | 5 | B |
| 33 | AD | 5 | C |
| 34 | AD; ischemia | 5 | C |
| 35 | AD | 4 | C |
| 36 | AD | 5 | C |
| 37 | AD | 4 | C |
| 38 | AD | 4 | C |
| 39 | AD | 5 | B/C |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romagnoli, M.; Porcellini, E.; Carbone, I.; Veerhuis, R.; Licastro, F. Impaired Innate Immunity Mechanisms in the Brain of Alzheimer’s Disease. Int. J. Mol. Sci. 2020, 21, 1126. https://doi.org/10.3390/ijms21031126
Romagnoli M, Porcellini E, Carbone I, Veerhuis R, Licastro F. Impaired Innate Immunity Mechanisms in the Brain of Alzheimer’s Disease. International Journal of Molecular Sciences. 2020; 21(3):1126. https://doi.org/10.3390/ijms21031126
Chicago/Turabian StyleRomagnoli, Martina, Elisa Porcellini, Ilaria Carbone, Robert Veerhuis, and Federico Licastro. 2020. "Impaired Innate Immunity Mechanisms in the Brain of Alzheimer’s Disease" International Journal of Molecular Sciences 21, no. 3: 1126. https://doi.org/10.3390/ijms21031126